

Summary
The serine/threonine protein phosphatase-5 (PP5) regulates multiple cellular signaling networks. A number of cellular factors, including heat shock protein-90 (Hsp90) promote the activation of PP5. However, it is unclear whether post-translational modifications also influence PP5 phosphatase activity. Here, we show an “on/off switch” mechanism for PP5 regulation. The casein kinase-1 δ (CK1δ) phosphorylates T362 in the catalytic domain of PP5, which activates and enhances phosphatase activity independent of Hsp90. Overexpression of the phosphomimetic T362E-PP5 mutant hyperdephosphorylates the substrates such as the co-chaperone Cdc37 and the glucocorticoid receptor in cells. Our proteomic approach identified the tumor suppressor von Hippel-Lindau protein (VHL) to interact and ubiquitinate K185/K199-PP5 for proteasomal degradation in a hypoxia- and prolyl hydroxylation-independent manner. Finally, VHL-deficient clear cell renal cell carcinoma (ccRCC) cell lines and patient tumors exhibit elevated PP5 levels. Down-regulation of PP5 causes ccRCC cells to undergo apoptosis, suggesting a prosurvival role for PP5 in kidney cancer.
Keywords: Serine/threonine phosphatase-5, molecular chaperone, co-chaperone, heat shock protein-90, clear cell renal cell carcinoma
eTOC
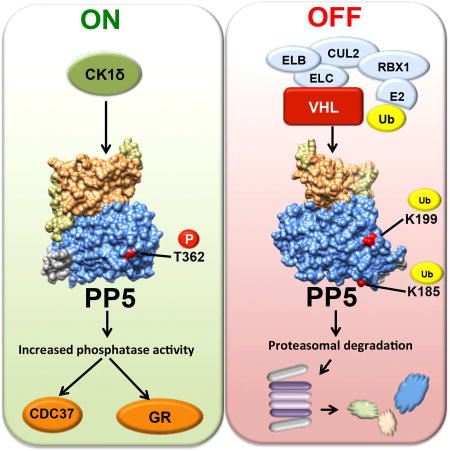
Dushukyan et al. show that casein kinase-1-δ phosphorylates and activates protein phosphatase-5 (PP5), whereas von Hippel-Lindau protein (VHL) ubiquitinates and degrades PP5 in the proteasome. Kidney cancer cells with mutations and inactivation of VHL have elevated PP5. Down-regulation of PP5 causes apoptosis, demonstrating a prosurvival function for PP5 in kidney cancer.
Introduction
The serine/threonine protein phosphatase-5 (PP5) belongs to the phosphoprotein phosphatase (PPP) family. Unlike other phosphatases within this family, PP5 is encoded by a single gene and its regulatory and catalytic domains are all contained within the same polypeptide (Shi, 2009). Recently, a positive correlation between PP5 expression levels and breast cancer metastasis has been identified (Golden et al., 2008a), indicating that PP5 may act as an oncogene during carcinogenesis. Also, recent work has also shown the importance of PP5 in colorectal cancer cell growth (Wang et al., 2015).
PP5 generally has low basal activity because the tetratricopeptide repeat (TPR) motif at its amino-terminus interacts with the αJ-helix in the carboxy-terminus. This prevents substrates from entering the active site of PP5 (Cliff et al., 2006; Haslbeck et al., 2015a; Kang et al., 2001; Ramsey and Chinkers, 2002; Yang et al., 2005). Activation of PP5 depends on binding of its TPR-domain to the molecular chaperone Heat Shock Protein-90 (Hsp90) and client substrates. Other cellular factors such as polyunsaturated fatty acids also activate PP5 (Chatterjee et al., 2010; Ramsey and Chinkers, 2002; Yang et al., 2005).
The majority of PP5 substrates are in complex with Hsp90 and include the glucocorticoid receptor (GR), tumor suppressor p53 and the co-chaperone Cdc37 (Vaughan et al., 2008; Zuo et al., 1998; Zuo et al., 1999). PP5 also functions as a co-chaperone of Hsp90 (Haslbeck et al., 2015b; Vaughan et al., 2008; Wandinger et al., 2006; Xu et al., 2012) and its dephosphorylation of Cdc37 in complex with Hsp90 activates kinase clients. Hsp90 and its co-chaperones are subject to post-translational modifications (PTMs) (reviewed here, (Mayer and Le Breton, 2015; Walton-Diaz et al., 2013; Woodford et al., 2016a)). However, it is unclear if PP5 is also subject to any PTMs and how this impacts its phosphatase activity in cells. In this study we identified CK1δ to phosphorylate T362 in the catalytic domain of PP5, which is involved in its activation and hyperactivity, independent of Hsp90 binding. We have also identified the tumor suppressor von Hippel-Lindau protein (VHL) to target K185/K199 for ubiquitination and proteasomal degradation of PP5 in a hypoxia independent manner. Mutation and inactivation of VHL has been associated with clear cell renal cell carcinoma (ccRCC) (Cancer Genome Atlas Research, 2013). In the absence of VHL, PP5 was upregulated and hyperphosphorylated in ccRCC cells. Small interfering RNA (siRNA) mediated silencing of PP5 induced apoptosis in VHL-null ccRCC, suggesting a prosurvival role for PP5 in these cells.
Results
CK1δ phosphorylates T362 in the catalytic domain of PP5
Previous work has shown that PP5 interacts with casein kinase 1 (CK1) (Partch et al., 2006). We used this information and showed that yeast, Hrr25 (homolog of mammalian serine/threonine kinase casein kinase 1 delta (CK1δ)) phosphorylates Ppt1 (yeast PP5) on both serine and threonine residues (Figure S1A). This was achieved by co-expressing C-terminally His6-tagged Ppt1 under its native promoter and C-terminally cMyc-tagged Hrr25 under the galactose-inducible promoter of GAL1 in yeast. Threonine and serine phosphorylation of Ppt1-His6 was detectable by immunoblotting using pan-anti-phosphothreonine-P6623 and anti-phosphoserine-P5754 (Sigma-Aldrich) antibodies (Figure S1A). Growing the yeast cells in galactose, hence over-expression of Hrr25-cMyc led to hyperphosphorylation of Ppt1 on both threonine and serine sites (Figure S1A). We next asked if similar phenomena also occur with the human CK1δ and human PP5. Amino-terminal FLAG-tagged PP5 (PP5-FLAG) was transiently expressed in HEK293 cells. Using anti-FLAG M2 affinity gel, PP5-FLAG was immunoprecipitated and co-immunoprecipitation of CK1δ was observed by immunoblotting (Figure 1A). A similar experiment was also conducted by transiently co-transfecting HEK293 cells with PP5-FLAG and N-terminally cMyc-tagged CK1δ. Immunoprecipitation of PP5-FLAG led to co-immunoprecipitation of CK1δ-cMyc (Figure 1B). These data show that CK1δ interacts with PP5.
Figure 1. CK1δ phosphorylates T362-PP5. See also Figure S1.

A) Empty vector (EV) or PP5-FLAG plasmids were transiently expressed and immunoprecipitated (IP) from HEK293 cells. Co-immunoprecipitation (Co-IP) of endogenous CK1δ was examined by immunoblotting. Empty vector (EV) was used as a control.
B) PP5-FLAG and CK1δ-cMyc were transiently co-expressed in HEK293 cells. PP5-FLAG was IP and Co-IP of CK1δ-cMyc was assessed by immunoblotting. Empty vector (EV) was used as a control.
C) Recombinant PP5-His6 was used in an in vitro kinase assay. CK1δ-GST phosphorylates only threonine residues on PP5-His6. Phosphorylation was examined by immunoblotting with anti-phosphoserine or phosphothreonine antibodies. SE (short exposure) and LE (long exposure) of the radiographic film.
D) Schematic representation of PP5 with highlighted CK1δ consensus sequence E/DXXT.
E) Potential CK1δ targeted threonine sites on PP5 are highlighted on the cartoon of representation of the PP5 protein, modeled with UCSF Chimera software (PDB:1WAO). Color-coded as in Figure 1D.
F) PP5-threonine residues within the CK1δ consensus sequence were mutated individually to alanine (A), transiently expressed, and IP from HEK293 cells. Threonine phosphorylation was detected by immunoblotting with anti-phosphothreonine antibody. This experiment was repeated with co-transfection of CK1δ-cMyc with the phospho-PP5-FLAG mutants in HEK293 cells. PP5-FLAG and its mutants were isolated and threonine phosphorylation was assessed by immunoblotting with anti-phosphothreonine antibody. SE (short exposure) and LE (long exposure) of the radiographic film.
G) Recombinant PP5-His6 and T362A-PP5-His6 were used as substrates of CK1δ-GST in an in vitro kinase assay. Phosphorylation was assessed by immunoblotting with anti-phosphoserine or phosphothreonine antibodies.
Our previous work has shown PP5-mediated dephosphorylation of the co-chaperone Cdc37 is essential for activation of the kinase clients of Hsp90 (Vaughan et al., 2008). We first established whether CK1δ is an Hsp90 client by treating HEK293 cells with 1 µM Hsp90 inhibitor ganetespib (GB) over time. This did not impact the stability of CK1δ (Figure S1B), therefore it is unlikely that CK1δ is a client of Hsp90. We next examined if CK1δ directly phosphorylates PP5 in an in vitro kinase assay. Bacterially expressed and purified PP5-His6 was bound to Ni-NTA agarose and then incubated with active CK1δ-GST (glutatione S-transferase). Using immunoblotting and anti-phosphothreonine-P6623 (Sigma-Aldrich) antibody, it was possible to detect threonine phosphorylation of PP5 (Figure 1C). Unlike in yeast, CK1δ does not phosphorylate any serine sites on PP5 (Figure 1C). There are seven mammalian CK1 isoforms, however CK1δ and CK1ε display the highest homology (Schittek and Sinnberg, 2014). We repeated our in vitro kinase assay with PP5-His6 and CK1ε-GST. Surprisingly, CK1ε did not phosphorylate PP5 on threonine or serine sites (Figure S1C).
Generally, the CK1 consensus phosphorylation site is S/Tp-X-X-S/T, where S/Tp refers to phospho-serine or phospho-threonine priming sites, X refers to any amino acid, and the underlined residues refer to the target site (Flotow et al., 1990). We did not identify any possible threonine phosphorylation site using this consensus motif in PP5. CK1 also phosphorylates a related unprimed site, D/E-X-X-S/T, where underlined residues refer to the phosphorylated amino acid (Flotow et al., 1990). We identified five threonine residues within this CK1 consensus site (Figure 1D); T33, T121, T171, T238 and T362. With the exception of T238, all the identified threonine sites are located on the surface of PP5 (Figure 1E). These sites were individually mutated to non-phosphorylatable alanine in the PP5-FLAG construct and transiently co-expressed with or without CK1δ-cMyc in HEK293 cells. PP5-FLAG was then immunoprecipitated with anti-FLAG M2 affinity gel and threonine phosphorylation was detected by immunoblotting using anti-phosphothreonine-P6623 (Sigma-Aldrich) antibody. Overexpression of CK1δ-cMyc increased threonine phosphorylation of wild-type PP5-FLAG and the non-phosphorylatable threonine mutants except for T362A-PP5-FLAG (Figures 1F and S1D). The threonine phosphorylation of this mutant was significantly lower, even with overexpression of CK1δ-cMyc (Figures 1F and S1D), suggesting that T362 is phosphorylated by CK1δ. We obtained further evidence by carrying out an in vitro kinase assay with bacterially expressed and purified wild-type PP5-His6 and the non-phosphorylatable mutant T362A-PP5-His6. We were unable to detect the threonine phosphorylation of this mutant by immunoblotting (Figure 1G) therefore confirming CK1δ targets and phosphorylates only T362 in PP5.
T362 phosphorylation activates and increases PP5 activity independent of Hsp90
To determine the impact of T362 phosphorylation on activation and activity of PP5, we initially tested the ability of the T362-PP5 phospho-mutants to dephosphorylate paranitrophenyl phosphate (pNPP), a commonly used small molecule for assaying nonspecific phosphatase activity (Oberoi et al., 2016). Our data showed that the bacterially expressed and purified wild-type-PP5-His6 had basal phosphatase activity and addition of Hsp90α stimulated this activity (Figures S2A and S2B). The phosphomimetic T362E-PP5-His6 or CK1δ mediated phosphorylation of T362-PP5-His6 stimulated PP5 activity in the absence of Hsp90α (Figures S2A and S2B). We repeated the same experiments instead using the non-phosphorylating T362A-PP5-His6. This mutant had a similar basal phosphatase activity as the wild-type-PP5-His6 suggesting that the mutation did not structurally affect the PP5 activity. However, addition of Hsp90α or CK1δ (attempting to phosphorylate T362A-PP5-His6 in vitro) did not stimulate the phosphatase activity (Figures S2A and S2B). We next used an in vitro dephosphorylation assay of phospho-S13-Cdc37, which is a bona fide substrate of PP5 (Vaughan et al., 2008). We first showed that addition of Hsp90α to wild-type PP5 leads to complete dephosphorylation of phospho-S13-Cdc37 after 30 min (Figures 2A and B). However, addition of Hsp90α to phosphomimetic T362E-PP5-His6 (Figure 2A) or phospho-T362-PP5-His6 (Figure 2B) led to dephosphorylation of Cdc37 after only 10 min. To determine whether activation of phospho-T362-PP5 is independent of Hsp90, we repeated the above experiment in the absence of Hsp90α. Our data revealed that wild-type PP5 is unable to dephosphorylate phospho-S13-Cdc37 in the absence of Hsp90α (Figures 2C and D). However, the phosphomimetic T362E-PP5-His6 (Figure 2C) or CK1δ mediated phosphorylation of T362-PP5 (Figure 2D) dephosphorylated phospho-S13-Cdc37 in vitro even in the absence of Hsp90.
Figure 2. CK1δ mediated phosphorylation of PP5 activates and increases the rate of phosphatase activity. See also Figure S2.

A) Dephosphorylation of phospho-S13-Cdc37 with a recombinant wild-type PP5-His6 and phosphomimetic T362E-PP5-His6, in the presence of Hsp90α. Rate of Cdc37 dephosphorylation was assessed by immunoblotting with a phospho-specific S13-Cdc37 antibody over time (minutes).
B) Recombinant wild-type PP5-His6 was phosphorylated by CK1δ in vitro and then used in the dephosphorylation of phospho-S13-Cdc37 in vitro. The assay was performed in presence of Hsp90α. PP5 activity was assessed with immunoblotting using a phospho-specific S13-Cdc37 antibody over time (minutes).
C) Dephosphorylation of phospho-S13-Cdc37 with recombinant wild-type PP5-His6 and phosphomimetic T362E-PP5-His6 was performed in the absence of Hsp90α. Activity was assessed with immunoblotting using a phospho-specific S13-Cdc37 antibody over time (minutes).
D) Recombinant wild-type PP5-His6 was phosphorylated by CK1δ in vitro and then used in the dephosphorylation of phospho-S13-Cdc37 in vitro without Hsp90α. PP5 activity was assessed with immunoblotting using a phospho-specific S13-Cdc37 antibody over time (minutes).
E) Recombinant PP5-His6 was used in an in vitro kinase assay with CK1δ-GST. PP5-His6 was immunoprecipitated (IP) and threonine phosphorylation of PP5 as well as co-immunoprecipitation (Co-IP) of CK1δ-GST were examined by immunoblotting with anti-phosphothreonine and anti-GST antibodies.
F) Recombinant wild-type PP5-His6 was phosphorylated by CK1δ in vitro in the presence (+) or absence (−) of ATP. PP5-His6 proteins were then used in the dephosphorylation of phospho-S13-Cdc37 in vitro without Hsp90α. PP5 activity was assessed with immunoblotting using a phospho-specific S13-Cdc37 antibody over time (minutes).
G) PP5-FLAG and T362-PP5 phosphomutants (T362A and T362E) were transiently transfected in HEK293 cells. Cdc37, phospho-S13-Cdc37, GR and phospho S211-GR protein levels were examined by immunoblotting. Empty vector (EV) was used as a control, GAPDH was used a loading control.
H) Wild-type PP5-FLAG, non-phosphorylating T362A-PP5-FLAG and the phosphomimetic T362E-PP5-FLAG were transiently expressed and IP from HEK293 cells. Co-IP of GR, Cdc37 and Hsp90 were examined by immunoblotting.
To ascertain whether CK1δ interaction with PP5 caused an increase in its phosphatase activity, CK1δ was incubated with PP5-His6 in the presence and absence of ATP. CK1δ has the ability to interact with PP5-His6 independent of ATP (Figure 2E). However only CK1δ incubation with PP5-His6 in the presence of ATP, hence phosphorylation of PP5, is capable of its activation. This leads to dephosphorylation of PP5 substrate, (i.e. phospho-S13-Cdc37) (Figure 2F). Taken together, these in vitro data suggest that CK1δ mediated phosphorylation of T362-PP5 causes activation and hyperactivity of PP5 phosphatase independent of binding to Hsp90.
To obtain further evidence for this observation we overexpressed wild-type PP5-FLAG and the phospho-mutants T362A and T362E in HEK293 cells. The dephosphorylation of PP5 substrates, phospho-S13-Cdc37 and phospho-S211-glucocorticoid receptor (GR), were examined by immunoblotting. Overexpression of wild-type PP5-FLAG led to dephosphorylation of phospho-S13-Cdc37 and phospho-S211-GR (Figure 2G). This effect was enhanced with overexpression of the phosphomimetic T362E-PP5-FLAG and unaffected (similar to the empty vector control) with T362A-PP5-FLAG, which is consistent with our in vitro data (Figure 2G). Finally, we confirmed that the interaction of the phospho-T362 mutants with Hsp90, Cdc37 and GR were not affected, and therefore our observation is not due to lack of interaction of PP5 mutants with the substrates (Figure 2H). Our in vitro and in vivo data here show that phosphorylation of T362-PP5 is involved in both activation and hyperactivity of PP5. Furthermore, based on our in vitro results, CK1δ mediated phosphorylation and activation of PP5 does not depend on binding to Hsp90.
VHL E3 ligase targets PP5 independent of hypoxia
To determine the binding partners of the PP5 that may be involved in its regulation, we immunoprecipitated the endogenous PP5 from HEK293 cells and identified its intracellular binding proteins by mass spectrometry (MS) analysis (described in Experimental Procedures). Our interactome data identified VHL as a binding partner of PP5 (Table S1). VHL forms a multi-protein complex VCB-Cul2 (VHL-Elongin C-Elongin B-Cullin-2) and Rbx1 that acts as a ubiquitin-ligase (E3) (Gossage et al., 2015; Kamura et al., 1999; Stebbins et al., 1999) and directs proteasome-dependent degradation of targeted proteins such as hypoxia-inducible factors (HIF1α or HIF2α) (Kamura et al., 2000). VHL is expressed as two known isoforms; VHL30, with an apparent molecular weight of ~24 to 30 kDa (VHL30), and VHL19, roughly 19 kDa in size (Iliopoulos et al., 1995). Both isoforms appear to retain tumor suppressor activity, however VHL30 is the commonly examined isoform. Here, we confirmed our interactome data by immunoprecipitating endogenous PP5 from HEK293 cells and detecting VHL30 (Figure 3A). The reciprocal immunoprecipitation of the endogenous VHL30 yielded PP5 (Figure 3B). Since VHL is the substrate recognition subunit of an E3 ubiquitin ligase, we overexpressed VHL30 in the VHL deficient clear cell renal cell carcinoma (ccRCC) cell line, 786-O, and observed down-regulation of endogenous PP5 by immunoblotting (Figure 3C). We next transiently transfected and expressed PP5-FLAG in the 786-O cell line. Using immunoprecipitation and immunoblotting, we were unable to detect PP5 ubiquitination in these cells (Figure 3D), even when treated with 50 nM proteasome inhibitor bortezomib (BZ) for 2 hr (Figure 3D). However, transient co-expression of PP5-FLAG and VHL30-His6 in 786-O cells for 16 hr and subsequent treatment with 50 nM BZ for 2 hr led to detection of distinct ubiquitination bands, suggesting multi-monoubiquitination of PP5-FLAG (Figure 3D). It is noteworthy that anti-ubiquitin antibody (P4D1) used in these experiments recognizes both mono and polyubiquitination. Taken together, these data suggest that VHL E3 ligase ubiquitinates and degrades PP5 in the proteasome.
Figure 3. VHL E3 ligase ubiquitinates PP5 independent of hypoxia. See also Figure S3.

A) Endogenous PP5 was immunoprecipitated (IP) from HEK293 cells and co-immunoprecipitation (Co-IP) of VHL30 was assessed by immunoblotting.
B) Endogenous VHL30 was IP from HEK293 cells and Co-IP of PP5 was examined by immunoblotting.
C) VHL30-FLAG or empty vector (EV) was transiently over-expressed in 786-O cells and endogenous PP5 protein levels were assessed by immunoblotting. GAPDH was used a loading control.
D) The 786-O cells transiently expressing PP5-FLAG, were treated with or without 50 nM proteasome inhibitor bortezomib (BZ) for 2 hr. PP5-FLAG was also co-expressed with VHL30-His6 with additional treatment of 50 nM BZ for 2 hr. PP5-FLAG was IP and its ubiquitination was assessed by immunoblotting.
E) Egln1-HA, Egln2-HA, Egln3-HA and empty vector pcDNA3.1 were transiently over-expressed in HEK293 cells. The expression of Egln1, 2 and 3 as well as PP5, and HIF1α were assessed by immunoblotting with anti-HA, anti-PP5 and anti- HIF1α antibodies. GAPDH was used a loading control.
F) Caki-1 cells cultured in normoxia and hypoxia (1%O2, 5%CO2, 94%N2). PP5 and HIF1α protein levels were examined by immunoblotting using anti-PP5 and anti-HIF1α antibodies. GAPDH was used a loading control.
G) HIF1α or HIF2α were silenced by small interfering RNA (siRNA) in HEK293 cells. HIF1α, HIF2α and PP5 protein levels were examined by immunoblotting using anti-HIF1α, anti-HIF2α and anti-PP5 antibodies. GAPDH was used a loading control.
Canonically, VHL substrates are recognized when hydroxylated by prolyl hydroxylases (PHDs) at specific proline sites (Ivan et al., 2001; Jaakkola et al., 2001). To determine whether hydroxylation was essential for VHL-mediated ubiquitination of PP5, we first overexpressed three EGLN genes (EGLN1, 2 and 3) encoding for three isoforms of PHDs in HEK293 cells. Overexpression of these genes did not affect the endogenous PP5 protein levels, but as expected markedly reduced HIF1α levels (Figure 3E). Treatment of HEK293 cells with the PHD inhibitor dimethyloxaloylglycine (DMOG) or hypoxia mimetic deferoxamine (DFX) or CoCl2 similarly did not affect PP5 protein levels (Figure S3A). As expected, these conditions all led to stabilization of HIF1α (Figure S3A). We obtained a similar result when we treated the Caki-1 ccRCC cell line (containing wild-type VHL gene) with DMOG, DFX or CoCl2 (Figure S3B). Finally, we examined PP5 protein levels in Caki-1 cells cultured in normoxia and hypoxia (1%O2, 5%CO2, 94%N2). While HIF1α levels increased under hypoxia, PP5 levels were similar to normoxia (Figure 3F). Finally, previous work has indicated that transcription of PP5 can be mediated by HIF1 (Zhou et al., 2004). We examined this possibility by using small interfering RNA (siRNA) to silence HIF1α or HIF2α in HEK293 cells. Our results showed that neither HIF1α nor HIF2α are involved in regulation of PP5 (Figure 3G). We conducted a similar experiment in 786-O ccRCC cells and silenced only HIF2α (HIF1α is down-regulated in these cells). Our data also showed that HIF2α is not involved in PP5 expression in VHL-null 786-O cells (Figure S3C). These data indicate that VHL ubiquitinates PP5 independent of prolyl hydroxylation and hypoxia.
VHL ubiquitinates K185/K199-PP5
Based on the data available on PhosphoSitePlus® (www.phosphosite.org), which is an online systems biology resource providing comprehensive information on PTMs of human proteins, we identified five ubiquitinated lysine sites (K32, K40, K185, K199 and K320) on PP5 (Figure 4A). These sites are all located on the surface of the PP5 protein (Figure 4B). We individually mutated these lysine sites to a non-ubiquitinating arginine residue and transiently expressed them in HEK293 cells (Figure 4C). Ubiquitination of the wild-type PP5 was detectable by immunoprecipitation and immunoblotting techniques (Figures 4C and S4A). However, ubiquitination of PP5 was significantly reduced in the K185R and K199R mutants (Figure 4C). Our data also showed that K185R and K199R-PP5-FLAG mutants were slightly more stable than the wild-type PP5-FLAG expressed in HEK293 cells (Figure S4B), whereas these mutants were expressed at the same levels as the wild-type PP5-FLAG in the VHL-null 786-O cells (Figure S4C).
Figure 4. VHL-mediated ubiquitination and proteasomal degradation of K185/K199-PP5. See also Figure S4.

A) Schematic representation of PP5 with highlighted lysine residues possibly subject to VHL mediated ubiquitination.
B) Potential ubiquitinating lysine residues are highlighted on the cartoon representation of the PP5 protein modeled with UCSF Chimera software (PDB:1WAO). Color-coded as in Figure 1D.
C) Wild-type PP5-FLAG and its potentially non-ubiquitinating lysine mutants were transiently expressed and immunoprecipitated (IP) from HEK293 cells. Ubiquitination of PP5 was examined by immunoblotting. Empty vector (EV) was used as a control.
D) Wild-type PP5-FLAG and its non-ubiquitinating K185/K199R mutant were transiently expressed and IP from HEK293 cells. Ubiquitination of PP5 was examined by immunoblotting anti-ubiquitin antibody. Empty vector (EV) was used as a control.
E) Recombinant PP5-His6 and K185/K199R double mutant were used in an in vitro ubiquitination assay with VCB-Cul2 (VHL30-Elongin C-Elongin B-Cullin-2) and Rbx1, which acts as an ubiquitin-ligase (E3). Ubiquitination of PP5 was assessed with immunoblotting.
F) PP5-FLAG or K185/K199R-PP5-FLAG double mutant were co-transfected with VHL30-His6 in 786-O cells and then after 24 hr treated with 50 nM BZ for 2 hr. PP5-FLAG or K185/K199R-PP5-FLAG was IP and ubiquitination was assessed by immunoblotting.
G) PP5-FLAG, K185-PP5-FLAG, K199R-PP5-FLAG, K185/K199R-PP5-FLAG and empty vector (EV) were individually and transiently expressed in HEK293 cells. They were IP and their interactions with VHL30 were assessed in the Co-IP by immunoblotting.
We next created a K185R/K199R-PP5-FLAG double mutant and transiently expressed it in HEK293 cells. Ubiquitination of this mutant was undetectable by immunoprecipitation and immunoblotting experiments (Figures 4D and S4D). To determine if VHL is responsible for ubiquitination of these sites, we used an in vitro ubiquitination assay kit (Millipore) with the VCB-Cul2 (VHL30-Elongin C-Elongin B-Cullin-2) complex (Figure S4E). As mentioned earlier, VHL is part of a multi-protein complex, VCB-Cul2 and Rbx1, acting as a ubiquitin-ligase (E3) and directing proteasome dependent degradation of targeted proteins. Next, bacterially expressed and purified wild-type PP5-His6 and K185R/K199R-PP5-His6 double mutant were used in our in vitro ubiquitination assay. Our data show that the recombinant wild-type PP5-His6 but not the K185R/K199R-PP5-His6 mutant were subject to ubiquitination. Based on the immunoblots and the appearance of the bands, our data suggest that PP5 is subject to multi-monoubiquitination (Figure 4E), therefore suggesting that VHL targets K185/K199-PP5 for ubiquitination. To gain further evidence of VHL-mediated ubiquitination of K185/K199-PP5 in cells, we transiently co-expressed the wild-type PP5-FLAG or K185R/K199R-PP5-FLAG with VHL30-His6 for 16 hr in VHL-null 786-O cells, then treated them with 50nM BZ for 2 hr. We were only able to detect the ubiquitination of the wild-type PP5 but not the K185R/K199R-PP5 double mutant (Figure 4F). The K185R-, K199R-PP5 single or double mutants interacted with the same affinity as the wild-type PP5 to VHL30 (Figure 4G). Therefore the reduced ubiquitination of these mutants could not be due to their inability to bind to VHL30.
Taken together, our data show that VHL E3 ligase ubiquitinates K185 and K199 residues in PP5.
PP5 down-regulation causes apoptosis in VHL null ccRCC cells
To gain further insight into the significance of PP5 stability and hyperactivity in ccRCC, we first examined PP5 protein levels in tumors and adjacent normal tissues from nine patients with ccRCC. Within 10 min of removal of tumors by radical or partial nephrectomy, tumors and adjacent normal tissues were dissected into 10 mm3 pieces followed by staining with haematoxylin and eosin (H&E) (Figure 5A) or protein extraction (Figure 5B). Our data showed that VHL30 is absent in the ccRCC tumors. Conversely PP5 and CK1δ were both upregulated in the tumors compared to the adjacent normal tissues (Figure 5B).
Figure 5. Downregulation of PP5 induces apoptosis in VHL deficient ccRCC cells. See also Figure S5.

A) Clear cell renal cell carcinoma (ccRCC) tumors, (T) and adjacent normal tissues (N) were stained with haematoxylin and eosin (H&E). Bar scale represents 200µm.
B) Proteins were extracted from above tumors and adjacent normal tissues and expression of PP5, CK1δ, VHL30 and Hsp90 was examined by immunoblotting. Hsp90 was used a loading control.
C) PP5, Cdc37 and phosphorylated S13-Cdc37, GR and phosphorylated S211-GR, CK1δ and VHL30 proteins from ccRCC cell lines 786-O, A-498 (VHL-deficient) and Caki-1 (VHL-containing) were assessed by immunoblotting. GAPDH was used as a loading control.
D) PP5 was silenced by siRNA in 786-O and Caki-1 cells. siCtrl represents the non-targeting siRNA control. Induction of apoptotic markers shown by immunoblotting using anti-cleaved caspase-3 and cleaved-PARP antibodies. GAPDH was used a loading control.
E) Targeted siRNA was used to silence PP5 in A498 cells. siCtrl represents the non-targeting siRNA control. Induction of apoptotic markers shown by immunoblotting using anti-cleaved caspase-3 and cleaved-PARP antibodies. PP5 protein levels were also examined by immunoblotting. GAPDH was used as a control.
F) PP5 was immunoprecipitated (IP) from ccRCC cell lines, Caki-1 and 786-O, lysates using anti-PP5 antibody or IgG (control). Threonine phosphorylation of PP5 was assessed by immunoblotting with anti-phosphothreonine antibody. GAPDH was used a loading control.
G) PP5 was isolated from the lysates of 786-O cell treated with 2 µM IC261 for 16 hr using anti-PP5 or IgG (control) antibodies. Threonine phosphorylation of PP5 was examined by immunoblotting using anti-phosphothreonine antibody. Anti-phosphoserine antibody was used as a control.
We confirmed this data by using established VHL-deficient (786-O and A-498) and VHL-containing (Caki-1) ccRCC cell lines. Our data showed that PP5 was upregulated in the 786-O and A-498 cell lines compared to Caki-1 cells. CK1δ levels displayed a similar expression pattern, while CK1ε levels were unchanged between the VHL-deficient and VHL-containing ccRCC cell lines (Figures 5C and S5A). This pattern was consistent with dephosphorylation of PP5 substrates, Cdc37 and GR. These were both more dephosphorylated in 786-O and A-498 cells compared to the VHL-containing Caki-1 cells (Figure 5C). We next used siRNA to silence PP5 in 786-O and Caki-1 cells and observed induction of the pro-apoptotic markers cleaved caspase-3 and cleaved-poly-ADP-ribose polymerase (PARP) only in the VHL-null 786-O cells (Figure 5D). We also showed that silencing of PP5 in 786-O cells did not affect the CK1δ protein levels (Figure S5B). Furthermore, siRNA silencing of PP5 in A-498 cells (VHL-null cell line) also led to induction of the apoptotic markers cleaved caspase-3 and cleaved-PARP (Figure 5E).
To evaluate the threonine phosphorylation status of PP5 in the VHL-null cells, we immunoprecipitated the endogenous PP5 from 786-O and Caki-1 cells and showed that PP5 from VHL-null 786-O cells was hyperphosphorylated on threonine residues (Figure 5F). IC261 is a potent specific inhibitor of CK1δ/ε, we next examined whether pharmacologic inhibition of CK1δ, reduces threonine phosphorylation of PP5. 786-O cells treated or untreated with 2 µM IC261 16 hr and PP5 was immunoprecipitated and as expected, IC261 treatment led to a marked reduction of PP5 threonine phosphorylation but not serine phosphorylation (Figure 5G). Taken together, our data show that PP5 is upregulated, and also phosphorylated by CK1δ in VHL-null ccRCC cells. Downregulation of PP5 in VHL-null cells causes apoptosis.
PP5 upregulation provides a pro-survival mechanism in VHL null ccRCC cells
We next examined whether pharmacologic inhibition of CK1δ, causing apoptosis in VHL-null cells; A498 and 786-O. We used Caki-1 cells as a control since this ccRCC cell line has the VHL gene. These cell lines were treated with different amounts of IC261, which is a potent specific inhibitor of CK1δ/ε. Increasing amounts of IC261 correlated with increased induction of the pro-apoptotic markers cleaved caspase-3 and cleaved-PARP only in A498 and 786-O (VHL-null) cells (Figure 6A). Another hallmark of apoptosis is the loss of cell membrane integrity, which can be monitored by annexin V/propidium iodide (AV/PI) staining. We treated A498, 786-O and Caki-1 cells with 0.5µM and 2.0µM IC261 for 16 hr prior to analysis of apoptosis by AV/PI staining. Treatment of the A498 and 786-O cells with 2.0µM IC261 resulted in cells progressing through the AV+/PI− apoptotic quadrant (Figure 6B and S6A). However, this effect was not observed in Caki-1 cells following the same treatment with IC261 (Figure 6B and S6A). We next examined the effects of IC261 on proliferation of ccRCC cell lines. A498, 786-O and Caki-1 cells were treated with different amounts of CK1δ inhibitor, IC261. After 72 hr, proliferation was measured by the 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assay. Our data revealed that 2.0µM IC261 significantly inhibited the proliferation of A498, 786-O cells compared to Caki-1 cells (Figure 6C). A hallmark of tumorigenic cells is their anchorage-independent growth property, which is measurable by soft agar assay. Treating the A498, 786-O cells with 2.0µM IC261 significantly reduced the anchorage-independent growth of these cells compared to the Caki-1 cell line (Figure 6D and S6B).
Figure 6. CK1δ inhibition induces apoptosis and reduced proliferation in VHL-null ccRCC cells. See also Figure S6.

A) ccRCC cell lines A498, 786-O and Caki-1 were treated with indicated amounts of CK1δ inhibitor IC261 for 16 hr and induction of apoptotic markers shown by immunoblotting using anti-cleaved caspase-3 and cleaved-PARP antibodies. GAPDH was used a loading control.
B) AV/PI graphs of Caki-1, A498 and 786-O cells untreated (0µM) or treated with 2µM IC261 for 16hr for 2 hr. The top left quadrants represent dead cells stained only with PI. The bottom right quadrants represent apoptotic cells stained only with AV. The top right quadrants represent cells stained with both PI and AV (secondary necrosis and late apoptosis). Percentage of each stained cell population is indicated. Dot plots shown are representative of one of three independent experiments.
C) MTT assay of A498, 786-O and Caki-1 cells treated with indicated amounts of IC261. The errors bars represent the SD of three independent experiments (****p < 0.0001).
D) Anchorage-independent growth of A498, 786-O and Caki-1 cells treated with indicated amounts of IC261 in soft agar. Colony number was quantified. The errors bars represent the SD of three independent experiments (***p < 0.0005).
E) Wild-type PP5-FLAG, non-phosphorylating T362A-PP5-FLAG and the phosphomimetic T362E-PP5-FLAG were transiently expressed in 786-O cells. Cells were then either untreated (−IC261) or treated (+IC261) with 2 µM IC261 for 16 hr and induction of apoptotic markers shown by immunoblotting using anti-cleaved caspase-3 and cleaved-PARP antibodies. GAPDH was used a loading control.
F) MTT assay of 786-O cells transiently expressing wild-type PP5-FLAG, T362APP5-FLAG and T362E-PP5-FLAG and then treated with indicated amounts of IC261. The errors bars represent the SD of three independent experiments (**p < 0.005 and ***p < 0.0005).
Although treatment of VHL-null cells with IC261 caused apoptosis, it is unclear if this effect was due to lack of PP5 phosphorylation and activity. We addressed this question by transiently expressing wild-type PP5-FLAG and its phospho-PP5 mutants T362A and T362E in 786-O cells. Treating the cells expressing empty vector (EV) or non-phosphorylatable T362A-PP5-FLAG mutant with 2.0µM IC261 led to induction of the pro-apoptotic markers cleaved caspase-3 and cleaved-PARP. This effect was significantly reduced upon expression of the wild-type PP5-FLAG and completely abrogated with the phosphomimetic T362E-PP5-FLAG mutant (Figure 6E). We further confirmed this data by using the MTT assay. Our data showed that treating the 786-O cells transiently expressing EV or non-phosphorylatable T362A-PP5-FLAG mutant with 2.0µM IC261 caused a marked reduction in cell proliferation where as this effect was not observed in 786-O cells expressing wild-type PP5-FLAG and phospho-mutant T362E-PP5 mutants treated with 2.0µM IC261 (Figure 6F and S6C).
Taken together, Pharmacologic inhibition of CK1δ caused apoptosis in ccRCC cells, and this effect is through lack of threonine phosphorylation of PP5 and reduced phosphatase activity.
Discussion
PP5 plays a key role in the regulation of both hormone- and stress-induced signaling networks that allow cells to respond appropriately to genomic stress (Golden et al., 2008b). Structural work and in vitro based assays have shown that PP5 activity is promoted by a number of cellular factors, including the molecular chaperone Hsp90 and fatty acids, both of which release autoinhibition by interacting with the TPR domain of PP5 (Ramsey and Chinkers, 2002; Yang et al., 2005). In this study we elucidated an alternative mechanism for activation and regulation of PP5 both in vitro and in vivo. Our data showed that the serine/threonine kinase CK1δ interacts with and phosphorylates T362 on PP5. This in turn activates PP5 independent of binding to Hsp90. We have also shown that CK1δ is not a client of Hsp90, since pharmacologic inhibition of Hsp90 did not affect the stability of CK1δ. Additionally, binding of CK1δ to PP5, without phosphorylation, is insufficient to activate PP5. Our cell-based assays have demonstrated that expression of the non-phosphorylating T362A-PP5 did not dephosphorylate PP5 substrates such as GR and Cdc37, whereas over-expression of the phosphomimetic T362E-PP5 had the opposite effect on those two substrates. Our findings clearly demonstrate that although phosphorylation of T362-PP5 does not affect the binding of PP5 to its substrates or even Hsp90, the phosphatase activity of PP5 is influenced by T362 phosphorylation (Figure 7).
Figure 7. Post-translational regulation of PP5.

CK1δ mediated phosphorylation of T362 in the catalytic domain of PP5, activates and enhances its phosphatase activity therefore dephosphorylating its substrates such as the co-chaperone Cdc37 and steroid hormone receptor GR. VCB-Cul2 (VHL-Elongin C-Elongin B-Cullin-2), Rbx1 (E3 ubiquitinligase) target and ubiquitinate K185 and K199 on PP5. This leads to proteasomal degradation of PP5.
What is the mechanism of phosphorylation-mediated PP5 activation? Threonine-362 is close to one of the six metal-coordinating sites, H352, but it is remote from the catalytic site. T362 is also at the center of an acidic patch located between D365, D364, and E416, E417. The latter residues are linked to the active site R400. Therefore, it is conceivable that phosphorylation or phosphomimetic mutation of T362 could either destabilize the domain as a whole or cause some local unfolding, possibly allosterically ‘opening’ the active site, therefore increasing the rate of phosphatase activity.
Our proteomic data has also identified the interaction of the tumor suppressor VHL with PP5. VHL is an E3 ligase that canonically recognizes its substrates as part of an oxygen-dependent prolyl hydroxylase (PHD) reaction, with HIFα being its most studied substrate (Clifford et al., 2001; Kondo et al., 2002; Maxwell et al., 1999). Our data revealed that PP5 is a hypoxia-independent target for VHL ubiquitination. We have also shown that VHL ubiquitinates K185/K199-PP5 to target this phosphatase for proteasome-mediated degradation (Figure 7). Our finding of a hypoxia-independent function of VHL is not unusual; recent work has shown that VHL also directly multi-monoubiquitinates Aurora kinase A (AURKA) independent of oxygen or PHD activity (Hasanov et al., 2017).
Mutation and inactivation of VHL is associated with the most common type of kidney cancer, known as ccRCC (Cancer Genome Atlas Research, 2013; Gossage et al., 2015). We further demonstrated that inactivation of VHL in tumors or established cell lines leads to increased PP5 protein levels. This appeared to be independent of HIF1α or HIF2α. Our findings also provided a prosurvival role for PP5 in VHL-null kidney cancer cells. Down-regulation of PP5 by siRNA activated apoptosis in ccRCC cells. This effect was apparent only in the VHL-null ccRCC cells. We also showed that pharmacologic inhibition of CK1δ by IC261 leads to apoptosis and decreased proliferation of ccRCC cells. This effect was completely abrogated by over-expressing the phosphomimetic T362E-PP5 mutant in IC261-treated VHL-null ccRCC cells. Our findings therefore strongly suggest that the apoptotic effect of CK1δ inhibition is through lack of phosphorylation and thus inactivation of PP5 in ccRCC cells. It is worth noting that IC261 is also a potent inhibitor of CK1ε, however CK1ε does not phosphorylate PP5. Although the apoptotic effect of IC261 may be the result of CK1ε inhibition, this possibility is highly unlikely because treating the 786-O cells overexpressing the phosphomimetic T362E-PP5 with IC261 did not induce apoptosis.
Based on previous studies, there is circumstantial evidence suggesting that aberrant expression of PP5 may aid tumor development and progression (Golden et al., 2008a; Wang et al., 2015). Our study here also suggests that upregulation of PP5 is essential for ccRCC survival, but how is this achieved by PP5? Our previous work has shown that PP5-mediated dephosphorylation of the co-chaperone Cdc37 is essential for activation of the kinase client proteins of Hsp90 (Vaughan et al., 2008). Several of these clients, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) receptors as well as the mammalian target of Rapamycin (mTOR) are important in ccRCC (Gossage et al., 2015; Linehan, 2012) and therefore potentially rely on the co-chaperone activity of PP5. Down-regulation of PP5 in these cells appears to be detrimental for the ccRCC survival, perhaps because of compromised function of these kinases. PP5 has been shown to be a negative regulator of the ASK1/MKK4/JNK signaling that promotes apoptosis (Zhou et al., 2004), and its overexpression, under hypoxia, in breast cancer has been shown to turn off this apoptotic signaling pathway. In contrast to the previous study (Zhou et al., 2004), we were unable to show HIF1 or HIF2 involvement in regulation of PP5.
Further investigation is warranted to identify the exact signaling pathways dependent on PP5 function in VHL-null ccRCC cells and whether downregulation of PP5 activates the intrinsic- or extrinsic- apoptotic pathway. These findings could ultimately have therapeutic implications.
Experimental Procedures
Plasmids, Yeast Strains and Yeast Growth Media
Detailed methodologies, a list of primers (Table S2), and media conditions for both yeast and mammalian cells are provided in the Supplemental Information.
Analysis of Human ccRCC Tumors
Tumor and adjacent normal tissues of the patients with conventional ccRCC were obtained with written informed consent from the Department of Urology at SUNY Upstate Medical University and approved by the Institutional Review Board. Patients identity was kept confidential therefore information on gender and age were not available. At the time of radical or partial nephrectomy, which was done with <10 minutes of renal ischemia, ccRCC tumors were dissected into approximately 8 mm3 pieces and protein was extracted and quantified as previously described in detail (Woodford et al., 2016b). The tissues were also stained with haematoxylin and eosin (H&E) and examined by a pathologist.
Protein Extraction, Immunoprecipitation and Immunoblotting
Protein extraction from yeast, mammalian cells and human tissues was carried out using methods previously described by (Mollapour et al., 2010; Woodford et al., 2016b). Detailed methodology is provided in the Supplemental Information.
In Vitro Cdc37 Dephosphorylation Assay
Rate of dephosphorylation of S13-Cdc37 by PP5 was monitored by mixing 5 µM purified phospho-S13-Cdc37 without or with 2.5 µM Hsp90α in a buffer containing 100 mM NaCl, 50 mMTris, pH7.5, 2 mM DTT, and 1 mM MnCl2. The reaction was started by adding 0.25 µM PP5 (wild-type or phospho-mutants or CK1δ-mediated phosphorylated form), and the samples were incubated at 30°C. Samp les were taken every 10 min for SDS/PAGE analysis. The phosphorylation state of S13-Cdc37 was examined by immunoblotting using phospho-Ser13–specific antibody (Abcam).
PP5 Activity Assay with pNPP
PP5 activity was assayed in vitro para-Nitrophenyl Phosphate, pNPP, assay previously described by (Oberoi et al., 2016). Detailed method is provided in the Supplemental Information.
In Vitro Ubiquitination of PP5
Detailed method for in vitro ubiquitination of PP5 is provided in the Supplemental Information.
Annexin V/PI Apoptosis Analysis
Apoptosis was detected by Annexin V/PI immunostaining assay as described by the manufacture kit (BioRad/AbD Serotec). Detailed procedure is presented provided in the Supplemental Information.
MTT Assay
A498, 786-O and Caki-1 cells were plated at 10,000 cells per well in 96-well plates. Cells were treated with 0.1, 0.2, 0.3, 0.5, 1.0 and 2.0 µM IC261. After 72 hr, an MTT assay was performed according to the manufacturer’s protocol (BioVision, Cat# K302-500). Detailed methodology is described in the Supplemental Information.
Soft Agar Colony Formation Assay
Soft agar colony formation assay was performed similar to previously described method (Borowicz et al., 2014). Detailed methodology is found in Supplemental Information.
Quantification and Statistical Analysis
The data presented are the representative or examples of three biological replicates unless it is specified. Data were analyzed with unpaired t-test. Asterisks in figures indicate significant differences (* P < 0.05, ** P < 0.005, *** P < 0.0005, and **** P < 0.0001). Error bars represent the standard deviation (S.D.) for three independent experiments, unless it is indicated.
Supplementary Material
HIGHLIGHTS.
-
-
Casein kinase-1 δ (CK1δ) phosphorylates T362 in the catalytic domain of PP5.
-
-
Phosphorylation activates and enhances phosphatase activity of PP5.
-
-
Von Hippel-Lindau protein (VHL) multi-monoubiquitinates PP5 on K185 and K199.
-
-
PP5 downregulation in VHL-null clear cell renal cell carcinoma causes apoptosis.
Acknowledgments
We would like to thank Dr. Gustavo de la Roza at the Department of Pathology, SUNY Upstate Medical University for his help and expert advice on the tumors. This work was partly supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R01GM124256 (M.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported with funds from the SUNY Upstate Medical University, Upstate Foundation, One Square Mile of Hope Foundation, Carol M. Baldwin Breast Cancer Fund (D.B., M.M.) and in part by the Urology Care Foundation Research Scholar Award Program and American Urological Association (M.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
N.D., D.M.D., R.A.S., M.R.W., D.L., M.D., A.J.B.W., A.W.T and M.M., performed experiments. D.M.D., M.R.W., R.A.S., J.D.C., C.K.V., T.A.H., G.B., D.B., M.M., designed experiments. M.M. wrote the manuscript. M.M. conceived the project.
References
- Borowicz S, Van Scoyk M, Avasarala S, Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK, Winn RA. The soft agar colony formation assay. J Vis Exp. 2014:e51998. doi: 10.3791/51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Wang L, Armstrong DL, Rossie S. Activated Rac1 GTPase translocates protein phosphatase 5 to the cell membrane and stimulates phosphatase activity in vitro. J Biol Chem. 2010;285:3872–3882. doi: 10.1074/jbc.M109.088427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff MJ, Harris R, Barford D, Ladbury JE, Williams MA. Conformational diversity in the TPR domain-mediated interaction of protein phosphatase 5 with Hsp90. Structure. 2006;14:415–426. doi: 10.1016/j.str.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Clifford SC, Astuti D, Hooper L, Maxwell PH, Ratcliffe PJ, Maher ER. The pVHL-associated SCF ubiquitin ligase complex: molecular genetic analysis of elongin B and C, Rbx1 and HIF-1alpha in renal cell carcinoma. Oncogene. 2001;20:5067–5074. doi: 10.1038/sj.onc.1204602. [DOI] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem. 1990;265:14264–14269. [PubMed] [Google Scholar]
- Golden T, Aragon IV, Rutland B, Tucker JA, Shevde LA, Samant RS, Zhou G, Amable L, Skarra D, Honkanen RE. Elevated levels of Ser/Thr protein phosphatase 5 (PP5) in human breast cancer. Biochim Biophys Acta. 2008a;1782:259–270. doi: 10.1016/j.bbadis.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden T, Swingle M, Honkanen RE. The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer metastasis reviews. 2008b;27:169–178. doi: 10.1007/s10555-008-9125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15:55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- Hasanov E, Chen G, Chowdhury P, Weldon J, Ding Z, Jonasch E, Sen S, Walker CL, Dere R. Ubiquitination and regulation of AURKA identifies a hypoxia-independent E3 ligase activity of VHL. Oncogene. 2017 doi: 10.1038/onc.2016.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck V, Drazic A, Eckl JM, Alte F, Helmuth M, Popowicz G, Schmidt W, Braun F, Weiwad M, Fischer G, et al. Selective activators of protein phosphatase 5 target the auto-inhibitory mechanism. Biosci Rep. 2015a;35 doi: 10.1042/BSR20150042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck V, Eckl JM, Drazic A, Rutz DA, Lorenz OR, Zimmermann K, Kriehuber T, Lindemann C, Madl T, Richter K. The activity of protein phosphatase 5 towards native clients is modulated by the middle- and C-terminal domains of Hsp90. Scientific reports. 2015b;5:17058. doi: 10.1038/srep17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos O, Kibel A, Gray S, Kaelin WG., Jr Tumour suppression by the human von Hippel-Lindau gene product. Nat Med. 1995;1:822–826. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Jr, Elledge SJ, Conaway RC, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Sayner SL, Gross KL, Russell LC, Chinkers M. Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry. 2001;40:10485–10490. doi: 10.1021/bi010999i. [DOI] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome research. 2012;22:2089–2100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Le Breton L. Hsp90: breaking the symmetry. Mol Cell. 2015;58:8–20. doi: 10.1016/j.molcel.2015.02.022. [DOI] [PubMed] [Google Scholar]
- Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, Lee MJ, Lee S, Morra G, Bourboulia D, Scroggins BT, et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi J, Dunn DM, Woodford MR, Mariotti L, Schulman J, Bourboulia D, Mollapour M, Vaughan CK. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1603059113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch CL, Shields KF, Thompson CL, Selby CP, Sancar A. Posttranslational regulation of the mammalian circadian clock by cryptochrome and protein phosphatase 5. Proc Natl Acad Sci U S A. 2006;103:10467–10472. doi: 10.1073/pnas.0604138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey AJ, Chinkers M. Identification of potential physiological activators of protein phosphatase 5. Biochemistry. 2002;41:5625–5632. doi: 10.1021/bi016090h. [DOI] [PubMed] [Google Scholar]
- Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014;13:231. doi: 10.1186/1476-4598-13-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, Panaretou B, Neckers L, Clarke PA, Workman P, et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell. 2008;31:886–895. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton-Diaz A, Khan S, Bourboulia D, Trepel JB, Neckers L, Mollapour M. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future medicinal chemistry. 2013;5:1059–1071. doi: 10.4155/fmc.13.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006;25:367–376. doi: 10.1038/sj.emboj.7600930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhu J, Dong M, Yu H, Dai X, Li K. Inhibition of protein phosphatase 5 (PP5) suppresses survival and growth of colorectal cancer cells. Biotechnology and applied biochemistry. 2015;62:621–627. doi: 10.1002/bab.1308. [DOI] [PubMed] [Google Scholar]
- Woodford MR, Dunn D, Miller JB, Jamal S, Neckers L, Mollapour M. Impact of Posttranslational Modifications on the Anticancer Activity of Hsp90 Inhibitors. Adv Cancer Res. 2016a;129:31–50. doi: 10.1016/bs.acr.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Woodford MR, Truman AW, Dunn DM, Jensen SM, Cotran R, Bullard R, Abouelleil M, Beebe K, Wolfgeher D, Wierzbicki S, et al. Mps1 Mediated Phosphorylation of Hsp90 Confers Renal Cell Carcinoma Sensitivity and Selectivity to Hsp90 Inhibitors. Cell reports. 2016b;14:872–884. doi: 10.1016/j.celrep.2015.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Mollapour M, Prodromou C, Wang S, Scroggins BT, Palchick Z, Beebe K, Siderius M, Lee MJ, Couvillon A, et al. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50(CDC37)-AHA1 chaperone machine. Mol Cell. 2012;47:434–443. doi: 10.1016/j.molcel.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Roe SM, Cliff MJ, Williams MA, Ladbury JE, Cohen PT, Barford D. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J. 2005;24:1–10. doi: 10.1038/sj.emboj.7600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Golden T, Aragon IV, Honkanen RE. Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem. 2004;279:46595–46605. doi: 10.1074/jbc.M408320200. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Dean NM, Honkanen RE. Serine/threonine protein phosphatase type 5 acts upstream of p53 to regulate the induction of p21(WAF1/Cip1) and mediate growth arrest. J Biol Chem. 1998;273:12250–12258. doi: 10.1074/jbc.273.20.12250. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Urban G, Scammell JG, Dean NM, McLean TK, Aragon I, Honkanen RE. Ser/Thr protein phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry. 1999;38:8849–8857. doi: 10.1021/bi990842e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.