

Abstract
The transient receptor potential melastatin 2 (TRPM2) channel, a Ca2+ permeable channel activated by cAMP, is expressed on pancreatic β-cells and is responsible for the regulation of insulin secretion. It is known that glucose-stimulated insulin secretion (GSIS) can be potentiated by glucagon like peptide-1 (GLP-1), and that the changes in the extracellular glucose concentration alter the levels of intracellular adenosine ATP and cAMP. The present study hypothesized that TRPM2 mediates the modulatory effect of GLP-1 on insulin secretion. The results demonstrated that silencing of TRPM2 eliminated GLP-1-enhanced insulin secretion, indicating the involvement of TRPM2 in this process. In addition, the results of current recordings of TRPM2 and measurement of the resulting insulin secretion in β-cells in the presence of GLP-1 and various concentrations of glucose suggest that GLP-1 regulates GSIS via the TRPM2 channel. Furthermore, inhibiting the activity or expression of TRPM2 attenuated GLP-1-induced GSIS. By using specific activators or inhibitors, the present study demonstrated that the two primary downstream effectors of the GLP-1 receptor, exchange protein directly activated by cAMP and protein kinase A, differentially influence GSIS and GLP-1-potentiated GSIS. In conclusion, the present study revealed the role of TRPM2 in GLP-1-regulated insulin secretion. The results of the present study provide a novel avenue for the prevention and treatment of diabetes and its complications.
Keywords: transient receptor potential melastatin 2, glucose-stimu- lated insulin secretion, glucagon like peptide-1, pancreatic β-cell
Introduction
Pancreatic β-cells secrete insulin via insulin-containing granules that translocate from the intracellular insulin reservoir to the cell surface membrane, fuse with the membrane and secrete insulin through exocytosis (1,2). Cytosolic Ca2+ ions serve a role in the process of insulin secretion and can be classified into the following two types: Endogenous and exogenous Ca2+. Endogenous Ca2+ is primarily derived from intracellular organelles, such as the endoplasmic reticulum (ER), which stores and releases Ca2+ into the cytoplasm, resulting in an increased cytosolic Ca2+ concentration. By contrast, exogenous Ca2+ increases intracellular Ca2+ concentration [(Ca2+)i] via an influx of Ca2+ through ion channels located on the cell surface membrane. Thus far, the Ca2+ channels known to be on the cell surface membranes of β-cells include voltage-dependent Ca2+ channels (VDCCs), including Cav1.2 and Cav1.3, the Ca2+ release-activated channel (CRAC) and transient receptor potential (TRP) family channels. A previous study revealed that Cav1.3 is associated with the regulation of insulin secretion in β-cells (3). The CRAC channel on the cell surface membrane of β-cells is activated by the release of Ca2+ from the ER; this channel replenishes the ER with Ca2+, and mediates ER-regulated membrane potential and insulin secretion (4). TRP family channels are non-selective cation channels in general, however some members are selective with with differential permeability to Ca2+ (5). There are ~30 types of TRP channels that have been reported to be expressed on β-cells, including TRP vanilloid, TRP ankyrin, TRP canonical and TRP melastatin (TRPM) (6). Among these channels, TRPM2, a Ca2+-permeable channel, was demonstrated to be associated with glucose metabolism and insulin secretion, yet little is known about the underlying mechanisms of these associations (7).
In recent years, glucagon like peptide 1 (GLP-1) and its analogues, also known as incretins, have attracted much attention for their anti-diabetic properties. A previous study demonstrated that incretins are secreted following a meal and promote insulin secretion, thereby minimizing the fluctuation of postprandial blood glucose concentrations (8). However, the ‘incretin effect’ in patients with type 2 diabetes is compromised. This is reflected in the attenuated increase in GLP-1 concentration following dining, accompanied by relatively normal incretin secretion and hypoglycemic function (9). Therefore, GLP-1 and its analogues have been proposed as promising therapeutic agents for treating type 2 diabetes (10,11). A previous study demonstrated that the binding of GLP-1 with the GLP-1 receptor (GLP-1R) causes little Ca2+ influx or insulin secretion when the concentration of glucose is low (12). By contrast, when the glucose concentration is high, the ATP/ADP ratio is increased, thus ATP-dependent K+ (KATP) channels close and the opening of voltage-gated Ca2+ channels is prolonged (13). Therefore, GLP-1 causes extensive Ca2+ influx and insulin secretion in the presence of a high concentration of glucose.
Upon the binding of GLP-1 with GLP-1R on the cell surface membrane, adenylyl cyclase activates and generates cAMP. Subsequently, cAMP activates protein kinase A (PKA), which in turn activates cAMP-response element binding protein (CREB). CREB binds to the cAMP response element on the insulin gene promoter. This signaling pathway results in the transcriptional activation of the insulin gene and promotes insulin biosynthesis (14,15). Additionally, glucose induces an increase in the intracellular ATP/ADP ratio, leading to K+ channel closures and membrane depolarization. Thus, extracellular Ca2+ enters into the cell through activated VDCCs and increases the [Ca2+]i (16). Evidence suggests that the activation of TRPM2 in β-cells also depends upon ATP (17). Providing that GLP-1 prolongs the opening of Ca2+ channels in an ATP-dependent manner and that the opening of TRPM2 is also ATP-dependent (12), the present study hypothesized that GLP-1 stimulates insulin secretion through activating TRPM2.
Although little is known about TRPM2, it has been hypothesized that TRPM2 is a type of non-selective cation channel that is permeable to Ca2+ (18–20). Multiple molecules can activate TRPM2, including ADP-ribose and hydrogen peroxide. In addition, the activity of TRPM2 is associated with a number of factors, including [Ca2+]i, pH, temperature and intracellular Cl− concentration (18,21–26). A previous study by our group demonstrated that TRPM2 is sensitive to temperature and exhibits Ca2+ permeability (27). Another previous study reported that TRPM2-knockout mice exhibited impaired insulin secretion and suffered from hyperglycemia (28). Additionally, TRPM2-deficient β-cells have been identified to exhibit suppressed intracellular Ca2+ signaling, in addition to a reduction in insulin secretion in response to glucose or incretins, such as GLP-1 (28). These findings support the theory that TRPM2 serves an important role in insulin secretion in β-cells.
As the driver of growth and differentiation of pancreatic β-cells, GLP-1 can activate adenylyl cyclase and promote cAMP production (29,30). In β-cells, cAMP activates the classic PKA signaling pathway and the exchange protein directly activated by cAMP (Epac) signaling pathway, which triggers the downstream signaling of mitogen activated protein kinase and results in the rapid phosphorylation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 4,5-bisphosphate 3-kinase-mediated phosphorylation of protein kinase B (31–34). In addition, the PKA and Epac signaling pathways are associated with Ca2+-dependent insulin granule exocytosis from β-cells (35,36). Furthermore, by interacting with Rim2, a target of the small G-protein Rab3, Epac mediates cAMP-dependent and PKA-independent insulin exocytosis (36–38). Therefore, the activation of PKA or Epac may cause TRPM2 activation, which modulates Ca2+ influx and insulin secretion. The aim of the present study was to investigate the role of TRPM2 in GLP-1-stimulated insulin secretion and explore the possible underlying mechanisms.
Materials and methods
Reagents
GLP-1 was purchased from EMD Millipore (Billerica, MA, USA). ADP-ribose, 2-APB and ESI-09 were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). H89 was purchased from Calbiochem® (EMD Millipore). The 8-pCPT and 6-Benz cAMP were purchased from BIOLOG Life Science Institute Laboratory and Biochemivcal Sales GmbH (Bremen, Germany).
Isolation and culture of primary pancreatic islets
The present study, involving primary animal cells, was approved by the Ethics Committee of Tianjin Metabolic Diseases Hospital affiliated to Tianjin Medical University (Tianjin, China). Rat pancreatic islets were isolated from healthy Sprague Dawley rats. A total of 80 6–8 week old SD rats, weighing 180–220 g, were purchased from Huafukang Bioscience Co., Inc. (Beijing, China). The animals were adaptively housed for one week in the Aminal Center of Tianjin Medical University under standard conditions (23±2°C; 65±5% humidity; 12 h light/dark cycle) with access to food and water ad libitum. Approximately 105 pancreatic β-cells were isolated from each rat. Briefly, the rats were sacrificed and the common bile duct was ligated near the hepatic portal. Collagenase V (1 mg/ml) was injected from the common bile duct into the pancreas to digest the pancreatic tissues. When the pancreas had fully swollen, it was removed and incubated at 37°C for 25 min. Following this, 40 ml cold Hanks' balanced salt solution was added into the common bile duct to terminate the digestion. The pancreatic tissues were dispersed by repeated pipetting, followed by filtration through a 150-µm sieve and centrifugation at 300 × g for 15 min at room temperature. The digested products were resuspended in 25% Ficoll solution and separated by Ficoll gradient centrifugation (23, 20 and 11%, from bottom to top) at 1,400 × g for 20 min at 4°C. The pancreatic islets in the 23–20 and 10–11% layers were collected and washed twice with 40 ml Hanks' buffer. The round pancreatic islets with a regular shape were collected under a light microscope. The islets were recovered and incubated in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA), 100 U/ml penicillin and 100 µg/ml streptomycin for 3 h at 37°C in a humidified atmosphere with 5% CO2. Following the isolation, insulin release experiments, electrophysiological measurements or viral infections were performed.
Silencing and overexpression of TRPM2
Adenoviruses containing green fluorescent protein (GFP) alone (pHBAd-MCMV-GFP), the TRPM2 gene (pHBAd-MCMV-GFP-TRPM2), the negative control gene (pHBAd-U6-GFP-scramble) or short hairpin RNA directed against TRPM2 (sense: 5′-AATTCGGACTAAGCTGGAGAAGTTCATTCAAGAGATGAACTTCTCCAGCTTAGTCCTTTTTTG-3′; pHBAd-U6-GFP-TRPM2) were designed and prepared by Hanbio Biotechnology Co., Ltd. (Shanghai, China). The isolated β-cells were seeded in 24-well plates at a density of 30,000 cells per well. Following adhesion to the plate, the cells were infected with the viruses (50:1) and successful transduction was detected by the presence of a GFP signal under a fluoresence microscope. Silencing and overexpression of TRPM2 was also confirmed by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analyses 24 h post-infection, and non-infected cells served as the control.
RT-qPCR
Total RNA was extracted from β-cells using a RNA extraction kit (BioTeke Corporation, Beijing, China) according to the manufacturer's protocol. The RNA was reverse transcribed to cDNA by incubating RNA samples with super M-MLV reverse transcriptase (BioTeke) and random primers at 42°C for 50 min. The level of TRPM2 mRNA in each sample was measured by using cDNA as the template and the following primers: TRPM2 forward, 5′-AAGTATGTCCGGGTCTCCC-3′ and reverse, 5′-TAACGGCCCAAATGAGAAGGTCACG-3′; β-actin forward, 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse, 5′-CTGTCACCTTCACCGTTCCAGTTT-3′. β-actin served as the internal reference. qPCR was performed on an Exicycler 96 Quantitative Thermal Block (Bioneer, Daejeon, Korea) using SYBR Green Master Mix (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China), and the amplification conditions were set as follows: 10 min at 95°C; 40 cycles of 10 sec at 95°C, 20 sec at 60°C and 30 sec at 72°C; and finally 5 min at 4°C. The level of TRPM2 mRNA was normalized to the non-infected (Control) cells using the 2−ΔΔCq method (39).
Western blot analysis
Proteins were extracted from β-cells using a protein extraction kit (WanLeibio, Inc., Shenyang, China) according to the manufacturer's protocol. The protein concentration was determined with a BCA assay kit (WanLeibio, Inc.), and 40 µg proteins from each sample were subjected to 7% SDS-PAGE. Subsequently, the separated proteins were transferred onto a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). Following blocking with 5% skimmed milk for 1 hat room temperature, the membrane was incubated with rabbit polyclonal anti-TRPM2 antibody (1:400 dilution; cat. no. BA3459; Wuhan Boster Biological Technology, Ltd., Wuhan, China) at 4°C overnight. Following washing with TBS-Tween-20 (0.15% v/v), the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:5,000; cat. no. WLA023, WanLeibio, Inc.) for 45 min at 37°C. Finally, the blots were developed with ECL reagent (WanLeibio, Inc.), and the signals were exposed to films. Thereafter, the membrane was stripped with stripping buffer (WanLeibio, Inc.) and re-blotted with an anti-β-actin antibody (1:1,000; cat. no. WL01845; WanLeibio, Inc.) to verify equal loading and transfer of the proteins. The films were scanned, and the relative levels of TRPM2 were calculated using Gel-Pro-Analyzer (Media Cybernetics, Inc., Rockville, MD, USA) with β-actin as the internal reference.
Insulin secretion assay
Selected pancreatic islets were transferred into glass tubes (10 pancreatic islets/tube). A total of 500 µl of modified Krebs-Ringer buffer (K-R buffer; 115 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4. 7H2O, 1.2 mM KH2PO4, 25 mM NaHCO3 and 10 mM HEPES) containing 3.3 mM glucose, 1 mg/ml bovine serum albumin (Sigma-Aldrich; Merck KGaA) was added into each tube, which was then incubated at 37°C with 5% CO2 and continuous agitation for 30 min. The islets either continued to be incubated with the low-glucose K-R buffer (control), or were exposed to K-R buffer containing one or more of the following reagents: 5.6 or 16.6 mM glucose, 30 mM KCl, 10 nM GLP-1, 100 µM ADP-ribose (TRPM2 activator), 10 µM 2-APB (TRPM2 inhibitor), 100 µM 6-Benz cAMP (PKA activator), 10 µM H89 (PKA inhibitor), 10 µM 8-pCPT (Epac activator), and 10 µM ESI-09 (Epac inhibitor). The islets were incubated at 37°C, and the concentration of insulin in the supernatant was measured at 0, 10 and 60 min post-exposure by ELISA using a commercial kit (cat. no. 035-94, Phoenix Pharmaceuticals, Inc., Burlingame, CA, USA). For the experiments involving PKA or Epac activator/inhibitor, untreated cells were used as the control.
Electrophysiological measurements
Following finding cells under an inverted light microscope, a patch clamp amplifier (Axopatch-1D; Molecular Devices, LLC, Sunnyvale, CA, USA) was used to measure the current across the cell surface membrane. The bath solution was K-R buffer (pH7.4, adjusted with NaOH), and the pipette solution contained 40 mM K2SO4, 50 mM KCl, 5 mM MgCl2, 0.5 mM EGTA and 10 mM HEPES, (pH 7.2, adjusted with KOH). The pCLAMP™ software (version 10.2) and Digidata® 1440A (both Molecular Devices, LLC) were adopted to give stimulatory commands (voltage clamped at a holding potential of −70 mV) and record data. The flow velocity of HEPES buffer (containing 2.2 mM glucose) was 5 ml/min and the electrical resistance was ~3 MΩ. The Ca2+ current and the changes in membrane capacitance were recorded throughout continuous depolarization.
Statistical analysis
The data are expressed as the mean ± standard deviation of repeated experiments. Comparisons of multiple groups were performed using analysis of variance followed by Tukey's test. P<0.05 was considered to indicate a statistically significant difference.
Results
TRPM2 channels are involved in GSIS
To investigate the role of TRPM2 in insulin secretion in pancreatic β-cells, primary β-cells were infected with adenoviruses containing a TRPM2 expression construct or shTRPM2. Overexpression or silencing of TRPM2 was achieved in the transduced β-cells, and mRNA expression was significantly increased and decreased compared with the control group in the overexpression and shTRPM2 groups, respectively (Fig. 1A). A similar trend was observed in protein expression (Fig. 1B).
Figure 1.
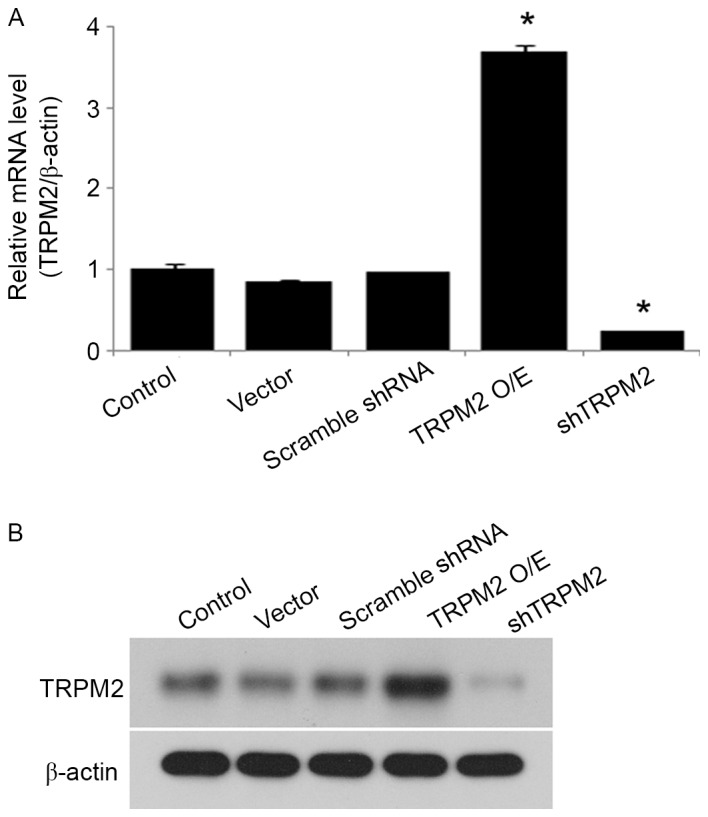
Successful transduction of adenoviral vectors into pancreatic β-cells. The pancreatic islets were infected with adenoviruses containing the pHBAd-MCMV vector, pHBAd-MCMV-TRPM2, pHBAd-U6-control shRNA (scramble shRNA) or pHBAd-U6-TRPM2 shRNA (shTRPM2). After 24 h, the expression levels of TRPM2 were examined by (A) reverse transcription-quantitative polymerase chain reaction analysis and (B) western blot analysis. n=3. *P<0.05 vs. the control group. TRPM2, transient receptor potential melastatin 2; O/E, overexpression; shRNA, short hairpin RNA; shTRPM2, shRNA directed against TRPM2.
To examine whether TRPM2 is involved in GSIS in primary β-cells, whole-cell patch clamp experiments and measurements of insulin secretion were conducted. TRPM2 currents were characterized in response to 100 µM ADP-ribose, a TRPM2 agonist, challenge in the cells transduced with scramble shRNA (typical current: gradual decline to the lowest value of −60 mV around 40 sec and remained at the lowest thereafter) or shTRPM2 (eliminated current) (Fig. 2A). During the exposure to a low (3.3 mM), normal (5.6 mM) and high (16.6 mM) concentration of glucose, the TRPM2 peak current (60 sec following ADP-ribose exposure) of the scramble shRNA-transduced cells increased significantly upon high-glucose stimulation compared with the shTRPM2-transduced cells (Fig. 2B). To determine whether high glucose-induced activation of TRPM2 channels is associated with GSIS and whether the modulatory effect is time-dependent, insulin secretion during the first 10 min (first phase) and the following 50 min (second phase) was assessed. Insulin secretion stimulated by a high glucose concentration was significantly reduced in shTRPM2 cells compared with scramble shRNA cells in the first and second phases (Fig. 2C and D, respectively), suggesting that the TRPM2 channels are involved in GSIS in β-cells.
Figure 2.

Glucose-stimulated insulin secretion is attenuated by TRPM2 knockdown in primary pancreatic islets. (A) A whole-cell patch clamp experiment was performed on pancreatic β-cells in the presence of 100 µM ADP-ribose, revealing that the current flow was eliminated in TRPM2-silenced cells. The plot is the representative current flow chart. (B) The peak current was recorded at three glucose concentrations. Only a high glucose concentration (16.6 mM) potentiated the current in the negative control islets, while TRPM2 silencing inhibited this effect. Measurements were taken in the (C) first (0–10 min) and (D) second (10–60 min) phases of insulin secretion. n=8. *P<0.05. shRNA, short-hairpin RNA; TRPM2, transient receptor potential melastatin 2; shTRPM2, shRNA directed against TRPM2.
GLP-1 potentiates GSIS via TRPM2, but TRPM2 activation is independent of K+-induced membrane depolarization
Glucose is known to close KATP channels and induce membrane depolarization, resulting in the opening of VDCCs and subsequent Ca2+ influx, which ultimately triggers the exocytosis of insulin-containing granules (40–42). To investigate whether a high K+ concentration influences the activation of TRPM2 during GSIS, the whole-cell TRPM2 current and second phase insulin secretion in the control and shTRPM2-transduced β-cells was examined. Exposure to 10 nM GLP-1 significantly elevated the peak current, whereas an additional 30 mM KCl did not produce a significant increase in the current compared with the cells treated with GLP-1 alone (Fig. 3A). In addition, a high K+ concentration challenge alone failed to induce a significant change in the current in the control cells (Fig. 3A). Silencing of TRPM2, however, significantly reduced the basal and GLP-1-stimulated currents in β-cells (Fig. 3A). These electrophysiology results suggest that GLP-1 is an agonist of TRPM2 and that the activity of the TRPM2 channel is independent of K+. In a low glucose concentration, KCl significantly enhanced insulin secretion regardless of the presence or absence of TRPM2 or GLP-1 (Fig. 3B). In a high glucose concentration condition, GLP-1 potentiated insulin secretion significantly, while shTRPM2 attenuated GSIS and GLP-1-potentiated GSIS (Fig. 3B). Additionally, cotreatment with GLP-1 and KCl did not significantly elevate GSIS compared with GLP-1 treatment alone (Fig. 3B). These observations indicate that TRPM2 is required for GSIS and GLP-1-potentiated GSIS, whereas a high K+ concentration stimulates insulin release under a low glucose concentration condition through another signaling pathway.
Figure 3.

GLP-1 potentiates glucose-stimulated insulin secretion via TRPM2 rather than through K+-induced insulin release. (A) Whole-cell currents were recorded in response to 5.6 mM glucose. When administered, 10 nM GLP-1 increased TRPM2 current with or without the addition of 30 mM KCl. (B) Insulin release was measured at a low (3.3 mM) and high (16.6 mM) glucose concentration. n=9. *P<0.05. #, &, §, ΔP<0.05 between the two groups with the same label. shRNA, short-hairpin RNA; TRPM2, transient receptor potential melastatin 2; shTRPM2, shRNA directed against TRPM2; ns, not significant; GLP-1, glucagon-like protein-1.
TRPM2 mediates GLP-1-potentiated GSIS
To validate the role of TRPM2 in GLP-1-potentiated GSIS, TRPM2 was overexpressed in β-cells, followed by whole-cell patch clamp experiment and measurement of insulin secretion at 60 and 90 sec. In the presence of 100 µM ADP-ribose, TRPM2 overexpression enhanced the ADP-ribose-induced current in the cells (Fig. 4A). Whereas 2-APB, a TRP channel inhibitor added at 60 sec, significantly inhibited the current in pHBAd-MCMV-GFP vector-transduced and TRPM2-overexpressing cells (Fig. 4B). These patch clamp experiment results indicate that exogenous TRPM2 is functional in β-cells. Next, the effect of TRPM2 overexpression on GSIS in the presence or absence of GLP-1 stimulation was examined. At a low glucose concentration, neither GLP-1 treatment nor TRPM2 overexpression potentiated insulin secretion (Fig. 4C). In addition, 2-APB exhibited no effect on insulin release under low glucose conditions (Fig. 4C). At a high glucose concentration, GLP-1 significantly increased insulin release and TRPM2 overexpression significantly stimulated GLP-1-potentiated GSIS (Fig. 4D). By contrast, 2-APB significantly eliminated GLP-1-potentiated GSIS in the control and TRPM2-overexpessing cells (Fig. 4D). These results highlight the important role of TRPM2 in GLP-1-potentiated GSIS.
Figure 4.

Manipulating the expression or activity of TRPM2 modulates GLP-1-potentiated glucose-stimulated insulin secretion. (A) Whole-cell currents were recorded and 2-APB was added at 60 sec to inhibit the TRPM2 channel in vector- and TRPM2-transduced β-cells. (B) The currents were recorded at 60 and 90 sec, and then analyzed to demonstrate the successful inhibition of the TRPM2 channel. The second phase of insulin secretion was measured at (C) low (3.3 mM) and (D) high (16.6 mM) glucose concentrations. GLP-1-induced insulin secretion was potentiated by TRPM2 overexpression and decreased by 2-APB in β-cells. n=8. *P<0.05. O/E, overexpression; GLP-1, glucagon-like protein-1; TRPM2, transient receptor potential melastatin 2.
Epac serves a role in GLP-1-potentiated GSIS, but GLP-1-potentiated GSIS does not act through PKA
To explore the underlying mechanisms of TRPM2 in GSIS, the roles of PKA and Epac in GLP-1-potentiated GSIS were examined. To achieve this a PKA activator (6-Benz cAMP), a PKA inhibitor (H89), an Epac activator (8-pCPT) and an Epac inhibitor (ESI-09) were used. A GLP-1-induced current was not inhibited by H89 in the presence of 5.6 mM glucose, and 6-Benz cAMP failed to increase the current (Fig. 5A). These results suggest that PKA is not be involved in the activation of TRPM2. By contrast, 8-pCPT significantly increased the current compared with the control, but there was no more increase following GLP-1 stimulation (Fig. 5B). Additionally, the GLP-1-stimulated current was significantly inhibited by ESI-09 and GLP-1 compared with treatment with GLP-1 alone (Fig. 5B). These data suggest that Epac serves an important role in the GLP-1-induced activation of TRPM2.
Figure 5.

Epac serves a role in GLP-1-potentiated GSIS, but GLP-1-potentiated GSIS does not act through PKA. The following drugs were used to examine the functions of PKA and Epac in GSIS and GLP-1-potentiated GSIS in β-cells: 6-Benz cAMP (PKA activator), H89 (PKA inhibitor), 8-pCPT (Epac activator) and ESI-09 (Epac inhibitor). Whole-cell currents in response to 5.6 mM glucose and (A) GLP-1, 6-Benz cAMP and H89, and (B) GLP-1, 8-pCPT and ESI-09 were recorded at 60 sec following glucose administration. An insulin secretion experiment was performed at a high glucose concentration (16.6 mM) in order to observe the second phase of insulin secretion in response to (C) GLP-1, 6-Benz cAMP and H89, and (D) GLP-1, 8-pCPT and ESI-09. n=9. *P<0.05. ns, not significant; GLP-1, glucagon-like protein-1; Epac, exchange protein directly activated by cAMP; GSIS, glucose-stimulated insulin secretion; PKA, protein kinase A; TRPM2, transient receptor potential melastatin 2.
In a high glucose concentration (16.6 mM) condition, 6-Benz cAMP exhibited a stronger efficacy compared with GLP-1 on potentiating insulin secretion, while H89 markedly reduced GSIS and GLP-1-potentiated GSIS (Fig. 5C). This result indicates that GLP-1 and PKA stimulate insulin secretion via different signaling pathways. Conversely, 8-pCPT stimulation significantly increased insulin secretion compared with the control, which was not enhanced by additional GLP-1 (Fig. 5D). Additionally, ESI-09 significantly inhibited GLP-1-induced insulin secretion compared with GLP-1-potentiated insulin secretion under high glucose conditions (Fig. 5D). These data are consistent with current knowledge and suggest that GLP-1 potentiates GSIS via the Epac rather than PKA signaling pathway.
Discussion
The present study demonstrated that TRPM2 channel opening and KATP channel closure promoted GSIS in pancreatic β-cells. Typically, the closure of the KATP channels results in membrane depolarization, which initiates the activation of VDCCs. Ca2+ influx through VDCCs leads to an increase in [Ca2+]i and Ca2+-induced Ca2+ release (CICR) from the ER. This elevated [Ca2+]i is an effective stimulus for insulin secretion. Therefore, insulin release in response to a high K+ concentration challenge exhibits independency from glucose metabolism. By contrast, in the presence of glucose, glucose metabolism causes an increase in the cytosolic ATP/ADP ratio, which induces the closure of KATP channels, resulting in membrane depolarization and periodic influx of Ca2+. In such circumstances, high K+ treatment cannot further enhance insulin secretion.
In response to an elevated ATP level upon glucose induction, adenylyl cyclase produces the second messenger cAMP, which serves a critical role in the activation of TRPM2. GLP-1R is a ligand-specific G-protein-coupled receptor. When activated by GLP-1, GLP-1R couples with a trimeric G-protein complex and facilitates the release of the activated Gαs subunit from the complex, and Gαs, in turn, activates membrane-bound adenylyl cyclase to produce cAMP (43). During this process, ATP acts as the substrate for the generation of cAMP. Thus, glucose is essential for GLP-1/TRPM2-mediated insulin secretion. In addition to cAMP, glucose stimulates the generation of cADP-ribose, another second messenger important for Ca2+ mobilization from the ER and subsequent insulin secretion from the pancreatic islets (44). cADP-ribose activates TRPM2 and is involved in the GLP-1-mediated Ca2+ signal for the regulation of insulin secretion (21,45). Therefore, cAMP and cADP-ribose may be involved in GLP-1-regulated GSIS via TRPM2.
The present study demonstrated that interfering with the expression or activity of TRPM2 influenced GLP-1-stimulated GSIS. These results provide support for the following hypothesis of the present study: That GLP-1 potentiates GSIS via the TRPM2 channel. The two primary downstream effectors of GLP-1R are PKA and Epac, and by using their agonists and antagonists the present study revealed that GLP-1R activated the TRPM2 channel via the cAMP-Epac signaling pathway rather than the PKA signaling pathway. Nonetheless, the activation of the Epac or PKA signaling pathways can result in the augmentation of GSIS, since their inhibitors were demonstrated to suppress GSIS.
Taken together, these results indicate potential mechanisms through which the Epac and PKA signaling pathways regulate GSIS. The binding of GLP-1 with GLP-1R leads to the phosphorylation of adenylyl cyclase and an elevated extracellular glucose concentration, which increases ATP as a consequence of intracellular glucose metabolism. The elevated ATP is converted to cAMP by adenylyl cyclase, and cAMP then promotes gene expression via PKA-mediated phosphorylation of CREB family activators for insulin biosynthesis. Additionally, Epac-mediated activation of the TRPM2 channel triggers Ca2+ influx, which serves an important role in the exocytosis of insulin-containing granules (46). In addition, the increase in [Ca2+]i acts as a stimulus for CICR from the ER through ryanodine receptors (RyRs). ER-released Ca2+ is an agonist of the inositol 1,4,5-triphosphate receptor (IP3R) and stimulates additional Ca2+ release from IP3R-regulated Ca2+ stores; RyR- and IP3R-mediated Ca2+ release is facilitated by the PKA signaling pathway (3,47). As a result, increases in insulin biosynthesis and exocytosis cause enhanced insulin secretion, and the PKA and Epac signaling pathways serve different roles in the process of GSIS.
In conclusion, the results of the present study demonstrated that the expression level and activity of TRPM2 affects GSIS. GLP-1 was demonstrated to enhance GSIS by activating TRPM2 via the Epac signaling pathway, whereas KATP closure-mediated activation of VDCCs facilitated insulin release in a glucose-independent manner. Thus, the present study highlights a novel area for the prevention and treatment of diabetes and its complications.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant nos. 81300664 and 81470187), the Natural Science Foundation of Tianjin (grant no. 14JCYBJC26200) and the Foundation of Tianjin Medical University (grant no. 279). The authors thank all clinical and laboratory staff in the Metabolic Diseases Hospital of Tianjin Medical University and the Department of physiology at Seoul National University College of Medicine for their dedication to this study.
References
- 1.Rorsman P, Eliasson L, Renström E, Gromada J, Barg S, Göpel S. The cell physiology of biphasic insulin secretion. News Pysiol Sci. 2000;15:72–77. doi: 10.1152/physiologyonline.2000.15.2.72. [DOI] [PubMed] [Google Scholar]
- 2.Efanov AM, Zaitsev SV, Berggren PO. Inositol hexakisphosphate stimulates non-Ca2+-mediated and primes Ca2+-mediated exocytosis of insulin by activation of protein kinase C. Proc Natl Acad Sci USA. 1997;94:4435–4439. doi: 10.1073/pnas.94.9.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reinbothe TM, Alkayyali S, Ahlqvist E, Tuomi T, Isomaa B, Lyssenko V, Renström E. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia. 2013;56:340–349. doi: 10.1007/s00125-012-2758-z. [DOI] [PubMed] [Google Scholar]
- 4.Dyachok O, Gylfe E. Store-operated influx of Ca(2+) in pancreatic β-cells exhibits graded dependence on the filling of the endoplasmic reticulum. J Cell Sci. 2001;114:2179–2186. doi: 10.1242/jcs.114.11.2179. [DOI] [PubMed] [Google Scholar]
- 5.Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- 6.Colsoul B, Vennekens R, Nilius B. Transient receptor potential cation channels pancreatic-β cells. Rev Physiol Biochem Pharmacol. 2011;161:87–110. doi: 10.1007/112_2011_2. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z, Zhang W, Jung DY, Ko HJ, Lee Y, Friedline RH, Lee E, Jun J, Ma Z, Kim F, et al. TRPM2 Ca2+ channel regulates energy balance and glucose metabolism. Am J Physiol Endocrinol Metab. 2012;302:E807–E816. doi: 10.1152/ajpendo.00239.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60:470–512. doi: 10.1124/pr.108.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang XL, Ye F, Li J, Zhu LY, Feng G, Chang XY, Sun K. Impaired secretion of glucagon-like peptide 1 during oral glucose tolerance test in patients with newly diagnosed type 2 diabetes mellitus. Saudi Med J. 2016;37:48–54. doi: 10.15537/smj.2016.1.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman JS. Improving glucagon-like peptide-1 dynamics in patients with type 2 diabetes mellitus. J Am Osteopath Assoc. 2012;112(1 Suppl 1):S2–S6. [PubMed] [Google Scholar]
- 11.Gavin JR., III Initiating a glucagon-like peptide-1 receptor agonist in the management of type 2 diabetes mellitus. J Am Osteopath Assoc. 2012;112(1 Suppl 1):S16–S21. [PubMed] [Google Scholar]
- 12.Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabetes Obes Metab. 2013;15:15–27. doi: 10.1111/j.1463-1326.2012.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahlouli S, Mokaddem A, Hamdache F, Riane H, Kamenche M. Fractal behavior of the pancreatic β-cell near the percolation threshold: Effect of the KATP channel on the electrical response. IEEE/ACM Trans Comput Biol Bioinform. 2016;13:112–121. doi: 10.1109/TCBB.2015.2415797. [DOI] [PubMed] [Google Scholar]
- 14.Toft-Nielsen MB, Madsbad S, Holst JJ. Determinants of the effectiveness of glucagon-like peptide-1 in type 2 diabetes. J Clin Endocrinol Metab. 2001;86:3853–3860. doi: 10.1210/jcem.86.8.7743. [DOI] [PubMed] [Google Scholar]
- 15.Oetjen E, Diedrich T, Eggers A, Eckert B, Knepel W. Distinct properties of the cAMP-responsive element of the rat insulin I gene. J Biol Chem. 1994;269:27036–27044. [PubMed] [Google Scholar]
- 16.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 17.Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, McNulty S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong CW, Kim TK, Ham HY, Nam JS, Kim YH, Zheng H, Pang B, Min TK, Jung JS, Lee SN, et al. Lysophosphatidylcholine increases neutrophil bactericidal activity by enhancement of azurophil granule-phagosome fusion via glycine. GlyR alpha 2/TRPM2/p38 MAPK signaling. J Immunol. 2010;184:4401–4413. doi: 10.4049/jimmunol.0902814. [DOI] [PubMed] [Google Scholar]
- 20.Wehrhahn J, Kraft R, Harteneck C, Hauschildt S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J Immunol. 2010;184:2386–2393. doi: 10.4049/jimmunol.0902474. [DOI] [PubMed] [Google Scholar]
- 21.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–1815. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 23.Beck A, Kolisek M, Bagley LA, Fleig A, Penner R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006;20:962–964. doi: 10.1096/fj.05-5538fje. [DOI] [PubMed] [Google Scholar]
- 24.Starkus JG, Fleig A, Penner R. The Calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J Physiol. 2010;588:1227–1240. doi: 10.1113/jphysiol.2010.187476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kühn FJ, Heiner I, Lückhoff A. TRPM2: A calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch. 2005;451:212–219. doi: 10.1007/s00424-005-1446-y. [DOI] [PubMed] [Google Scholar]
- 26.Naziroğlu M. New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res. 2007;32:1990–2001. doi: 10.1007/s11064-007-9386-x. [DOI] [PubMed] [Google Scholar]
- 27.Pang B, Shin DH, Park KS, Huh YJ, Woo J, Zhang YH, Kang TM, Lee KY, Kim SJ. Differential pathways for calcium influx activated by concanavalin A and CD3 stimulation in Jurkat T cells. Pflugers Arch. 2012;463:309–318. doi: 10.1007/s00424-011-1039-x. [DOI] [PubMed] [Google Scholar]
- 28.Uchida K, Dezaki K, Damdindor B, Inada H, Shiuchi T, Mori Y, Yada T, Minokoshi Y, Tominaga M. Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes. 2011;60:119–126. doi: 10.2337/db10-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egan JM, Bulotta A, Hui H, Perfetti R. GLP-1 receptor agonists are growth and differentiation factors for pancreatic islet beta cells. Diabetes Metab Res Rev. 2003;19:115–123. doi: 10.1002/dmrr.357. [DOI] [PubMed] [Google Scholar]
- 30.Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, Pipeleers D, Ling Z, Drucker DJ. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4:391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Gomez E, Pritchard C, Herbert TP. cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediate Raf-independent activation of extracellular regulated kinase in response to glucogon-like peptide-1 in pancreatic beta-cells. J Biol Chem. 2002;277:48146–48151. doi: 10.1074/jbc.M209165200. [DOI] [PubMed] [Google Scholar]
- 32.Arnette D, Gibson TB, Lawrence MC, January B, Khoo S, McGlynn K, Vanderbilt CA, Cobb MH. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem. 2003;278:32517–32525. doi: 10.1074/jbc.M301174200. [DOI] [PubMed] [Google Scholar]
- 33.Buteau J, Roduit R, Susini S, Prentki M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increase transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia. 1999;42:856–864. doi: 10.1007/s001250051238. [DOI] [PubMed] [Google Scholar]
- 34.Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- 35.Renstrom E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic β-cells. J Physiol. 1997;502:105–118. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem. 2003;278:8279–8285. doi: 10.1074/jbc.M211682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujimoto K, Shibasaki T, Yokoi N, Nashima Y, Matsumoto M, Sasaki T, Tajima N, Iwanaga T, Seino S. Piccolo, a Ca2+ sensor in pancreatic beta-cells. Involvement of cAMP-GEFII. Rim2. Piccolo complex in cAMP-dependent exocytosis. J Biol Chem. 2002;277:50497–50502. doi: 10.1074/jbc.M210146200. [DOI] [PubMed] [Google Scholar]
- 38.Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFFII-Rim2 complex in incretin-protentiated insulin secretion. J Biol Chem. 2001;276:46046–46053. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- 39.Overbergh L, Vig S, Coun F, Mathieu C. Chapter 4: Quantitative polymerase chain reaction. In: Patrinos GP, editor. Molecular Diagnostics. 3rd. Academic Press; 2017. pp. 41–58. [DOI] [Google Scholar]
- 40.Damdindorj B, Dezaki K, Kurashina T, Sone H, Rita R, Kakei M, Yada T. Exogenous and endogenous ghrelin counteracts GLP-1 action to stimulate cAMP signaling and insulin secretion in islet β-cells. FEBS Lett. 2012;586:2555–2562. doi: 10.1016/j.febslet.2012.06.034. [DOI] [PubMed] [Google Scholar]
- 41.Holz GG, Heart E, Leech CA. Synchronizing Ca2+ and cAMP oscillations in pancreatic beta-cells: A role for glucose metabolism and GLP-1 receptors? Focus on ‘regulation of cAMP dynamics by Ca2+ and G protein-coupled receptors in the pancreatic beta-cell: A computational approach’. Am J Physiol Cell Physiol. 2008;294:C4–C6. doi: 10.1152/ajpcell.00522.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shigeto M, Katsura M, Matsuda M, Ohkuma S, Kaku K. Low, but physiological, concentration of GLP-1 stimulates insulin secretion independent of the cAMP-dependent protein kinase pathway. J Pharmacol Sci. 2008;108:274–279. doi: 10.1254/jphs.08090FP. [DOI] [PubMed] [Google Scholar]
- 43.Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takasawa S, Nata K, Yonekura H, Okamoto H. Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science. 1993;259:370–373. doi: 10.1126/science.8420005. [DOI] [PubMed] [Google Scholar]
- 45.Kim BJ, Park KH, Yim CY, Takasawa S, Okamoto H, Im MJ, Kim UH. Generation of nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes. 2008;57:868–878. doi: 10.2337/db07-0443. [DOI] [PubMed] [Google Scholar]
- 46.Fridlyand LE, Philipson LH. Coupling of metabolic, second messenger pathways and insulin granule dynamics in pancreatic beta-cells: A computational analysis. Prog Biophys Mol Biol. 2011;107:293–303. doi: 10.1016/j.pbiomolbio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park S, Dong X, Fisher TL, Dunn S, Omer AK, Weir G, White MF. Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J Biol Chem. 2006;281:1159–1168. doi: 10.1074/jbc.M508307200. [DOI] [PubMed] [Google Scholar]