
Fig. 2.
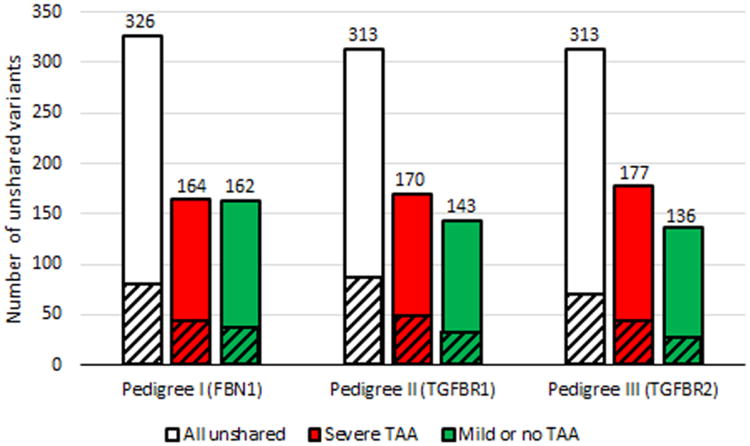
Distribution of unshared rare variants between first degree relatives with divergent TAA phenotypes. - Cross-hatched areas represent the number of variants predicted to be damaging by 4 of 4 bioinformatics programs or frameshift, nonsense, stop loss, splice site, or initiation codon variants