

 The first application of synthetic halogen bond donors as catalysts in a Michael addition reaction is described.
The first application of synthetic halogen bond donors as catalysts in a Michael addition reaction is described.
Abstract
Only a few studies on the use of halogen bonding in catalysis have been published so far. Herein, (benz)imidazolium-based halogen bond donors are used as catalysts in a Michael addition reaction. The most potent catalyst, a rigid atropisomer featuring two iodobenzimidazolium moieties, provided a rate acceleration versus a reference compound of ca. 50.
Noncovalent attractive interactions between the electrophilic region of polarized halogen substituents and Lewis bases – the so-called halogen bonds – have been established in the last few decades as a valuable tool in solid phase chemistry1–3 and have recently also been increasingly investigated in solution.4,5 Overall, the interaction is quite similar to hydrogen bonding and may reach a similar strength in solution, but is more directional.6,7 Recent trends in the exploration of XB in solution are related to fundamental investigations,8 anion recognition,9 molecular self-assembly10,11 and organocatalysis.12–14
The use of halogen bonding in organocatalysis is still at an early stage.15 In 2009, Bolm et al. reported the reduction of quinoline derivatives with Hantzsch ester, catalysed by iodoperfluoroalkanes.16 In 2013, our group described the use of multidentate polyfluorinated iodoterphenyls as organocatalysts in a halide abstraction benchmark reaction (i.e., the reaction of 1-chloroisochroman with a ketene silyl acetal).13 In addition, two halogen-bonding catalyzed Diels–Alder reactions have been reported: the reaction of cyclopentadiene with methyl vinyl ketone12 as well as an aza-Diels–Alder reaction.17 Hence, even though the examples mentioned above are not comprehensive, there are still only a handful of reactions which are catalyzed by halogen bond donors.
In the beginning of 2017, Breugst and co-workers presented a mechanistic study of a Michael addition reaction catalyzed by elemental iodine (Scheme 1).18
Scheme 1. Michael addition benchmark reaction.

These investigations provided strong indications towards halogen bonding being at the origin of the mode of action of iodine. However, even though elemental iodine is a strong halogen bond donor, it is obviously not susceptible to modification. We thus wondered whether this catalysis is also possible with organic halogen bond donors, which, in the long term, would offer more variability. Thus, herein we investigate the use of previously established monodentate and bidentate (benz)imidazolium-based halogen bond donors as catalysts in the Michael reaction between indole (A) and trans-crotonophenone (B) (Scheme 1).
Previous research12 suggests that the role of the counterion of the cationic halogen bond donors is also very important in this kind of catalysis. Due to the weak activities of cationic XB donors with triflate counterions in a Diels–Alder benchmark reaction,12 we decided to use noncoordinating BArF4– (tetrakis[3,5-bis(trifluoro-methyl)phenyl]borate) counterions, as their interference with neutral Lewis bases in solution is unlikely. The substrate in the current study, trans-crotonophenone, is a slightly more electron-poor analogue of methyl vinyl ketone, and thus we expected somewhat similar activities of our cationic XB donors.
To determine and compare the catalytic properties of the halogen bond donors shown in Fig. 2, the Michael addition benchmark reaction (Scheme 1) was monitored via1H-NMR spectroscopy. Dichloromethane was chosen as the solvent, and catalyst loadings were at 20 mol%.
Fig. 2. Catalysts applied in the benchmark reaction.

First, we looked at the activity of the corresponding non-halogenated reference compounds. As halogen bonding – similar to hydrogen bonding – is a comparably weak interaction, the halogen bond donors may also interact via other interactions with the substrate, and it is important to rule these out as the driving force of the catalysis as best as possible. Possible such interactions are for instance π-stacking involving the cationic heteroaromatic core of the halogen bond donor or hydrogen bonding of its backbone protons. Since all non-halogen bonding interactions are also feasible for the non-halogenated compounds, a comparison of their performance with the halogen bond donors is the most direct way to measure the relevance of halogen bonding.
Non-halogenated compounds 1 and 2, despite being relatively strong hydrogen bond donors due to the electron-withdrawing heteroarene moiety, are virtually inactive in the chosen Michael reaction. The conversion of monodentate imidazolium derivative 1 (Fig. 1, purple line) and its bidentate variant 2 (yellow line) are shown in Fig. 1. The catalyst candidate 1 showed no sign of activity, and only 6% of product C are obtained with bidentate compound 2 after 36 hours (see also Table 1).
Fig. 1. Conversion versus time profile of the benchmark reaction with various catalysts (see Fig. 2).

Table 1. Conversion to product C after 36 h and the relative reaction rate krel (measured by the initial slope).
| Catalyst | Conversion (%) | k rel |
| 1 | 0 | 0.2 |
| 2 | 6 | 1 |
| 3 | 9 | 1 |
| 4 | 9 | 1 |
| 5 | 14 | 3 |
| 6 | 32 | 4 |
| 7 | 63 | 8 |
| 8 | 57 | 12 |
| 9 | 68 | 50 |
Next, we studied the influence of the heteroarene backbone using a monodentate halogen bond donor. Since iodine substituents form stronger Lewis acids compared to the lighter halogens, iodoimidazolium derivatives might have been the obvious choice. However, as noted above, elemental iodine is also very active in this reaction (76% conversion at a 5 mol% catalyst loading in CH2Cl2 after only 3 minutes at room temperature).18 Thus, traces of iodine, potentially produced by the decomposition of the halogen bond donor, could influence the kinetic study. Even though we have not obtained any indication of decomposition via NMR spectroscopy for any of the halogen bond donors reported herein, it is still difficult to rule out with certainty tiny amounts of iodine. As a consequence, brominated compounds were investigated first, as these issues may be satisfactorily solved for a potential liberation of elemental bromine (see below). Brominated halogen bond donor 3 led to a conversion of approximately 9% after 36 hours (Fig. 1, orange line), which is markedly higher than the one of its non-brominated reference compound 1. The different activity is also obvious from the relative reaction rate (determined by the initial slope of conversion versus time), which is about 5-fold higher for 3 relative to 1. The benzimidazolium core features a stronger electron withdrawing effect than the imidazolium backbone, and thus catalyst 5 is expected to be more active than compound 3. Indeed, a slightly higher conversion of approximately 14% was observed for halogen bond donor 5 (Fig. 1, grey line). This, together with the higher rate acceleration of the benchmark reaction by 5, may be attributed to a more polarized bromine substituent by the more electronegative benzimidazolium core.
To rule out an activation of the reaction by elemental bromine19 (generated by potential slight decomposition of the halogen bond donor), the experiments were repeated in the presence of cyclohexene (in an equal amount as the catalyst). As we had previously shown in a halogen bonding activated halide abstraction reaction,20 cyclohexene instantly quenches the activity of elemental bromine. The addition of cyclohexene to the benchmark reaction with catalyst 3 or 5 showed no significant influence. Any effect of cyclohexene on the reaction was also ruled out by its addition to the blank reaction. These experiments provide strong indications that the observed catalytic activity of 3 and 5 (as well as of 6, see below) is indeed due to the action of halogen bonding.
Bidentate halogen bond donors are expected to provide stronger catalytic activity, and thus several such species were subsequently tested. Somewhat surprisingly, activation was observed with the bidentate chlorinated halogen bond donor 4 (Fig. 1, red line), even though chlorine substituents are much less polarizable than the bromine and iodine ones. The activation strength is comparable to the activity of the monodentate bromine substituted catalysts 3, with a conversion of around 9% after 36 hours. A catalytic activity of chlorinated halogen bond donors was also reported in an aza-Diels–Alder reaction by the group of Takeda.17
For comparison, we tested the analogous brominated catalyst 6, albeit with triflate as the counterion since the corresponding BArF4–-salt could not be prepared.21 As triflate is more coordinating and thus more competitive than BArF4–, a detrimental influence on the catalytic activity is expected. On the other hand, bromine substituents are much more polarizable than their chlorine analogues, and the latter effect seems to dominate, as the conversion in the presence of catalyst 6 (Fig. 1, pale blue line) was markedly higher compared to 4. Over a period of 36 hours, compound 6 provided a yield of product C of approximately 32%.
Finally, we further enhanced the strength of the halogen bond donors by introducing iodine substituents. Based on previous studies and the comparison of catalysts 3 and 5, it was expected that the benzimidazolium-based compound 8 would be more active than its imidazolium analogue 7. Indeed, catalyst 8 provided a stronger initial acceleration of the reaction rate (Table 1). However, the lines of conversion-over-time for 7 (Fig. 1, dark blue) and 8 (green line) cross at approximately 24 hours. Currently, we cannot explain this behaviour satisfactorily. After 12 hours, the halogen bond donor 8 provided approximately 43% conversion compared to 34% by catalyst 7. At the end of the reaction period, the initially less active compound 7 resulted in a 10% higher conversion compared to its analogue 8.
In a halide abstraction test reaction, we could demonstrate that a syn-preorganized halogen bond donor featuring the cation of 9 was distinctively more active than Lewis acids like 7 or 8.14 Previously, however, this compound was only available with triflate counterions since several attempts of exchanging the anions failed due to decomposition. Lately, we could establish a successful method of anion exchange to obtain catalyst 9 (Scheme 2, see also ESI†).12
Scheme 2. Anion exchange between triflate and BArF4– salt.

When compound 9 was used as a catalyst in the benchmark reaction, a very fast product formation was observed (Fig. 1, dark grey line). Its relative initial rate acceleration – compared to the reference compound 2 – is about 50-fold. After three hours, almost 60% conversion was achieved. Subsequently, the reaction rate decreased markedly, and the conversion seemed to reach some kind of plateau at approximately 70% conversion.
All these findings showed that halogen bond donors, especially the iodinated ones, are active as catalysts, but overall less potent than elemental iodine. In a computational study, we aimed to (a) obtain a model of a likely transition state of the halogen bond catalysis and (b) see whether the relative strengths of the catalysts may be reproduced in silico. Consequently, the Michael reaction was modelled with DFT using the M062X functional22 with D3 dispersion corrections by Grimme23 and the triple-zeta TZVPP basis set.24 Transition states were obtained for the uncatalyzed reaction and the transformations in the presence of molecular iodine or model halogen bond donor 7 (with methyl groups on nitrogen and without counterions for reasons of computational costs). The latter transition state – which features a bidentate coordination of the halogen bond donating moieties to the carbonyl group – is shown in Fig. 3.
Fig. 3. Transition state of the Michael reaction catalyzed by the cation of halogen bond donor 7 (as obtained by DFT). Figure generated with CYLview,25 viewed along the newly-forming C–C bond. O–I distances are 2.62 and 2.64 Å.
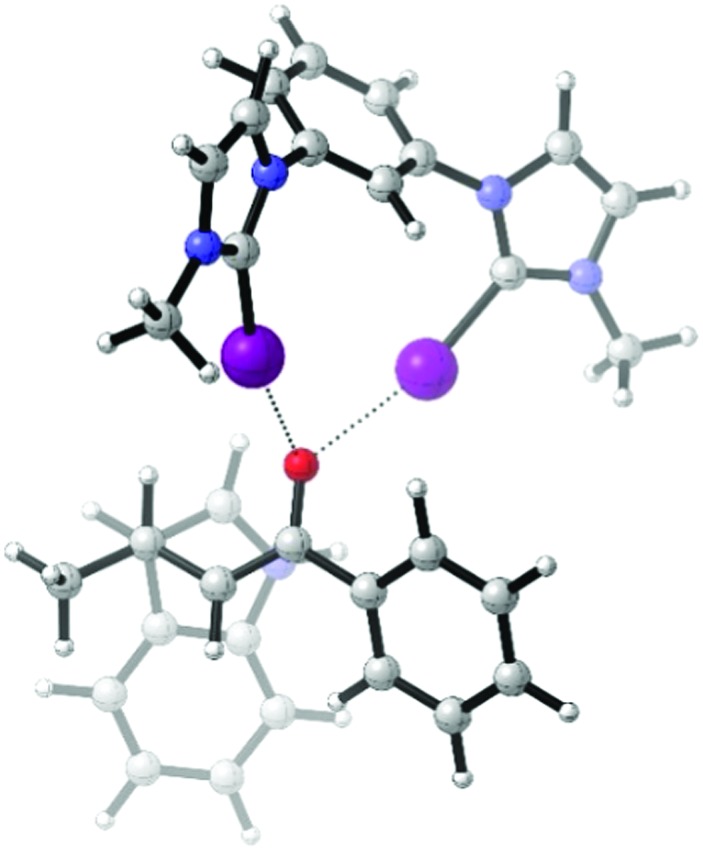
A comparison of the computed reaction barriers suffers from the intrinsic problem that 7 is dicationic while elemental iodine is a neutral species, and thus the Lewis acidity of 7 will be overestimated in the gas phase (indeed, barriers of approx. 34 kcal mol–1, 30 kcal mol–1 and 17 kcal mol–1 were obtained for the blank reaction and the catalyses by iodine and 7, respectively). The inclusion of an implicit solvation model (SMD with parameters for dichloromethane)26 attenuated this drastic difference to some degree (with barriers of 31 kcal mol–1, 25 kcal mol–1 and 21 kcal mol–1) but still maintained the wrong trend between the catalyses by iodine and 7. Thus, modelling the catalytic activity of these halogen bond donors via standard DFT methods seems very challenging.
In conclusion, (only) the second example12 demonstrating the activation of a carbonyl group by synthetic halogen bond donors27 as catalysts was presented. Comparison experiments demonstrated that other possible interactions like anion–π are not the main mode of catalysis. For the brominated halogen bond donors, a potential involvement of elemental bromine as a decomposition product could be ruled out. The most potent halogen bonding catalyst leads to a rate acceleration – relative to a barely active reference compound – by a factor of about 50.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (638337).
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc07175b
References
- Brammer L., Minguez Espallargas G., Libri S. CrystEngComm. 2008;10:1712. [Google Scholar]
- Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G. Chem. Rev. 2016;116:2478. doi: 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen K. CrystEngComm. 2008;10:1107. [Google Scholar]
- Erdelyi M. Chem. Soc. Rev. 2012;41:3547. doi: 10.1039/c2cs15292d. [DOI] [PubMed] [Google Scholar]
- Beale T. M., Chudzinski M. G., Sarwar M. G., Taylor M. S. Chem. Soc. Rev. 2013;42:1667. doi: 10.1039/c2cs35213c. [DOI] [PubMed] [Google Scholar]
- Walter S. M., Kniep F., Rout L., Schmidtchen F. P., Herdtweck E., Huber S. M. J. Am. Chem. Soc. 2012;134:8507. doi: 10.1021/ja2119207. [DOI] [PubMed] [Google Scholar]
- Libri S., Jasim N. A., Perutz R. N., Brammer L. J. Am. Chem. Soc. 2008;130:7842. doi: 10.1021/ja8020318. [DOI] [PubMed] [Google Scholar]
- Metrangolo P., Panzeri W., Recupero F., Resnati G. J. Fluorine Chem. 2002;114:27. [Google Scholar]
- Mele A., Metrangolo P., Neukirch H., Pilati T., Resnati G. J. Am. Chem. Soc. 2005;127:14972. doi: 10.1021/ja054862h. [DOI] [PubMed] [Google Scholar]
- Vanderkooy A., Taylor M. S. J. Am. Chem. Soc. 2015;137:5080. doi: 10.1021/jacs.5b00754. [DOI] [PubMed] [Google Scholar]
- Massena C. J., Wageling N. B., Decato D. A., Martin Rodriguez E., Rose A. M., Berryman O. B. Angew. Chem., Int. Ed. 2016;55:12398. doi: 10.1002/anie.201605440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbauer S. H., Walter S. M., Schindler S., Rout L., Kniep F., Huber S. M. Chem. Commun. 2014;50:6281. doi: 10.1039/c4cc03124e. [DOI] [PubMed] [Google Scholar]
- Kniep F., Jungbauer S. H., Zhang Q., Walter S. M., Schindler S., Schnapperelle I., Herdtweck E., Huber S. M. Angew. Chem., Int. Ed. 2013;52:7028. doi: 10.1002/anie.201301351. [DOI] [PubMed] [Google Scholar]
- Jungbauer S. H., Huber S. M. J. Am. Chem. Soc. 2015;137:12110. doi: 10.1021/jacs.5b07863. [DOI] [PubMed] [Google Scholar]
- Bulfield D., Huber S. M. Chem. – Eur. J. 2016;22:14434. doi: 10.1002/chem.201601844. [DOI] [PubMed] [Google Scholar]
- Bruckmann A., Pena M. A., Bolm C. Synlett. 2008:900. [Google Scholar]
- Takeda Y., Hisakuni D., Lin C. H., Minakata S. Org. Lett. 2015;17:318. doi: 10.1021/ol503426f. [DOI] [PubMed] [Google Scholar]
- von der Heiden D., Bozkus S., Klussmann M., Breugst M. J. Org. Chem. 2017;82:4037. doi: 10.1021/acs.joc.7b00445. [DOI] [PubMed] [Google Scholar]
- The reaction was completed after 30 min with 20 mol% of Br2 as catalyst
- Kniep F., Walter S. M., Herdtweck E., Huber S. M. Chem. – Eur. J. 2012;18:1306. doi: 10.1002/chem.201103071. [DOI] [PubMed] [Google Scholar]
- Decomposition of the cation was observed
- Zhao Y., Truhlar D. G. Theor. Chem. Acc. 2008;120:215. [Google Scholar]
- Grimme S., Antony J., Ehrlich S., Krieg H. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Weigend F., Ahlrichs R., Peterson K. A., Figgen D., Goll E., Stoll H., Dolg M. Phys. Chem. Chem. Phys. J. Chem. Phys. 2005;2003;7119:3297. 11113. [Google Scholar]
- Legault C. Y., CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org.
- Marenich A. V., Cramer C. J., Truhlar D. G. J. Phys. Chem. B. 2009;113:6378. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- “Synthetic” refers to XB donors with an organic backbone
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.