
Fig. 8.
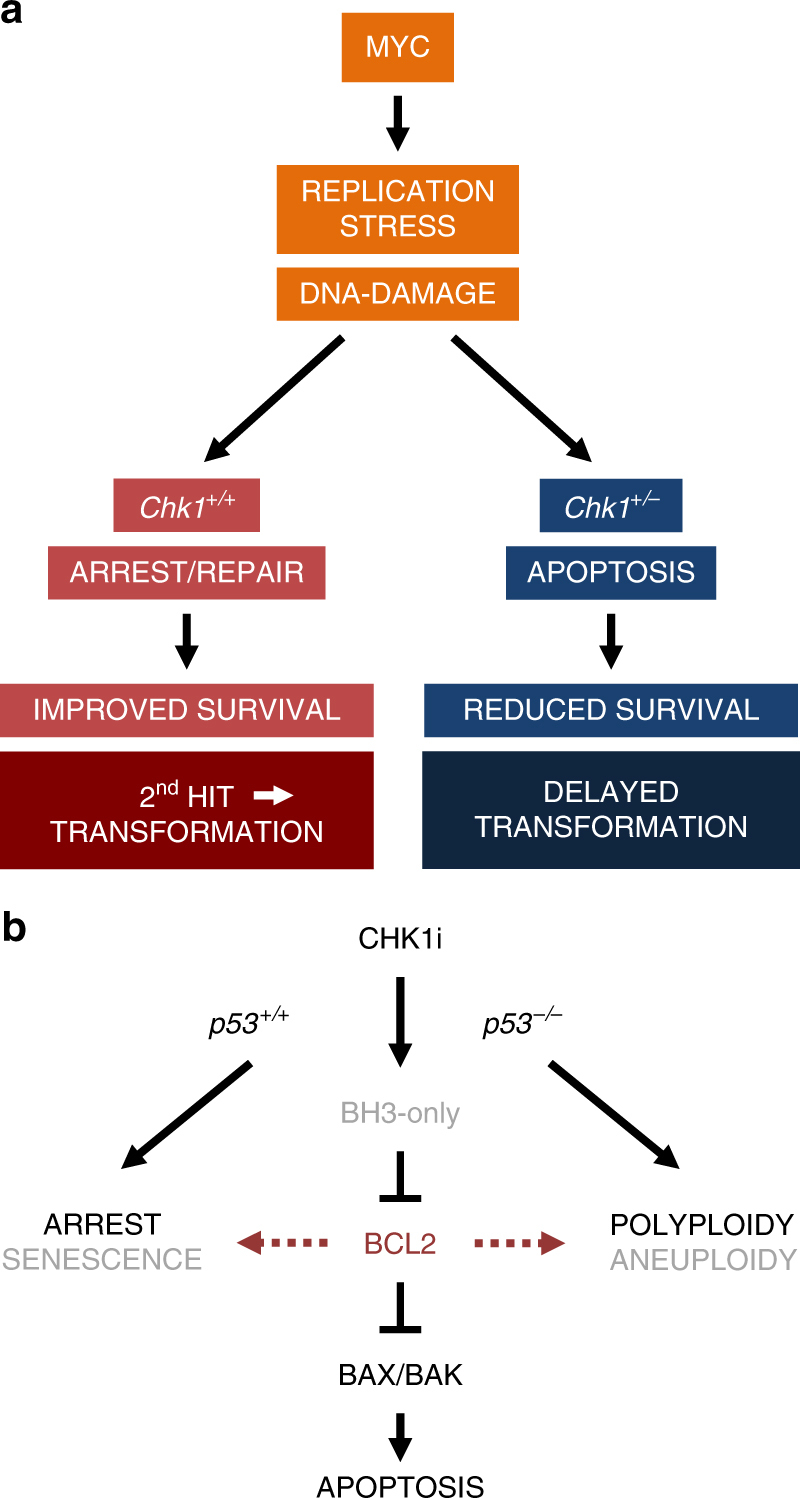
Proposed model of CHK1 as a facilitator of transformation and drug target. a Lymphocytes rely on CHK1 function to control replication fidelity to avoid DNA damage. In response to oncogenic stress, e.g., the one caused by MYC, replication stress and subsequent DNA damage increase the dependence on CHK1 for successful transformation that, in this model, depends on additional genetic alterations (2nd hit). A reduced CHK1 expression level curtails the cell´s ability to effectively deal with oncogene-driven replication stress, leading to increased DNA damage and apoptosis rates, thereby delaying transformation. b CHK1 inhibitors induce cell death in Burkitt lymphoma and ALL cells by engaging the BCL2-regulated BAX/BAK-dependent apoptosis pathway, kick-started by yet to be defined BH3-only proteins. Cell fate in response to inhibitor treatment is fine-tuned by p53 status and intrinsic apoptosis thresholds, controlled by the BCL2 family. Impaired cell death, e.g., due to BCL2 overexpression can deviate the cellular response to p21-mediated cell death, and, potentially senescence, when p53 is functional. Yet, in the absence of p53, cell death and cell cycle incompetence facilitates polyploidy of cancer cells exposed to CHK1 inhibitors. This can drive the selection of complex aneuploidy karyotypes and treatment failure