

Abstract
The clinical development of checkpoint inhibitor-based immunotherapy has ushered in an exciting era of anticancer therapy. Durable responses can be seen in patients with melanoma and other malignancies. Although monotherapy with PD-1 or PD-L1 agents are typically well tolerated, the risk of immune-related adverse events increases with combination regimens. The development of predictive biomarkers is needed to optimise patient benefit, minimise risk of toxicities, and guide combination approaches. The greatest focus has been on tumour-cell PD-L1 expression. Although PD-L1 positivity enriches for populations with clinical benefit, PD-L1 testing alone is insufficient for patient selection in most malignancies. In this Review, we discuss the status of PD-L1 testing and explore emerging data on new biomarker strategies with tumour-infiltrating lymphocytes, mutational burden, immune gene signatures, and multiplex immunohistochemistry. Future development of an effective predictive biomarker for checkpoint inhibitor-based immunotherapy will integrate multiple approaches for optimal characterisation of the immune tumour microenvironment.
Introduction
The immune system is important in cancer cell surveillance and elimination, and immune evasion of cancer cell populations by various mechanisms is considered one of the hallmarks of cancer.1 The cancer immunity cycle described by Chen and Mellman2 describes the foundation for strategies involved in augmenting antitumour immune responses. These strategies include steps such as: cancer antigen release and presentation by dendritic cells, priming and activation of peripheral immune cells, trafficking and infiltration of T cells to the tumour compartment, and tumour-cell recognition and immune-mediated cell death. The steps after priming and activation of peripheral immune cells result in what has been described as the T-cell inflamed phenotype, which includes the local production of chemokines, interferon signalling, and expansion of CD8+ cytotoxic T cells.3 Mechanisms of tolerance are common, such as upregulation of PD-L1 and IDO in response to interferon γ,4 which diminishes the ability for immune-mediated tumour eradication (figure). Immunotherapies are thought to be most effective in patients with this T-cell inflamed phenotype.
Figure. Immune response in the tumour microenvironment.
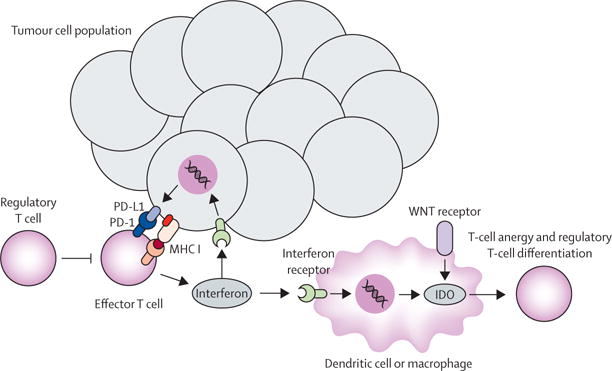
After an immune response directed against tumour cells, immune tolerance can develop in the tumour microenvironment. Various mechanisms have been described including upregulation of tumour cell PD-L1 and dendritic cell and macrophage IDO expression in response to interferon γ signalling, upregulation of additional checkpoints (eg, LAG3), and enhanced regulatory T-cell function. These events serve both as potential therapeutic targets and predictive biomarkers. MHC I=major histocompatibility complex I.
High-dose interleukin 2 and adoptive T-cell transfer have shown that durable clinical benefit can be achieved with immunotherapy in patients with advanced malignancies.5,6 Focus has now shifted to targeted manipulation of immune checkpoints. The CTLA-4 antibody ipilimumab was the first approved checkpoint inhibitor after it improved overall survival in patients with advanced melanoma in two randomised phase 3 trials.7,8 However, objective responses are low with ipilimumab monotherapy and 22% of patients with advanced melanoma survived at least 3 years after therapy, based on pooled data from past ipilimumab studies.9 Greater clinical activity has been shown in melanoma when either the PD-1 or PD-L1 checkpoint is targeted. The anti-PD-1 agents pembrolizumab and nivolumab are now approved by the US Food and Drug Administration (FDA) for patients with advanced unresectable melanoma and non-small-cell lung cancer (NSCLC). Objective responses are seen in 40–45% of patients with melanoma who were given pembrolizumab or nivolumab in the first-line setting and 20% of patients with NSCLC after failure of chemotherapy.10–14 Nivolumab is also FDA approved as second-line therapy for patients with metastatic renal-cell carcinoma, of whom 25% achieved an overall response.15 FDA approvals have been announced for nivolumab in patients with refractory Hodgkin’s lymphoma and for the anti-PD-L1 agent atezolizumab in patients with advanced bladder cancer. Furthermore, promising clinical activity of these anti-PD-1 and anti-PD-L1 therapies, as well as the anti-PD-L1 agents durvalumab and avelumab, has now been shown in a wide range of solid tumour and haematological malignancies.16
The CheckMate 067 trial,13 which compared nivolumab plus ipilimumab with ipilimumab monotherapy and nivolumab monotherapy in patients with metastatic melanoma, confirmed higher antitumour activity with combination immune checkpoint blockade than monotherapy. In CheckMate 067, 181 (58%) of 314 patients given the combination regimen achieved objective responses, and progression-free survival was longer than that in the ipilimumab monotherapy and nivolumab monotherapy groups. Data emerging for combined therapy with nivolumab plus ipilimumab in other disease types, such as small-cell lung cancer and renal-cell carcinoma, have also shown enhanced clinical activity.17,18 However, the risk of immune-related adverse events, such as dermatitis, colitis, and hepatitis, substantially increases with combination checkpoint blockade.13 In the CheckMate 067 trial, severe immune-related adverse events (grades 3 or 4) occurred in 172 (55%) of 313 patients given nivolumab plus ipilimumab, compared with 51 (16%) of 313 patients receiving nivolumab monotherapy, and 85 (27%) of 311 patients receiving ipilimumab monotherapy.13
The establishment of predictive biomarkers for checkpoint immunotherapy is therefore of utmost importance to maximise therapeutic benefit. One or more biomarker approaches that have high positive and negative predictive values are needed to assist oncologists in treatment recommendations for patients. Here, positive predictive value is referring to the number of correctly predicted responders or survivors divided by the total number of patients with a positive biomarker result, whereas negative predictive value is referring to the number of correctly predicted non-responders or non-survivors divided by the total number of patients with a negative biomarker result. Establishing predictive biomarkers is especially important for more aggressive treatment strategies, such as the nivolumab plus ipilimumab combination, in which the risk of severe (but manageable) toxicities is as high as the proportion of patients with an overall response. Biomarkers could be used to stratify patients between single-agent and combination immunotherapy or to prioritise when immunotherapy is given (first line vs salvage). Also, in patients predicted to not respond to current checkpoint immunotherapies, avoidance of unnecessary toxicities and use of alternative treatment strategies would have a major impact on patient care. So far, multiple biomarker strategies have emerged that focus on identifying aspects of the T-cell inflamed phenotype and so-called tumour foreignness (eg, mutational load, neoantigens) as approaches that are associated with clinical outcomes for anti-CTLA-4 and anti-PD-1 or anti-PD-L1 therapies. This Review investigates the progress of biomarkers as aids to checkpoint inhibitor immunotherapy in cancer (table).
Table.
Leading tumour biomarker strategies under development for checkpoint immunotherapy
| Details of approach | Malignancies studied | Improved clinical outcome association | |
|---|---|---|---|
| PD-L110,12,13,19–25 | Immunohistochemistry-based assessment of the proportion of PD-L1-positive tumour cells, immune cells, or both | Multiple tumour types | Positive PD-L1 tumour status |
| Tumour-infiltrating lymphocyte26–28 | Immunohistochemistry-based assessment of T cells at invasive tumour margin or tumour parenchyma | Melanoma; multiple tumour types | Increased CD8+ tumour-infiltrating lymphocyte density |
| T-cell receptor clonality27 | Involves next-generation sequencing of T-cell receptor β chain | Melanoma | Restricted, clonal T-cell receptor β chain |
| Mutational burden29–37 | Whole or targeted exome sequencing to assess non-synonymous somatic mutations | Melanoma, NSCLC, bladder cancer | High mutational count |
| Neoantigen burden31–33,37 | Predicted neoantigens derived from whole-exome sequencing data | Melanoma, NSCLC | High neoantigen count |
| Immune gene signatures38,39 | Assessment of gene expression from the tumour microenvironment using an automated platform | Melanoma | Interferon γ or T-cell inflamed profile |
| Multiplex immunohistochemistry27 | Direct assessment of multiple protein markers on tumour cells and immune cells, including spatial relationships | Multiple tumour types | Physical interaction with PD-1-positive and PD-L1-positive cells; others likely to be determined |
PD-L1=programmed death-ligand 1. NSCLC=non-small-cell lung cancer. PD-1=programmed death-1.
PD-L1 expression
Direct assessment of PD-L1 expression on tumour cells is a logical biomarker for the prediction of treatment response to anti-PD-1 or anti-PD-L1 therapies. Initial data from the phase 1 study19 on the use of nivolumab in patients with melanoma, NSCLC, renal-cell carcinoma, prostate cancer, or colorectal cancer supported a potential role for measuring tumour-cell PD-L1 expression by immunohistochemistry on tumour biopsy specimens. Using a threshold of 5% PD-L1-positive tumour cells to define PD-L1 positivity, nine (36%) of 25 patients with PD-L1-positive disease showed an objective response to nivolumab, whereas none of the patients with PD-L1-negative disease had an objective response. Subsequent studies have generally shown higher proportions of patients with an objective response with anti-PD-1 or anti-PD-L1 therapies in patient populations with PD-L1-positive disease.20 Improved progression-free survival and overall survival have also been shown in patients with advanced melanoma and NSCLC when comparing PD-L1-positive versus PD-L1-negative subgroups.10,12,13 Notably, companion PD-L1 immunohistochemistry diagnostic assays are approved by the FDA for use in patients with advanced NSCLC and bladder cancer, but PD-L1 positivity is only a requirement for treatment with pembrolizumab in patients with NSCLC. However, patients whose disease is PD-L1-negative by immunohistochemistry can still achieve clinical benefit with anti-PD-1 or anti-PD-L1 therapies. Indeed, objective responses in patients with PD-L1-negative tumours have been observed in most studies, usually ranging from 11% to 20%, and proportions of patients with an overall response as high as 41% with nivolumab monotherapy, and 54% with nivolumab plus ipilimumab in the CheckMate 067 melanoma study.13,20 These data indicate that the negative predictive value of anti-PD-1 or anti-PD-L1 therapies is suboptimal and is as low as 58% for nivolumab, and 45% for nivolumab plus ipilimumab regimen for patients with melanoma (based on CheckMate 067 data).
The poor reliability of PD-L1 immunohistochemistry as a biomarker for anti-PD-1 or anti-PD-L1 therapies is probably the result of multiple variables. First, PD-L1 expression is regulated by various mechanisms, including the MAPK and PI3K or Akt pathways, transcriptional factors HIF1, STAT3, and NFkB, and epigenetic factors.21 It can also be expressed by other immune cells in the tumour microenvironment. Aside from copy number gains of the PD-L1 gene (CD274) that potentially leads to constitutive expression, as seen in Hodgkin’s lymphoma,22 PD-L1 expression can be transient, and intrapatient and even intratumour heterogeneity in PD-L1 tumour expression can exist.23 Therefore, tumour sampling at one timepoint or at only one tumour site or a portion of one tumour might not accurately reflect the state of the PD-1 or PD-L1 axis in a patient. A second important variable is the poor uniformity in the PD-L1 immunohistochemistry anti bodies and different thresholds for PD-L1 positivity that are being used.24 For example, the 22C3 anti-PD-L1 antibody clone was used to assess PD-L1 expression in the pembrolizumab studies, whereas the anti-PD-L1 immunohistochemistry antibody 28−8 clone used in nivolumab studies. Positivity thresholds for PD-L1 expression for the studies vary, with some using a value of 1% or more of tumour cells, and others using a value of 50% or more. However, no studies have reported a threshold for which the positive predictive value or negative predictive value approaches 100%. Another important aspect is that PD-L1 immunohistochemistry alone does not take into account factors that could impede the anti-PD-1 or anti-PD-L1 therapy response, such as whether or not active immune-cell engagement of the PD-1 or PD-L1 axis occurs in the tumour micro-environment, or whether other concurrent suppressive immune pathways (eg, IDO, FoxP3+ regulatory T cells, and lymphocyte-activation gene 3 [LAG3]) are present.
Despite these limitations, PD-L1 immunohistochemistry does play an important role in the stratification of patients included in anti-PD-1 or anti-PD-L1 therapy trials. Ensuring an equal distribution of patients with PD-L1-positive tumours in comparative cohorts has been necessary to avoid the introduction of bias because of biological differences. Data indicate that PD-L1 expression status might help guide therapy when multiple treatment options are available. For example, data on patients with advanced NSCLC in the phase 1 study of pembrolizumab (KEYNOTE-001)12 showed that the proportion of patients with an objective response was 45% in patients with PD-L1-positive tumours (defined as PD-L1 positivity by immunohistochemistry in ≥50% of tumour cells), compared with 11% in those with PD-L1-negative tumours (defined as PD-L1 positivity by immunohistochemistry in <1% of tumour cells). On the basis of data similar to that from the KEYNOTE-001 trial, patients with NSCLC whose tumours are PD-L1-negative might benefit equally or more from an alternative therapeutic approach, such as chemotherapy or a different immunotherapy strategy.10 This idea is supported by the subgroup analysis on PD-L1 status reported in the CheckMate 057 trial10 of second-line nivolumab versus docetaxel in patients with non-squamous NSCLC, in which longer progression-free survival and similar overall survival were seen in patients given docetaxel who were PD-L1 negative. Notably, PD-L1 positivity has also been proposed as a biomarker for choosing anti-PD-1 monotherapy over the combination regimen nivolumab plus ipilimumab on the basis of initial data from the CheckMate 067 trial.13 In this trial, the median progression-free survival was the same (14 months) in patients with PD-L1-positive melanoma given either nivolumab or nivolumab plus ipilimumab.13 However, updated data from the trial has now shown longer progression-free survival (and higher proportions of patients with an overall response) with nivolumab plus ipilimumab than with nivolumab alone in patients with PD-L1-positive melanoma.25 On the basis of these findings, PD-L1 immunohistochemistry alone is not yet an adequate biomarker for routine clinical use in deciding which patients to offer anti-PD-1 or anti-PD-L1 therapy to, and which patients would benefit equally from monotherapy versus combination anti-PD-1 or anti-PD-L1 therapies.
Tumour-infiltrating lymphocytes
Lymphocyte infiltration in tumour biopsy samples has been associated with improved survival in retrospective studies of patients with a range of cancers such as colorectal cancer, melanoma, and NSCLC.40–42 Similarly, the presence of ectopic lymph node-like structures within solid tumour masses, such as colorectal cancer and melanoma metastases, might predict better patient survival.43 Data have also shown that patients with stage III NSCLC given chemoradiation have longer progression-free survival and overall survival when CD8+ tumour-infiltrating lymphocyte density is high in pretreatment biopsy samples compared with those with a low CD8+ tumour-infiltrating lymphocyte density.44 The immune recognition of these tumours is thought to result in a host-immune response or T-cell inflamed tumour phenotype, which improves disease control through immune mechanisms, and might serve as a prognostic biomarker. The presence of the T-cell inflamed tumour micro environment has also been associated with clinical benefit from immunotherapies such as the MAGE-A3 vaccine and high-dose interleukin 2.3 Therefore, baseline tumour-infiltrating lymphocyte status could also serve as a predictive biomarker for checkpoint inhibitor immunotherapy.
In a phase 2 study26 of ipilimumab in patients with metastatic melanoma, baseline tumour-infiltrating lymphocyte status was not associated with clinical activity (either complete or partial response, or stable disease lasting ≥24 weeks). However, increases in tumour-infiltrating lymphocyte density in tumour biopsy samples collected after the second dose of ipilimumab were associated with significantly greater clinical activity with ipilimumab than samples without increases in lymphocyte density. Subsequently, Tumeh and colleagues27 analysed the relationship between tumour-infiltrating lymphocytes and response to pembrolizumab in patients with melanoma enrolled on the KEYNOTE-001 study. Tumour-infiltrating lymphocyte density was quantified both in the tumour parenchyma and at the invasive tumour margin. Pretreatment tumour samples showed higher CD8+ (but not CD4+) T-cell densities at the invasive margin and within the tumour parenchyma in responding patients than in patients with disease progression. Similar to the observation with ipilimumab, an increase in CD8+ T-cell density was seen in serial biopsy samples of tumours during anti-PD-1 treatment in the responding group, but not in the disease progression group. Another study28 of patients with melanoma given anti-PD-1 therapy showed a modest association between CD8+, CD3+, and CD45RO+ T-cell densities in pretreatment samples of responders versus non-responders (response was defined as complete or partial response or stable disease lasting >6 months). After anti-PD-1 treatment, the associations were more significant. Although these findings are intriguing, baseline CD8+ T-cell density overlapped between the patients with a response and those with disease progression, which hinders the establishment of an absolute cutoff as a clinically useful predictive biomarker.
T-cell receptor clonality
Tumeh and colleagues27 further investigated whether baseline tumour-infiltrating lymphocytes had a narrow T-cell receptor repertoire focused on a tumour-specific immune response and whether this narrow repertoire correlated with response to pembrolizumab. Next-generation sequencing was done on pretreatment melanoma tumours to capture all uniquely rearranged variable T-cell receptor β-chain regions. Of the 23 patients with available response and sequencing data receiving pembrolizumab treatment, 12 (52%) patients had an objective response and 11 (48%) had disease progression. T-cell receptor β chain usage was more restricted (ie, a more clonal, less diverse population) in the responding patient group than in those with disease progression. Furthermore, pretreatment and post-treatment biopsy samples showed a ten-times increase in these clones after anti-PD-1 therapy in the responding group compared with the disease progression group, which implies a tumour-specific response to therapy for these patients. Notably, baseline T-cell receptor clonality did not highly correlate with tumour-infiltrating lymphocyte density, which suggests that some patients whose tumours have a low tumour-infiltrating lymphocyte density might still benefit from anti-PD-1 therapy if the tumour-infiltrating lymphocyte population has restricted T-cell receptor clonality specific to the tumour antigen. This hypothesis needs to be further validated in a large patient population and might require identification of the recognised tumour antigens before such an approach could be applied as a biomarker.
Mutational or neoantigen burden
Preclinical studies have identified neoantigens produced by somatic mutations in passenger genes of tumour cells as primary drivers of antitumour adaptive immune responses.45,46 Rooney and colleagues47 showed that immune cytolytic activity, measured by intra-tumoural perforin 1 and granzyme B gene expression (presumably produced by effector lymphocytes), is associated with higher mutational count, and they predicted antigenic neoepitopes in a range of solid tumour malignancies. Their findings support the idea that tumour types with high mutational burdens will be more responsive to immunotherapy strategies. Indeed, melanoma and lung cancer are predicted to have the greatest number of neoantigens and are responsive to checkpoint immunotherapies.48 Further support for the role of somatic mutations and neoantigens in immune activity is provided by phase 2 data29 of pembrolizumab in patients with mismatch repair-deficient colorectal cancer (in whom the mutational burden was >20 times higher than repair proficient cancer) versus mismatch repair-proficient colorectal cancer. In the updated data30 of 53 patients with advanced colorectal cancer, the proportion of patients with an objective response was 50% in patients with mismatch repair-deficient tumours versus 0% in patients with mismatch repair-proficient tumours. Progression-free survival and overall survival were also longer in patients with mismatch repair-deficient tumours than mismatch repair-proficient tumours and responses were durable.30
The use of mutational or neoantigen burden has also been studied as a predictive biomarker in patients given checkpoint inhibitor immunotherapies. In a study by Snyder and colleagues31 of 64 patients with advanced melanoma given ipilimumab or tremilimumab (also a CTLA-4 inhibitor), a mutational load of more than 100 non-synonymous somatic mutations based on tumour whole-exome sequencing was associated with long-term clinical benefit (defined as radiographic evidence of freedom of disease or other evidence of stable disease or decreased volume of disease for >6 months). This mutational load cutoff was associated with longer overall survival compared with patients with a lower mutational load (p=0·04 in the discovery set and p=0·10 in the validation set by log rank test). Furthermore, a neoepitope signature based on major histocompatibility complex (MHC) class I presentation was highly associated with clinical outcome with overlap in neoepitopes predicted to occur in many responding patients. A similar study of 110 patients with melanoma given ipilimumab and analysed by whole-exome sequencing showed that mutational and neoantigen load (>100 non-synonymous somatic mutations or neoantigens) were associated with clinical benefit from ipilimumab.32 Clinical benefit was defined as complete or partial response or stable disease with overall survival longer than 1 year, according to Response and Evaluation Criteria in Solid Tumors (RECIST) criteria. However, the study showed that, of the 75 179 unique neoantigens identified, only 28 (0·04%) occurred in more than one patient who showed clinical benefit.32 Using the neoepitope signature developed by Snyder and colleagues,31 clinical benefit was not predicted by the predetermined neoepitope panel in this patient cohort. These findings suggest that most neoantigens associated with immunotherapy benefit are patient specific.
With regards to anti-PD-1 or anti-PD-L1 therapy, Rizvi and colleagues33 showed that higher mutational and neoantigen burdens were associated with durable clinical benefit (partial or stable response lasting >6 months) in a study of patients with NSCLC given pembrolizumab. High mutational burden (≥178 non-synonymous mutations) and neoantigen burden were both associated with significantly longer progression-free survival. Similar results have been shown in a study by Johnson and colleagues34 of 65 patients with advanced melanoma given either nivolumab, pembrolizumab, or atezolizumab. High mutational load (measured by hybrid capture-based next-generation sequencing) was associated with response to therapy and long median progression-free survival and overall survival. In addition, the phase 2 study35 of atezolizumab for locally advanced and metastatic urothelial carcinoma showed a higher mutational load by targeted exome sequencing in patients achieving a complete or partial response than those with stable disease or progressive disease as their best response. However, another study36 of 38 patients with advanced melanoma who were given either pembrolizumab or nivolumab showed that high mutational burden correlated with overall survival, but not with objective response to therapy. This finding suggests that high mutational or neoantigen burden might be important in measuring immune antitumour activity, and it might serve as a prognostic factor or a predictor of clinical benefit (or both) with checkpoint immunotherapy depending on the patient population that is being studied.
Low neoantigen intratumour heterogeneity might also be important—in addition to the total mutational or neoantigen tumour burden—for immunotherapy response. McGranahan and colleagues37 showed that in seven primary NSCLC tumours, neoantigen heterogeneity varied considerably, with an average of 44% of neoantigens found only in subsets of tumour regions. Furthermore, they37 analysed The Cancer Genome Atlas (TCGA) data on NSCLC adenocarcinoma and showed that a combination of high mutational burden (upper quartile of entire cohort of NSCLC in TCGA) and low neoantigen intratumour heterogeneity (<1%) to be more significantly associated with longer survival time (irrespective of treatment) than either variable alone (notably, this association was not observed in the NSCLC squamous cell cohort). Using a similar approach with data from the study by Rizvi and colleagues33 of pembrolizumab in patients with NSCLC, McGranahan and colleagues37 found durable clinical benefit in patients whose tumours had high mutational burden and low intratumour neoantigen heterogeneity (<1%) compared with patients with high mutational burden alone (p=0·006). Similarly, longer progression-free survival (p=0·0017) and overall survival (p=0·008) were observed when mutational burden and intratumoral heterogeneity were accounted for compared with mutational burden alone.37 For example, 12 (92%) of 13 patients with melanomas showing a low neoantigen subclonal fraction (<5%) and high mutational burden (≥70, which was the median clonal neoantigen level of the cohort) had durable clinical benefit with pembrolizumab. These characteristics appeared to have a stronger association with durable clinical benefit than mutational burden alone, which was originally used by Rizvi and colleagues.33
Peripheral blood markers
Testing of peripheral blood markers is a non-invasive source of potential biomarkers in patients receiving immune checkpoint therapies. Although associations with clinical benefit and survival have been noted, none so far have been validated as predictive biomarkers in prospective studies. For ipilimumab studies, improved overall and progression-free survival was associated with baseline values including low absolute neutrophil count (<7500 cells/μL), low neutrophil to lymphocyte ratio (<3), low absolute monocyte count (<650 cells/μL), low frequency of myeloid-derived suppressor cells (<5·1%), high frequency of FoxP3+ regulatory T cells (≥1·5%), high frequency of lymphocytes (≥10·5%), and high eosinophil count (≥50 cells/μL).49,50 Dynamic changes with treatment have also been associated with clinical benefit to ipilimumab in patients with melanoma, including decreasing concentrations of FoxP3+ regulatory T cells, increasing absolute lymphocyte counts, and increasing eosinophil counts.51–53 Some overlapping findings have been observed in anti-PD-1 therapy studies. In a retrospective study54 of various available data, including 607 patients with melanoma given pembrolizumab, baseline elevated eosinophil count (≥1·5%) and elevated lymphocyte count (≥17·5%) were both associated with improved overall survival. In a phase 1/2 study55 of nivolumab plus multipeptide vaccine in patients with advanced melanoma, decline in regulatory T-cell populations during treatment and low baseline antigen-specific CD8+ T-cell populations (recognising NY-ESO-1 and MART-1) were associated with patients who had either an objective response or stable disease. In a similar study56 of patients with resected stage IIIC or IV melanoma, higher baseline regulatory T-cell (p=0·0583) and myeloid-derived suppressor cell (p=0·1718) populations were seen in patients who relapsed than those who didn’t relapse.
Assessment of peripheral T-cell populations— particularly the T-cell receptor gene sequences or reactivity to neoantigens—could have a potential role as a predictive biomarker. In a pilot study by Postow and colleagues,57 the pretreatment peripheral blood T-cell receptor repertoire diversity assessed using the ImmunTraCkeR test was correlated to patient outcomes with ipilimumab treatment (n=12 patients in study cohort). Increased T-cell receptor gene richness (ie, a repertoire containing many different V–J rearrangements) and evenness (ie, evenly distributed frequencies) were significantly associated with clinical benefit (response or stable disease lasting ≥9 months). However, neither was associated with a significant difference in overall survival, which might be a result of the small sample size. Alternatively, autologous blood lymphocytes can be tested for T-cell recognition of patient-specific neoantigens predicted from tumour whole-exome sequencing. This method was used in studies with ipilimumab (patients with melanoma) and pembrolizumab (patients with NSCLC).31,33 In patients with a response to anti-CTLA-4 and anti-PD-1 therapies, predicted human leucocyte antigen-restricted peptides were synthesised and used to screen ex vivo autologous T-cell reactivity using high-throughput approaches. The individual peptide sequences responsible for T-cell activation were then identified. Notably, T cells recognising these neoantigens were an exceptionally small proportion of the overall population of peripheral T cells at baseline, but the frequency of this population substantially increased during therapy. Although this approach shows proof of principle for antitumour neoantigen-specific T-cell recognition, its application to broader patient populations might be limited by technical complexities.
Immune gene signatures
A wider assessment of active innate and adaptive immune responses within the tumour microenvironment by gene expression profiling might effectively predict clinical benefit to checkpoint inhibitor strategies. A retrospective analysis38 of patients with advanced melanoma given ipilimumab in a phase 2 clinical trial (CA184004) provided evidence that gene expression profiling could indeed be a useful predictive biomarker. In this analysis,38 total RNA was extracted and analysed in 50 pretreatment tumour biopsy specimens. Patients were categorised as having clinical activity (objective response or protracted stable disease) or no clinical activity with ipilimumab. Pathway analyses of the genes that were substantially different between the patient groups identified the top functional category as inflammatory response. Expression of 22 immune-related genes had at least a 2·5-times increase, including markers for cytotoxic T cells (eg, CD8A, granzyme B, perforin 1), Th1 cytokines or chemokines, MHC class II (HLA-DQA1), and other immune-related genes (eg, NKG7, IDO1). Greater pretreatment and post-treatment expression values (eg, CXCL11, CXCR3) were associated with longer overall survival.
Immune gene signatures, especially those induced by interferon γ, might be robust biomarkers for predicting clinical benefit to anti-PD-1 or anti-PD-L1 therapies. This theory is supported by PD-L1 expression data as already described, and data from Johnson and colleagues,58 showing that high MHC class II (HLA-DR) expression was associated with improved clinical response, longer progression-free survival, and longer overall survival in patients with melanoma given anti-PD-1 or anti-PD-L1 therapy compared with patients with low MHC class II expression. Data presented by Ribas and colleages39 on a retrospective analysis of an interferon inflammatory immune gene signature and response to anti-PD-1 therapy in patients with advanced melanoma has shown further promise. In this study,39 19 patients enrolled on the KEYNOTE-001 trial were chosen for a discovery set, of whom 11 had an objective response to pembrolizumab (according to RECIST). Pretreatment tumour was analysed for a custom immune gene expression panel. An interferon γ score was developed that was based on a 10-gene signature, which was then expanded to a 28-gene signature in a validation set involving 62 patients with melanoma. The genes included those encoding interferon γ (IFNG), granzyme A and B (GZMA and GZMB), and perforin 1 (PFR1), IDO1, LAG3, and other immune-related genes (panel). Both the 10-gene and 28-gene scores showed significant correlation with best overall response and progression-free survival (a non-significant association with overall survival was also observed). Optimisation of the interferon-γ score cutoff on the basis of a receiver operating characteristic curve was able to achieve a positive predictive value of 59% for responder status and a negative predictive value of 90% for non-responder status.
Multiplex immunohistochemistry
Direct assessment of both tumour and immune-cell phenotypes and their spatial relationships by multiplex immunohistochemistry techniques provides information on the immune state of the tumour microenvironment that might be superior or complementary to gene expression profiling. These techniques involve serial staining of tumour slides with individual primary antibodies for the proteins of interest and detection by either chromogenic or immunofluorescence methods.59 Current approaches allow for the assessment of up to four chromogen colours and up to five fluorescent dyes using standard fluorescent microscopes (or up to eight fluorescent dyes with a multispectral camera). This multispectral method appears to have the greatest potential for clinical applications. For example, a multispectral immunohistochemistry platform was developed to analyse CD3, CD8, FoxP3, CD163, and PD-L1 on melanoma tumour slides to predict which patients would successfully generate tumour-infiltrating lymphocytes for adoptive cell therapy.60 The presence of CD8+ T cells alone was insufficient to predict successful tumour-infiltrating lymphocyte growth. However, the CD8+ T cell to CD3+FoxP3+ regulatory T-cell ratio was strongly associated with successful tumour-infiltrating lymphocyte growth (p=0·006; positive predictive value of 91%, negative predictive value of 86%). By incorporation of PD-L1+ (all cells), the negative predictive value increased to 100%. Similar applications to other immunotherapies are possible with this approach.
The use of multiplex chromogenic and immunofluorescence methods were reported by Tumeh and colleagues27 on baseline melanoma tumour samples collected from patients who received pembrolizumab in the KEYNOTE-001 trial. The physical interaction between PD-1+ and PD-L1+ cells, as determined by the relative proximity between these two cell populations, was associated with response to anti-PD-1 therapy (p=0·005 vs non-responders). The same investigators then showed that CD8 positivity was significantly associated with PD-L1 expression, on the basis of Spearman’s correlation analysis at both the tumour and the invasive margin. Similarly, the samples with high CD8 and PD-L1 expression were significantly associated (p<0·001) with the response group whereas those with low CD8 and PD-L1 expression were associated with the progression group (p<0·001). Colocalisation of PD-1+ and CD8+ was shown to be high in individual cells in the tumour microenvironment using multiplex immunofluorescence. The authors also report on the ability to characterise PD-L1-expressing cells with stains for SOX10 (melanoma cell nuclear transcription factor) and PD-L1. Cells staining for both markers were identified as PD-L1-positive tumour cells, whereas PD-L1-positive cells negative for SOX10 were characterised as lymphocytes (high nucleus to cytoplasm ratio) or macrophages (low nucleus to cytoplasm ratio).
Combined biomarker strategies
Strategies that combine two or more methods for capturing the immune status of the tumour micro-environment might be more effective as a composite predictive biomarker for immune checkpoint inhibitor therapy. High tumour PD-L1 expression can be present even when tumour-infiltrating lymphocyte counts are low, and tumours with high tumour-infiltrating lymphocyte density might not express PD-L1.61,62 In both scenarios, clinical activity of anti-PD-1 or anti-PD-L1 monotherapy might be low but could be inaccurately predicted to be high if either PD-L1 status or tumour-infiltrating lymphocyte density alone were used as a biomarker. Similarly, not all high mutational or neoantigen burden tumours show signs of pre-existing immune activity, which is thought to be one of the prerequisites for immunotherapy approaches.3,47 Multiple concurrent immune-suppressive mechanisms can also be present in the tumour microenvironment, including CTLA-4, PD-L2, LAG3, IDO1, and interleukin 10,63,64 which are likely to become important targets for identification as novel combination therapy strategies become available.
Gene expression profiling approaches, such as the interferon γ score, incorporates multiple immune variables that might be able to accurately predict responders and non-responders to various immunotherapies. However, mRNA concentrations of PD-L1 (and other checkpoints or immune factors) might not be as reliable as immunohistochemistry because of post-transcriptional regulation. Alternatively, the mRNA might be expressed by other cell populations such as dendritic cells and thus have different implications. Similarly, the multiplex immunohistochemistry techniques allow for quantification of multiple proteins and cell populations within the tumour microenvironment, but have limitations associated with the number of markers that can be examined at one time, and the need for validation steps each time the panel of markers is changed (eg, to exchange LAG3 for IDO1).
The potential benefit from a combined biomarker approach is supported by biomarker data presented at the 2016 ASCO Annual Meeting65 and 2015 European Cancer Congress66 on patients with NSCLC who participated in the phase 1/2 trial of durvalumab. Pretreatment biopsy specimens were analysed for PD-L1 immunohistochemistry and immune gene expression (mRNA analysis). Of the 100 genes tested, IFNG was most correlated with response to treatment. The proportion of patients with an objective response for the 200 evaluable patients with NSCLC was 16%. In patients whose tumours were PD-L1-positive (threshold of 25%), the proportion of patients with an objective response was 27%. A similar proportion of patients with an objective response (33%) was seen in the patients with interferon γ-expressing tumours. In patients with both dual PD-L1-positive and interferon γ-positive tumours, the proportion of patients with an objective response was 46%. Notably, patients with dual negative tumours—representing 20% (40 of 200 patients) of the evaluable population—had a proportion of patients with an objective response of 3%. Overall survival was longest in patients whose tumours were interferon γ-positive, particularly in the dual PD-L1-positive group. Although further confirmation is needed, these provocative findings provide substantial hope that biomarker strategies with strong positive and negative predictive values can be developed for routine clinical use to assist in the checkpoint immunotherapy-based management of patients with diverse malignancies.
Conclusion
Thus far, use of PD-L1 immunohistochemistry alone has not been sufficient for ruling in or ruling out the use of anti-PD-1 or anti-PD-L1 expression-based therapies. Characterisation of the tumour microenvironment immune state needs to be improved, including the presence of recognised tumour antigens, effector T-cell function, and immune suppressive mechanisms. Because of the potential for redundancy, further investigation into the relationships between PD-1 and PD-L1 expression, tumour-infiltrating lymphocytes, the T-cell receptor repertoire, and mutational or neoantigen burden should be aimed at creating an optimised model for predicting response to anti-PD-1 or anti-PD-L1-based therapies. Furthermore, specific mechanisms of T-cell exclusion, such as activation of the WNT/β-catenin signalling pathway as shown by Spranger and colleagues,67 should be included in future biomarker development. These models might need to be specific to individual tumour types, because immune responses do not appear to be uniform across all malignancies. Not only will it be beneficial to predict which patients will not respond to PD-1 or PD-L1 monotherapy or PD-1 or PD-L1 therapy in combination with anti-CTLA-4 agents, thus avoiding potential toxicities, but an integrated biomarker design should also be able to guide novel immunotherapy combination strategies to overcome therapeutic resistance. Biomarker-driven prospective studies are warranted to confirm these biomarker approaches before routine clinical use.
Panel: 28-gene panel associated with clinical benefit in patients with melanoma treated with pembrolizumab39.
IL2RG
HLA-DRA
CCR5
SLAMF6
CXCR6
PTPRC
CD3E
CXCL13
CD3D
CXCL9
GZMK
CXCL10
CD2
CCL5
IFNG
IDO1
ITGAL
NKG7
HLA-E
LAG3
TAGAP
GZMA
GZMB
STAT1
CIITA
PRF1
PDCD1
CXCL11
Search strategy and selection criteria.
We identified references for this Review through searches of PubMed using the search terms “biomarker”, “predictive”, “mutation”, “tumor infiltrating lymphocytes”, “TCR repertoire”, “immunotherapy”, “PD-1”, “PD-L1”, and “CTLA-4”. Articles were also identified through searches of the authors’ own files. Only papers and presentations or abstracts published in English between Jan 1, 2008, and June 30, 2016, were included for review. The final reference list was generated on the basis of originality and relevance to the broad scope of this Review.
Footnotes
Declaration of interests
GTG reports personal fees from Novartis and Merck. LMW reports personal fees from Merck, Genentech, Jounce Pharmaceuticals, Celldex Pharmaceuticals, and Merrimack Pharmaceuticals. MBA reports personal fees from Bristol-Myers Squibb, Merck, Genentech/Roche, Novartis, Pfizer, and Astra Zeneca.
Contributors
All authors have participated in writing this Review and agree to its submission.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–16. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–57. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 9.Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 13.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 15.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112:1421–27. doi: 10.1038/bjc.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammers HJ, Plimack ER, Infante JR, et al. Expanded cohort results from CheckMate 016: a phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC) Proc Am Soc Clin Oncol. 2015;33 doi: 10.1200/JCO.2016.72.1985. abstr 4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellmann MD, Gettinger SN, Goldman JW, et al. CheckMate 012: safety and efficacy of first-line (1L) nivolumab (nivo; N) and ipilimumab (ipi; I) in advanced (adv) NSCLC. Proc Am Soc Clin Oncol. 2016;34 abstr 3001. [Google Scholar]
- 19.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahoney KM, Atkins MB. Prognostic and predictive markers for the new immunotherapies. Oncology. 2014;28(suppl 3):39–48. [PubMed] [Google Scholar]
- 21.Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27:409–16. doi: 10.1093/annonc/mdv615. [DOI] [PubMed] [Google Scholar]
- 22.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–19. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mansfield AS, Murphy SJ, Peikert T, et al. Heterogeneity of programmed cell death ligand 1 expression in multifocal lung cancer. Clin Cancer Res. 2016;22:2177–82. doi: 10.1158/1078-0432.CCR-15-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel SP, Kurzrock R. PD-L1 Expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 25.Wolchock JD, Chiarion-Sileni V, Gonzalez R, et al. Updated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). 2016 ASCO Annual Meeting; Chicago, IL. June 3–7, 2016; Abstr 9505. [Google Scholar]
- 26.Hamid O, Schmidt H, Nissan A, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen PL, Roh W, Reuben A, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827–37. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le DT, Uram JN, Wang H, et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. Proc Am Soc Clin Oncol. 2016;34 abstr 103. [Google Scholar]
- 31.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–28. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson DB, Frampton GM, Rioth MJ, et al. Hybrid capture-based next-generation sequencing (HC NGS) in melanoma to identify markers of response to anti-PD-1/PD-L1. Proc Am Soc Clin Oncol. 2016;34 abstr 105. [Google Scholar]
- 35.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–69. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji RR, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–31. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribas A, Robert C, Hodi FS, et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. Proc Am Soc Clin Oncol. 2015;33 abstr 3001. [Google Scholar]
- 40.Huh JW, Lee JH, Kim HR. Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch Surg. 2012;147:366–72. doi: 10.1001/archsurg.2012.35. [DOI] [PubMed] [Google Scholar]
- 41.Thomas NE, Busam KJ, From L, et al. Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J Clin Oncol. 2013;31:4252–59. doi: 10.1200/JCO.2013.51.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng DQ, Yu YF, Ou QY, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes for clinical therapeutic research in patients with non-small cell lung cancer. Oncotarget. 2016;7:13765–81. doi: 10.18632/oncotarget.7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Messina JL, Fenstermacher DA, Eschrich S, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep. 2012;2:765. doi: 10.1038/srep00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tokito T, Azuma K, Kawahara A, et al. Predictive relevance of PD-L1 expression combined with CD8+ TIL density in stage III non-small cell lung cancer patients receiving concurrent chemoradiotherapy. Eur J Cancer. 2016;55:7–14. doi: 10.1016/j.ejca.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–91. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 46.Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–04. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 49.Ferrucci PF, Ascierto PA, Pigozzo J, et al. Baseline neutrophils and derived neutrophil-to-lymphocyte ratio: prognostic relevance in metastatic melanoma patients receiving ipilimumab. Ann Oncol. 2016;27:732–38. doi: 10.1093/annonc/mdw016. [DOI] [PubMed] [Google Scholar]
- 50.Martens A, Wistuba-Hamprecht K, Geukes-Foppen M, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22:2908–18. doi: 10.1158/1078-0432.CCR-15-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delyon J, Mateus C, Lefeuvre D, et al. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013;24:1697–703. doi: 10.1093/annonc/mdt027. [DOI] [PubMed] [Google Scholar]
- 52.Ku GY, Yuan J, Page DB, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116:1767–75. doi: 10.1002/cncr.24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simeone E, Gentilcore G, Giannarelli D, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63:675–83. doi: 10.1007/s00262-014-1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weide B, Martens A, Hassel JC, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0127. published online May 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weber JS, Kudchadkar RR, Yu B, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–18. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gibney GT, Kudchadkar RR, DeConti RC, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. 2015;21:712–20. doi: 10.1158/1078-0432.CCR-14-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Postow MA, Manuel M, Wong P, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson DB, Estrada MV, Salgado R, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuan J, Hegde PS, Clynes R, et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J Immunother Cancer. 2016;4:3. doi: 10.1186/s40425-016-0107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng Z, Puri S, Moudgil T, et al. Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes from melanoma. J Immunother Cancer. 2015;3:47. doi: 10.1186/s40425-015-0091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kluger HM, Zito CR, Barr ML, et al. Characterization of PD-L1 expression and associated T-cell infiltrates in metastatic melanoma samples from variable anatomic sites. Clin Cancer Res. 2015;21:3052–60. doi: 10.1158/1078-0432.CCR-14-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–74. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsushita H, Sato Y, Karasaki T, et al. Neoantigen load, antigen presentation machinery, and immune signatures determine prognosis in clear cell renal cell carcinoma. Cancer Immunol Res. 2016;4:463–71. doi: 10.1158/2326-6066.CIR-15-0225. [DOI] [PubMed] [Google Scholar]
- 64.Taube JM, Young GD, McMiller TL, et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: implications for PD-1 pathway blockade. Clin Cancer Res. 2015;21:3969–76. doi: 10.1158/1078-0432.CCR-15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Higgs BW, Morehouse C, Streicher K, et al. Relationship of baseline tumoral IFNγ mRNA and PD-L1 protein expression to overall survival in durvalumab-treated NSCLC patients. Proc Am Soc Clin Oncol. 2016;34 abstr 3036. [Google Scholar]
- 66.Higgs BW, Robbins PB, Blake-Haskins JA, et al. High tumoral IFNγ mRNA, PD-L1 protein, and combined IFNγ mRNA/PD-L1 protein expression associates with response to durvalumab (anti-PD-L1) monotherapy in NSCLC patients. European Cancer Congress 2015; Vienna. Sept 25-29; Abstr 15LBA. [Google Scholar]
- 67.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–35. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]