
Figure 2. DMEs of the whole system are more evenly distributed than the individual component DMEs.
In the experimental system, CI acts as a tight repressor. The distributions of fluorescence are shown in the absence of CI (red) and in the presence of CI (blue). Each distribution was obtained by measuring fluorescence of two independent measurements of 500,000 cells by flow cytometry, which were then pooled together. The dashed lines separate three categories of phenotypes – ‘no expression’ phenotypes (corresponding to repressed wildtype); ‘high expression’ phenotypes (corresponding to the wildtype in the absence of CI); and ‘intermediate’ phenotypes. No expression and high expression categories are defined to include >99.9% of the wildtype fluorescence distribution in the presence and in the absence of CI, respectively. The Shannon entropy (S) is used to estimate how uniform each distribution is across the entire range of possible expression levels. The associated standard deviation (±) is given for each S value. Blue numbers are percentage of counts in each category in the presence of CI. Numbers in parentheses are percentage of counts excluding the estimated percentages of uniquely transformed individuals carrying the wildtype genotype (see Materials and methods). The naïve additive convolution prediction for each system library and the associated predictions for the frequency of mutants in each category are shown in grey. Pearson’s Chi-squared test was used to assess the difference between the observed and the convolution-predicted frequency of mutants in each category (low: χ2(2)=8.20; p<0.05; intermediate: χ2(2)=32.26; p<0.0001; and high mutation frequency library: χ2(2)=74.51; p<0.0001). The distributions of the effects of mutations for the cis-element, the trans-element, and the whole system in the absence of CI are shown in Figure 2—figure supplement 1. Figure 2—figure supplement 2 shows distributions of the effects of 150 single point mutations in the cis- and the trans-elements. Statistical significance of the differences in entropy values between the mutant libraries is shown in Figure 2—source data 3. Flow cytometry measurements of 20 individual isolates from each library are shown in Figure 2—figure supplements 3, 4 and 5, the analysis of which was used to demonstrate that gene expression noise is constant (Figure 2—source data 1). Convolutions for each mutation probability performed with the knowledge of the genetic regulatory structure of the system are shown in Figure 2—figure supplement 6, while Figure 2—figure supplement 7 provides an explanation of how convolutions were performed. The outcome of the test for how sensitive the shapes of distributions are to the number of sampled individuals is shown in Figure 2—source data 4, while the confirmation that the mutagenesis protocol resulted in expected distributions of the number of mutations are shown in Figure 2—source data 2.

Figure 2—figure supplement 1. DMEs for cis-element, trans-element, and system libraries in the absence of CI.

Figure 2—figure supplement 2. Distribution of single mutation effects in 150 random system double mutants and their corresponding single mutants.

Figure 2—figure supplement 3. Mutant isolates from the low mutation probability libraries.

Figure 2—figure supplement 4. Mutant isolates from the intermediate mutation probability libraries.

Figure 2—figure supplement 5. Mutant isolates from the high mutation probability libraries.

Figure 2—figure supplement 6. Mathematical predictions that account for the genetic regulatory structure accurately describe the system DME.
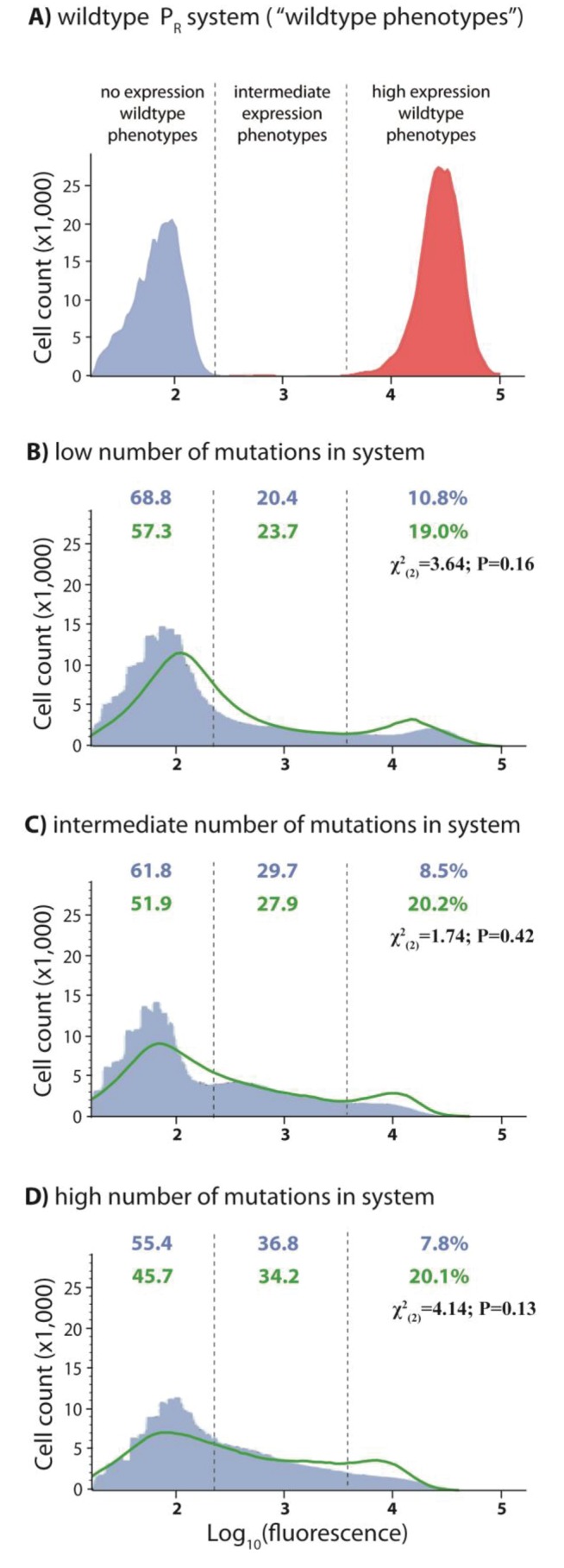
Figure 2—figure supplement 7. Predicting the system DME based on convolving component DMEs.
