

Abstract
The general mechanism involved in Tat activation of RNA Polymerase II (RNAP II) elongation of the integrated HIV-1 was elucidated over 20 years ago. This mechanism involves Tat binding to the TAR RNA element that forms at the 5′ end of viral transcripts and recruiting a general RNAP II elongation factor termed P-TEFb. This elongation factor consists of CDK9 and Cyclin T1, and when recruited by Tat to TAR RNA, CDK9 was proposed to phosphorylate the carboxyl terminal domain of RNAP II and thereby activate elongation. Research in the past two decades has shown that the mechanism of Tat action is considerably more complicated than this simple model. In metabolically active cells, CDK9 and Cyclin T1 are now known to be largely sequestered in a RNA-protein complex termed the 7SK RNP. CDK9 and Cyclin T1 are released from the 7SK RNP by mechanisms not yet fully elucidated and along with Tat, bind to TAR RNA and orchestrate the assembly of a Super Elongation Complex (SEC) containing several additional proteins. CDK9 in the SEC then phosphorylates multiple substrates in the RNAP II complex to activate elongation. Importantly for therapeutic strategies, CDK9 and Cyclin T1 function are down-regulated in resting CD4+ T cells that harbor latent HIV-1, and their up-regulation is required for reactivation of latent virus. Current strategies for a functional cure of HIV-1 infection therefore are likely to require development of latency reversal agents that up-regulate CDK9 and Cyclin T1 function in resting CD4+ T cells.
Keywords: HIV-1, Tat, P-TEFb, CDK9, Cyclin T1, HIV latency
Following HIV-1 entry into the host cell, the viral genomic RNA is reversed transcribed into cDNA and integrated into a cellular chromosome. RNA polymerase II (RNAP II) is then recruited to the HIV-1 promoter located in the viral 5′ Long Terminal Repeat (LTR) sequences and the viral proviral genome can be transcribed back into genomic RNA. Over 30 years ago, it was observed that expression of an HIV-1 LTR reporter plasmid was nearly 1,000-fold higher in HIV-infected cells than in control cells1. This potent stimulatory activity of viral gene expression is due to the activity of the HIV-1 Tat protein. There has been intense research over the past three decades into the mechanism of action of Tat and its importance to HIV-1 replication. The first decade of this research was rather contentious as several distinct mechanisms of Tat action were proposed. After this initial decade, however, a consensus emerged about the general mechanism whereby Tat stimulates viral gene expression. Research conducted in the past two decades has elucidated in increasingly greater detail the mechanisms involved in Tat action. This review will first describe studies that made key contributions to our understanding of Tat. As these mechanistic studies were being conducted, it was also observed that Tat function and therefore HIV-1 replication are regulated in CD4+ T cells and monocytes/macrophages – the two predominant cell types infected by HIV-1 in vivo. These studies of the regulation of Tat function will be reviewed, and we will end with a discussion of the significance of this regulation to current efforts to develop a functional cure for infection.
Mechanism of Tat activation of HIV-1 gene expression
The cis-regulatory sequences in the viral LTR that respond to Tat were mapped to sequences downstream from the site of RNAP II transcriptional initiation2, and the major function of Tat was found to stimulate transcriptional elongation rather than initiation3. Using a genetic approach, the cis-regulatory sequences that respond to Tat were shown to function in vivo as a stem-loop RNA element encompassing nucleotides +1 to +59, and this RNA element is termed TAR RNA4. Biochemical experiments subsequently showed that Tat binds directly to TAR RNA in vitro5, and this binding in vivo requires a cellular TAR RNA loop-binding factor6.
After the step in gene expression that is most strongly activated by Tat was elucidated – RNAP II elongation via Tat binding to TAR RNA – the key issue became the identification of cellular factors that mediate this function of Tat. A key finding was that a recombinant Tat protein specifically bound in vitro to a cellular protein kinase activity that was termed TAK for Tat-associated kinase7. TAK was subsequently shown to hyperphosphorylate the carboxyl terminal domain (CTD) of RNAP II, a pattern of phosphorylation implicated in activation of RNAP II elongation8. An analysis of multiple wild type and mutant Tat proteins demonstrated that Tat binding to TAK in vitro is strictly correlated with Tat function in vivo8. Shortly after the publications describing TAK, it was shown that TAK is identical to a general RNAP II elongation factor known as P-TEFb9, and the catalytic subunit of TAK/P-TEFb was identified as CDK9 (originally named PITALRE), a member of the cyclin–dependent protein kinase family9,10. The cyclin regulatory subunit of CDK9 was then identified as Cyclin T111,12, which was shown to be the previously genetically defined factor required for Tat binding to TAR RNA in vivo6. This cellular kinase composed of Cyclin T1 and CDK9 that mediates Tat function is now termed P-TEFb.
Mechanism of Tat activation found to be complex
The studies described above led to a simple model which proposed that Tat merely recruits CDK9 and Cyclin T1 to TAR RNA at the 5′ end viral transcripts, whereupon CDK9 phosphorylates the CTD of RNAP II and elongation is stimulated. However, work from numerous laboratories over the past two decades has revealed that the mechanism of Tat action is much more complex than this simple model. Research in the transcription field identified two multiprotein complexes termed NELF and DSIF that associate with the RNAP II complex and function to inhibit elongation13. Studies in the HIV-1 system found that when P-TEFb and Tat are recruited to the TAR RNA element, CDK9 phosphorylates subunits of both the NELF and DSIF complexes, as well as the CTD of RNAP II, resulting in a potent activation of transcriptional elongation14,15.
Biochemical studies further revealed that P-TEFb exists in three distinct complexes. The first complex is “Core P-TEFb” and it is composed of CDK9, CyclinT1, and Brd4; Brd4 contains two bromodomains which bind to histone acetyl-lysine residues in active chromatin. The second P-TEFb complex is the “Super Elongation Complex” (SEC) composed of CDK9, Cyclin T1, ELL1/ELL2, AFF4, and ENL/AF9 [reviewed in16]. The third P-TEFb complex is the “7SK RNP” and it is composed of 7SK RNA, CDK9, Cyclin T1, HEXIM1/2, LARP7, and MEPCE; CDK9 is not catalytically active in the 7SK RNP [reviewed in17].
Tat can utilize CDK9/Cyclin T1 in each of these three P-TEFb complexes by binding directly to CDK9 and Cyclin T1. Tat captures the Core P-TEFb complex by displacing Brd4 from CDK9/Cyclin T118. Tat participates in assembly of the SEC at TAR RNA19,20, and Tat can extract active CDK9/Cyclin T1 from the 7SK RNP21–23. Recent evidence suggests that the 7SK RNP is recruited to the HIV-1 promoter and following transcription of TAR RNA, Tat extracts Cyclin T1/CDK9 from the RNP through binding to TAR RNA24,25. A current model of Tat activation of HIV-1 LTR-directed transcriptional elongation is shown in Figure 1. As shown in this model, cis-regulatory elements in the viral LTR direct binding of the RNAP II complex; these regulatory elements include binding sites for NF-κB, NFAT (not shown), and Sp1.
Figure 1. Mechanism of action of Tat.
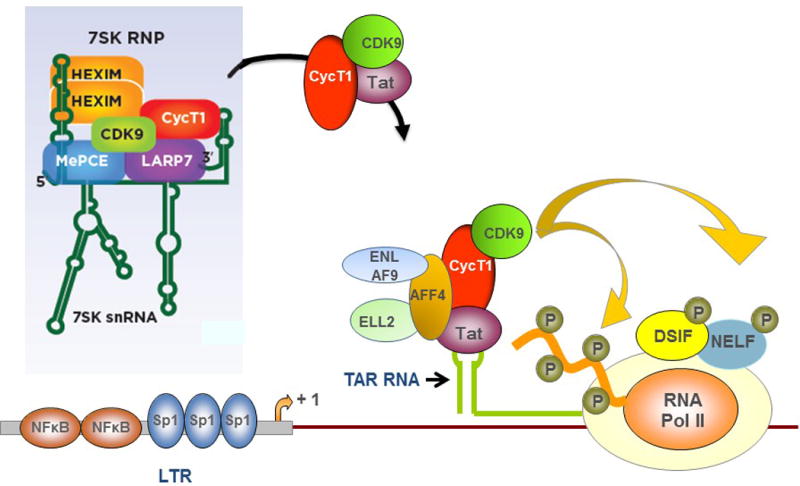
Cis-regulatory elements (e.g., NF-κB, Sp1) in the HIV-1 LTR direct binding of RNAP II to the viral promoter, transcriptional initiation occures, and TAR RNA at the 5′ end of the nascent viral transcript is produced. Tat binds to TAR RNA along with Cyclin T1 and CDK9 and this orchestrates the assembly of the Super Elongation Complex consisting of ENL/AF9/ELL2/AFF4. CDK9 phosphorylates subunits of DSIF and NELF and thereby overcomes their inhibition of RNAP II elongation. The CTD of RNAP II is hyperphosphorylated by CDK9. Cyclin T1 and CDK9 can be extracted from the 7SK P-TEFb RNP by Tat as indicated.
Regulation of P-TEFb in cells relevant to HIV-1 infection
In primary resting CD4+ T cells, it was observed that Cyclin T1 protein levels are low and cellular activation up-regulates Cyclin T1 by a post-transcriptional mechanism26–28. Four miRNAs are expressed at high levels in resting CD4+ T cells and appear to repress translation of Cyclin T1 mRNA in these cells: miR-27b, miR-29b, miR-150, and miR-22329. Notably, agents that minimally activate resting CD4+ T cells, such as a combination of cytokines (TNF-α, IL-2, IL-6) or the PKC agonist prostratin, up-regulate Cyclin T1 and CDK9 kinase activity without inducing T cell proliferation or T cell activation markers26,28. This point is highly relevant to HIV cure research and is discussed below.
Unlike Cyclin T1, CDK9 is generally expressed at a high basal level in resting CD4+ T cells26. However, phosphorylation of threonine 186 in the T-loop of CDK9, a post-translational modification essential for kinase activity, is low to absent in these cells30. Upon T cell activation, there is a rapid phosphorylation of the CDK9 T-loop30 and CDK7 appears to be the kinase responsible for this phosphorylation31. The phosphatase PPM1A regulates T-loop dephosphorylation in CD4+ T cells, as siRNA depletion of PPM1A in resting CD4+ T cells leads to elevated T-loop phosphorylation and enhanced HIV-1 gene expression32. Three other phosphatases are capable of dephosphorylating the CDK9 T-loop in transformed cell lines: PP2B, PP1α, PPM1G25,33. The relative importance of these three CDK9 T-loop phosphatases in primary CD4+ T cells is not known.
In monocytes isolated from healthy blood donors, Cyclin T1 levels are low and are up-regulated as the cells differentiate to macrophages34,35. MiR-198 has been identified as a miRNA that can repress Cyclin T1 protein expression in monocytes36. Similar to resting CD4+ T cells, CDK9 T-loop phosphorylation is low in monocytes and is up-regulated during macrophage differentiation35. Upon extended culture time of primary macrophages, the Cyclin T1 protein that was induced during differentiation is down-regulated by proteasome-mediated proteolysis34, and this down-regulation can be accelerated by IL-1037. After its shut-off in late-differentiated macrophages, Cyclin T1 can be re-induced through post-transcriptional mechanisms by pathogen-associated molecular patterns (PAMPs) or HIV-1 infection38,39.
The 7SK P-TEFb complex is present at very low levels in resting CD4+ T cells and monocytes, reflecting the low levels Cyclin T1 and CDK9 T-loop phosphorylation in these cells26,40–42. Following T cell activation or macrophage differentiation, there is a large increase in the level of the 7SK P-TEFb complex26,41,42. Signaling pathways have been identified that when activated cause release of Core P-TEFb from the 7SK RNP in metabolically active cells, with the subsequent activation of HIV-1 proviral transcription. T-cell receptor stimulation of Jurkat CD4+ T cell leads to dissociation of CDK9 and Cyclin T1 from the 7SK RNP and reactivation of a latent HIV-1 provirus43. Activation of PKC can also lead to dissociation of Core P-TEFb and activation of proviral transcription44.
Tat function and a functional HIV cure
Current combination anti-retroviral therapy (cART) is effective in suppressing HIV-1 replication in individuals with access to cART. However, a number of toxicities are associated with cART, including hyperlipidemia, bone loss, cardiovascular complications, and neuropathy. When cART is discontinued, a latent viral reservoir spontaneously reactivates in the vast majority of infected individuals and they must resume cART. Therefore, cART is currently required for the lifetime of infected individuals. As a consequence of these issues with cART, there is a major effort to develop therapeutic strategies to disrupt the latent viral reservoir and enhance the immune system’s ability to clear HIV-1. It is hoped that a strategy, termed “shock and kill,” can lead to a functional cure of infection. It has been proposed that latency reversal agents (LRAs) can be developed to “shock” latent HIV-1 to become transcriptionally active and therefore begin to express viral antigens. The “kill” involves methods to enhance the immune system’s ability to attack HIV-infected cells, perhaps the result of a therapeutic vaccine, which in combination with cART can clear reactivated virus. It is hoped that this strategy will reduce the size of the reservoir, if not eliminate it, and boost the immune system of infected individuals so that viral replication can be contained without the need for continued cART, resulting in a functional cure of infection.
The best understood component of the latent reservoir is that of memory CD4+ T cells that contain a transcriptionally silent but replication-competent provirus. As these latent viruses do not express viral RNA or protein, they are invisible to the immune system. As described above, P-TEFb and therefore Tat function are down-regulated in resting CD4+ T cells. Furthermore, it has been shown in a primary CD4+ T cell model of HIV-1 latency that P-TEFb must be up-regulated for reactivation of latent virus45. Therefore, the “shock” component of a shock and kill strategy will require the induction of P-TEFb.
It is critical that LRAs which induce P-TEFb and other cellular factors required for viral reactivation do not lead to generalized immune activation. Several cellular factors have been shown to function as signal-depedent activators for the initiation of RNAP II transcription of the viral LTR: NF-κB, NFAT, STAT5, and AP-146. LRAs capable of inducing these factors with only minimal effects on immune activation will be valuabe for the “shock,” as an increase in RNAP II initiation will contribute of high levels of viral reactivation if P-TEFb function can also be induce. An induction of P-TEFB without generalized immune activation is likely to be achievable as a combination of cytokines (TNF-α, IL-2, IL-6) or the PKC agonist prostratin have been shown to induce P-TEFb in resting CD4+ T cells without inducing the T cell activation marker CD25 or cellular proliferation26,28. A synthetic prostratin derivative has been developed that is potent in reactivating latent HIV-1 in patient cells ex vivo and does not induce significant levels of CD2547. Additional PKC agonists termed Ingenol esters have been shown to up-regulated P-TEFb and reactivate latent HIV-1 in patient cells ex vivo48–50.
An important component of the LRA “shock” to latent HIV-1 is an activity that can relieve repressive chromatin, such as LRAs that function as histone deacetylase inhibitors (HDACis)51. Interestingly, broad spectrum HDACis (vorinostat and panobinostat) that reactivate latent HIV-1 through relief of repressive chromatin also up-regulate P-TEFb through increased levels of T-loop phosphorylation, suggesting that some HDACis have multiple mechanisms of action as LRAs52,53.
Going forward, high-throughput screens for small molecules that up-regulate P-TEFb in resting CD4+ T cells can be used to develop novel LRAs. Microscopy-based assays with antisera against Cyclin T1 and the CDK9 T-loop have potential as robust assays for such screens. Additionally, release of Core P-TEFb from the 7SK RNP contributes to an increase in RNAP II elongation of the HIV-1 proviruses, and therefore small molecules that stimulate this release have potential as LRAs.
Acknowledgments
Work in the Author’s laboratory was supported by National Institutes of Health grants AI116173 and P30AI1036211 (Baylor-UTHouston CFAR). I thank Carolyn Rice for comments on the manuscript.
Abbreviations
- cART
combination anti-retroviral therapy
- cDNA
complementary DNA
- CTD
carboxyl terminal domain
- DSIF
DRB sensitivity inducing factor
- HDACi
histone deacetylase inhibitor
- HIV-1
human immunodeficiency virus type 1
- LRA
latency reversing agent
- LTR
long terminal repeat
- NELF
negative elongation factor
- P-TEFb
positive transcriptional elongation factor b
- RNAP II
RNA Polymerase II
- RNP
ribonucleoprotein
- SEC
super elongation complex
Footnotes
Conflicts of Interest
The author has no conflicts of interest.
References
- 1.Sodroski JG, Rosen CA, Wong-Staal F, Popovic M, Arya SK, Gallo RC, et al. Trans-Acting Transcriptional Regulation of Human T-Cell Leukemia Virus Type III Long Terminal Repeat. Science. 1985;227:171–3. doi: 10.1126/science.2981427. [DOI] [PubMed] [Google Scholar]
- 2.Sodroski JG, Patarca R, Rosen CA, Haseltine WA. Location of the trans-activating region on the genome of human T-cell lymphotropic virus III. Science. 1985;229:74–7. doi: 10.1126/science.2990041. [DOI] [PubMed] [Google Scholar]
- 3.Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330:489–93. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 4.Feng S, Holland EC. HIV-1 tat trans-activation requires the loop sequence within tar. Nature. 1988;334:165–7. doi: 10.1038/334165a0. [DOI] [PubMed] [Google Scholar]
- 5.Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, et al. Human immunodeficiency virus 1 tat protein binds trans- activation-responsive region (TAR) RNA in vitro. Proc Natl Acad Sci USA. 1989;86:6925–9. doi: 10.1073/pnas.86.18.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madore SJ, Cullen BR. Genetic analysis of the cofactor requirement for human immunodeficiency virus type 1 Tat function. J Virol. 1993;67:3703–11. doi: 10.1128/jvi.67.7.3703-3711.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrmann CH, Rice AP. Specific interaction of the human immunodeficiency virus Tat proteins with a cellular protein kinase. Virology. 1993;197:601–8. doi: 10.1006/viro.1993.1634. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann CH, Rice AP. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl- terminal domain of the large subunit of RNA polymerase II: candidate for a Tat cofactor. J Virol. 1995;69:1612–20. doi: 10.1128/jvi.69.3.1612-1620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall NF, Marshall TAB, et al. Transcriptional elongation factor p-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997;11:2622–32. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Gold MO, Tang DN, Lewis DE, Aguilar-Cordova E, Rice AP, et al. TAK, an HIV Tat-associated kinase, is a member of the cyclin-dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proceedings of the National Academy of Sciences USA. 1997;94:12331–6. doi: 10.1073/pnas.94.23.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem. 1998;273:13855–60. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- 12.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A Novel CDK9-Associated C-type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell. 1998;92:451–62. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu Rev Biochem. 2012;81:119–43. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21:227–37. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 15.Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–95. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13:543–7. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- 17.McNamara RP, Bacon CW, D’Orso I. Transcription elongation control by the 7SK snRNP complex: Releasing the pause. Cell Cycle. 2016;15:2115–23. doi: 10.1080/15384101.2016.1181241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. 2007;104:13690–5. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, et al. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–38. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, et al. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–51. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, Price DH. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007;35:4347–58. doi: 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, et al. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulte A, Czudnochowski N, Barboric M, Schonichen A, Blazek D, Peterlin BM, et al. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem. 2005;280:24968–77. doi: 10.1074/jbc.M501431200. [DOI] [PubMed] [Google Scholar]
- 24.D’Orso I, Jang GM, Pastuszak AW, Faust TB, Quezada E, Booth DS, et al. Transition Step during Assembly of HIV Tat:P-TEFb Transcription Complexes and Transfer to TAR RNA. Mol Cell Biol. 2012;32:4780–93. doi: 10.1128/MCB.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNamara RP, McCann JL, Gudipaty SA, D’Orso I. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 2013;5:1256–68. doi: 10.1016/j.celrep.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sung TL, Rice AP. Effects of prostratin on Cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology. 2006;3:66. doi: 10.1186/1742-4690-3-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J Virol. 1998;72:9881–8. doi: 10.1128/jvi.72.12.9881-9888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghose R, Liou LY, Herrmann CH, Rice AP. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J Virol. 2001;75:11336–43. doi: 10.1128/JVI.75.23.11336-11343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiang K, Sung TL, Rice AP. Regulation of Cyclin T1 and HIV-1 Replication by MicroRNAs in Resting CD4+ T Lymphocytes. J Virol. 2012;86:3244–52. doi: 10.1128/JVI.05065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramakrishnan R, Dow EC, Rice AP. Characterization of Cdk9 T-loop phosphorylation in resting and activated CD4(+) T lymphocytes. J Leukoc Biol. 2009;86:1345–50. doi: 10.1189/jlb.0509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19:1108–15. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budhiraja S, Ramakrishnan R, Rice AP. Phosphatase PPM1A negatively regulates P-TEFb function in resting CD4T+ T cells and inhibits HIV-1 gene expression. Retrovirology. 2012;9:52. doi: 10.1186/1742-4690-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008;22:1356–68. doi: 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liou LY, Herrmann CH, Rice AP. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J Virol. 2002;76:10579–87. doi: 10.1128/JVI.76.21.10579-10587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong C, Kwas C, Wu L. Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J Virol. 2009;83:3518–27. doi: 10.1128/JVI.02665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung TL, Rice AP. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009;5:e1000263. doi: 10.1371/journal.ppat.1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Rice AP. Interleukin-10 inhibits HIV-1 LTR-directed gene expression in human macrophages through the induction of cyclin T1 proteolysis. Virology. 2006;352:485–92. doi: 10.1016/j.virol.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 38.Liou LY, Herrmann CH, Rice AP. Human immunodeficiency virus type 1 infection induces cyclin T1 expression in macrophages. J Virol. 2004;78:8114–9. doi: 10.1128/JVI.78.15.8114-8119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liou LY, Haaland RE, Herrmann CH, Rice AP. Cyclin T1 but not cyclin T2a is induced by a post-transcriptional mechanism in PAMP-activated monocyte-derived macrophages. J Leukoc Biol. 2006;79:388–96. doi: 10.1189/jlb.0805429. [DOI] [PubMed] [Google Scholar]
- 40.Haaland RE, Herrmann CH, Rice AP. Increased association of 7SK snRNA with Tat cofactor P-TEFb following activation of peripheral blood lymphocytes. AIDS. 2003;17:2429–36. doi: 10.1097/00002030-200311210-00004. [DOI] [PubMed] [Google Scholar]
- 41.Budhiraja S, Famiglietti M, Bosque A, Planelles V, Rice AP. Cyclin T1 and CDK9 T-Loop Phosphorylation Are Downregulated during Establishment of HIV-1 Latency in Primary Resting Memory CD4+ T Cells. J Virol. 2013;87:1211–20. doi: 10.1128/JVI.02413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartholomeeusen K, Fujinaga K, Xiang Y, Peterlin BM. HDAC inhibitors that release Positive Transcription Elongation Factor b (P-TEFb) from its Inhibitory Complex also activate HIV Transcription. J Biol Chem. 2013 doi: 10.1074/jbc.M113.464834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YK, Mbonye U, Hokello J, Karn J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J Mol Biol. 2011;410:896–916. doi: 10.1016/j.jmb.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res. 2012;40:9160–70. doi: 10.1093/nar/gks682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–37. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014;454–455:328–39. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, et al. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci U S A. 2013;110:11698–703. doi: 10.1073/pnas.1302634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandelo JD, Bartholomeeusen K, da Cunha RD, Abreu CM, Glinski J, da Costa TB, et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology. 2014;462–463:328–39. doi: 10.1016/j.virol.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spivak AM, Bosque A, Balch AH, Smyth D, Martins L, Planelles V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4(+) T Cells from Aviremic Patients. Antimicrob Agents Chemother. 2015;59:5984–91. doi: 10.1128/AAC.01077-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang G, Mendes EA, Kaiser P, Wong DP, Tang Y, Cai I, et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS Pathog. 2015;11:e1005066. doi: 10.1371/journal.ppat.1005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lusic M, Giacca M. Regulation of HIV-1 latency by chromatin structure and nuclear architecture. J Mol Biol. 2015;427:688–94. doi: 10.1016/j.jmb.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 52.Jamaluddin MS, Hu PW, Danels YJ, Siwak ES, Rice AP. The Broad Spectrum Histone Deacetylase Inhibitors Vorinostat and Panobinostat Activate Latent HIV in CD4+ T cells in part through Phosphorylation of the T-Loop of the CDK9 Subunit of P-TEFb. AIDS Res Hum Retroviruses. 2016;32:169–73. doi: 10.1089/aid.2015.0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramakrishnan R, Liu H, Rice AP. SAHA (Vorinostat) Induces CDK9 Thr-186 (T-Loop) Phosphorylation in Resting CD4 T Cells: Implications for Reactivation of Latent HIV. AIDS Res Hum Retroviruses. 2014;31:137–41. doi: 10.1089/aid.2013.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]