

Abstract
For more than two decades, de novo protein design has proven to be an effective methodology for modeling native proteins. De novo design involves the construction of metal-binding sites within simple and/or unrelated α-helical peptide structures. The preparation of α3D, a single polypeptide that folds into a native-like three-helix bundle structure, has significantly expanded available de novo designed scaffolds. Devoid of a metal-binding site (MBS), we incorporated a 3Cys and 3His motif in α3D to construct a heavy metal and a transition metal center, respectively. These efforts produced excellent functional models for native metalloproteins/metalloregulatory proteins and metalloenzymes. Morever, these α3D derivatives serve as a foundation for constructing redox active sites with either the same (e.g., 4Cys) or mixed (e.g., 2HisCys) ligands, a feat that could be achieved in this preassembled framework. Here, we describe the process of constructing MBSs in α3D and our expression techniques.
Keywords: De novo protein design, Three-helix bundle, Metal-binding site, Metalloprotein, Metalloregulatory protein, Metalloenzyme, Protein expression
1 Introduction
De novo protein design offers a methodology for modeling the metal centers of metalloproteins and metalloenzymes [1-3]. This approach involves the construction of a desired metal-binding site(s) in a peptide scaffold with a sequence that is not found in nature, thus allowing scientists to uncover physical properties that may remain hidden from direct studies of native proteins. The most commonly used scaffolds have an α-helical fold and have previously been engineered to contain heme, nonheme iron, and zinc centers [4]. Much of our efforts have focused on building a 3Cys site in the TRI and Coil-Ser (CS) peptide system [3, 5]. This thiol-rich site is accomplished through the self-association of a single TRI or CS peptide into a three-stranded coiled tertiary structure (3SCC) (Fig. 1a) [6]. Our work with the 3SCC scaffolds has generated excellent spectroscopic, structural, and functional models for native metalloregulatory proteins that bind toxic heavy metals, including arsenic, cadmium, mercury, and lead [3]. Moreover, in an effort to recapitulate the activity of metalloenzymes bound to a transition metal, TRI constructs with a 3His site had also been developed and shown to possess copper nitrite reductase activity [7, 8] and zinc carbonic anhydrase [9, 10].
Fig. 1.
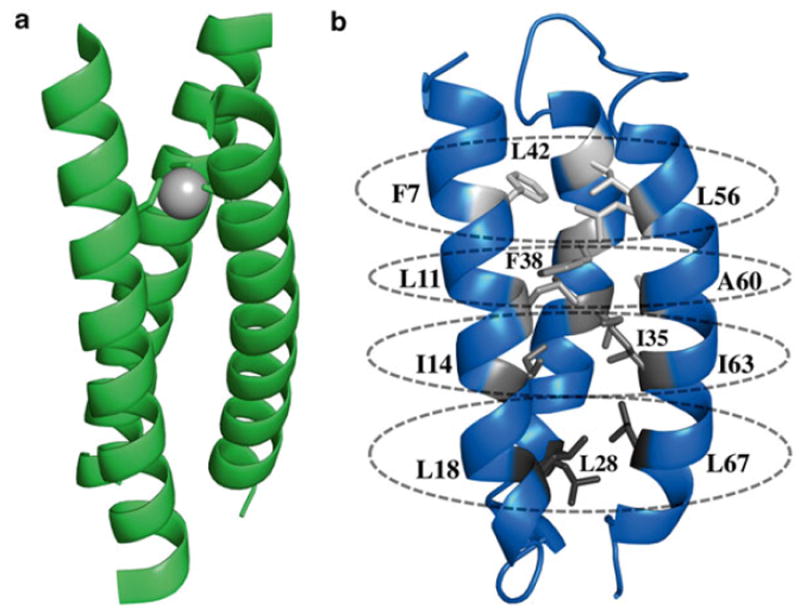
Structures of de novo designed peptides. (a) X-ray crystal structure of As(III) bound CSL9C (PDB 2JGO), a three-stranded coiled coil scaffold. (b) Solution structure of α3D. Apolar residues of α3D divided into four layers, as indicated by varying shades of gray. The first layer comprises F7, L42, and L56 at the N-terminal end of the bundle. Subsequent layer contains L11, F38, and A60. The third layer has all isoleucine residues at the 14th, 35th, and 63rd positions. The C-terminal layer composes of L18, L28, and L67. These layers were predicted to provide a 3Cys metal-binding site
DeGrado and coworkers expanded available de novo designed scaffolds through the preparation of a native-like peptide, α3D [11] (Fig. 1b). This scaffold is a single polypeptide chain that preassembles into an antiparallel three-helix bundle, a major advancement in de novo protein design. Lacking a metal-binding site, our first approach aimed to introduce a 3Cys site in α3D. Through the substitutions of apolar residues, as shown in Fig. 1b, four locations (categorized as layers) were identified that could accommodate this design. Based on Nuclear Magnetic Resonance (NMR) analysis on α3D, the fourth layer, which is composed of L18, L28, and L67, was predicted to be the most amenable to mutations. Chakraborty et al. prepared α3DIV (Fig. 2a), an α3D derivative with a 3Cys site at the C-terminal end of the bundle [12]. The authors showed that α3DIV binds heavy metals Cd, Hg, and Pb in the expected mode, serving as a spectroscopic and functional model for metalloregulatory proteins that contain an MS3 center. The NMR structure of α3DIV was also solved, which revealed that the overall fold of α3D was not significantly perturbed after the removal of stabilizing Leu residues [13]. Subsequently, a 3His zinc metal site was also incorporated in the fourth layer, generating α3DH3 [14] (Fig. 2b, Table 1).
Fig. 2.
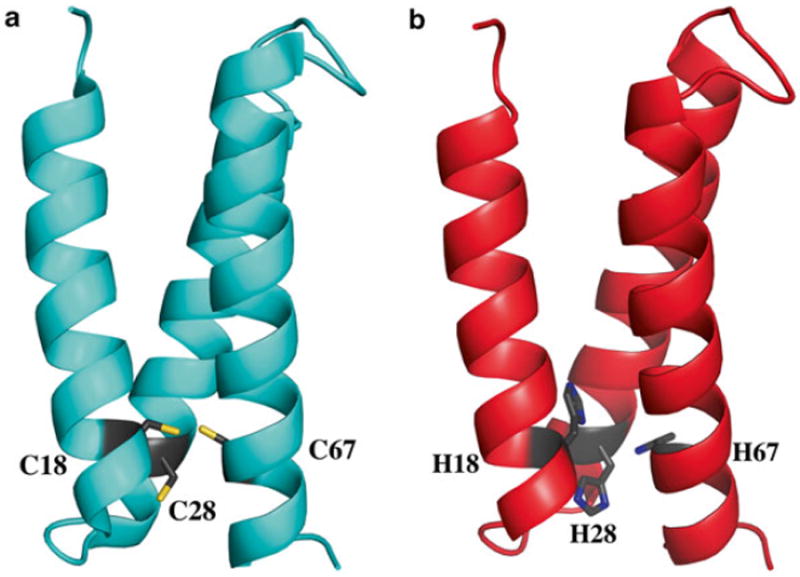
Subsequent α3D derivatives for heavy and transition metal binding. (a) Solution structure of α3DIV, which exhibits a 3Cys site at positions 18, 28, and 67 that coordinates Cd, Hg, and Pb. (b) Model of a 3His α3D derivative, α3DH3, which was demonstrated to bind Zn and perform the function of carbonic anhydrase. This model was constructed from the α3DIV structure
Table 1.
Amino acid sequence of α3D constructs
| Construct | Sequence | Molecular weight (Da) | PDB code |
|---|---|---|---|
| α3D | MGSWAEFKQR LAAIKTR LQAL GGS EAELAAFEKE IAAFESE LQAY KGKG | 7977.2 | 2A3D |
| NPEVEALRKE AAAIRDE LQAYRHN | |||
|
| |||
| α3DIV | MGSWAEFKQR LAAIKTR CQAL GGS EAECAAFEKE IAAFESE LQAY KGKG | 7946.9 | 2MTQ |
| NPEVEALRKE AAAIRDE CQAYRHN | |||
|
| |||
| α3DH3a | MGSWAEFKQR LAAIKTR HQAL GGS EAEHAAFEKE IAAFESE LQAY KGKG | 8283.5 | |
| NPEVEALRKE AAAIRDE HQAYRVNGSGA | |||
Bolded residues indicate change from the sequence of α3D
See Note 1
Construct α3DH3 was extended with a glycine-serine-glycinealanine (GSGA) tail in an attempt to increase its overall stability after the incorporation of bulky His residues inside the core, without perturbing the overall framework of α3D (see Note 1). This tail can also be modified to glycine-serine-glycine-cysteine (GSGC) with an A77C mutation. Both derivatives resulted in high expression yields of 100 mg/L, and from chemical denaturation studies, the GSGA construct increased the Gibbs free energy of unfolding (∆GU) of α3DH3 to 3.1 from 2.5 kcal/mol compared to α3DIV. Moreover, α3DH3 was shown to bind Zn and perform the CO2 hydrolysis associated with carbonic anhydrase. Overall, these efforts increased in scope the use of α3D as a viable framework for modeling the metal centers of native proteins. They provide the opportunity to tackle redox active sites with either the same ligands (e.g., 4Cys) or mixed ligands (e.g., 2HisCysMet) [15], which can be achieved in this preassembled scaffold. This chapter presents our design and expression techniques in preparing α3D derivatives.
2 Materials
Prepare all solutions using MQ or double distilled H2O. Prepare and store solutions at room temperature, unless noted otherwise. Prepare all solutions on a sterile lab bench, cleaned with 10 % bleach and followed by 70 % ethanol. Autoclave all the necessary glassware.
2.1 Modeling Using PyMOL
Access to a computer console connected to the Internet that contains a more recent version of PyMOL (1.3–1.7) is required [16]. A payment is required to obtain a license for PyMOL (http://www.pymol.org), but a free version for students and educators is available (http://pymol.org/edu/?q=educational/). Once a computer is equipped with PyMOL, download the structure of α3D (PDB code 2A3D) and/or α3DIV (PDB code 2MTQ) from the RCSB Protein Data Bank (RCSB PDB) (http://www.rcsb.org/pdb/home/home.do) by entering the PDB code in the search box and downloading the PDB text file under the Download Files tab. A computer mouse with at least three customizable buttons is ideal for visualizing structures on PyMOL.
2.2 Transformation Components
A synthetic gene that contains the DNA sequence of the designed α3D derivative cloned into pET-15b.
One-shot (50 μL) BL21(DE3) chemically competent Escherichia coli cells.
LB agar plates: Plates are prepared on a sterilized lab bench and under a flame provided by an isopropanol lamp. Suspend 4.0 g LB agar powder in 250 mL beaker containing 100 mL water and autoclave using a liquid program. Allow the solution to cool to touch and then add 100 μL of a 100 mg/mL ampicillin (amp) solution. Pour LB agar solution in 100 × 15 mm petri dishes, allow to solidify, and store plates upside down in 4 °C.
SOC media, which can be prepared or commercially purchased. SOB media: Dissolve 0.20 g tryptone, 0.05 g yeast extract, and 0.005 g NaCl in 9.8 mL H2O. Autoclave this solution using a liquid program and allow to cool to room temperature. Subsequently, add 100 μL of 1.0 M MgCl2 and 100 μL of 1.0 M MgSO4 to the SOB solution. A 1.0 mL SOC stock media is prepared by adding 20 μL of 20 % glucose (w/v) into 980 μL SOB media. Store leftover SOB and SOC media in 4 °C or −20 °C for short or long storage, respectively.
2.3 Protein Expression Components
An autoinduction media [17] is the preferred expression media (see Note 2) and prepared in 6 L batches (3 × 2 L solutions), which contains a rich media and a sugar solution. In a 4 L flask, suspend 48 g yeast extract powder and 24 g tryptone powder in 1.8 L H2O. For a 6 L rich media, prepare the sugar solution by adding in a 2 L flask containing 600 mL H2O 13.8 g KH2PO4 (monobasic), 62.0 g K2HPO4 (dibasic), 5.0 mL glycerol, 0.5 g glucose, and 2.0 g lactose. Autoclave the rich and sugar solutions using a short liquid program (see Note 3). The autoinduction media is prepared by aliquoting 0.2 L of the sugar solution into a 1.8 L of rich media (see Note 4).
LB media: Suspend 10 g tryptone powder, 5 g yeast extract powder, and 10 g NaCl in a 2 L flask containing 1.0 L H2O. Autoclave using a liquid program.
2.4 Protein Purification Components
Lysis buffer: 1X PBS and 2 mM DTT. To prepare a 1 L 10X PBS buffer, dissolve in 800 mL H2O 80 g NaCl, 2.0 g KCl, 14.4 Na2HPO4, and 2.4 KH2PO4. Adjust pH to 7.4 and autoclave using a liquid program. For a 100 mL 1X lysis buffer solution, add 10 mL of 10X PBS solution into 90 mL H2O and dissolve 30.9 mg DTT. Prepare the lysis buffer solution fresh every expression.
A centrifuge for 1 L and 50 mL cell cultures.
A sonicator and a steel cup that can hold 100–200 mL volume.
A water bath set at 55 °C.
A pH electrode and lyophilizer.
A reverse-phase C18 HPLC. Solvents: The polar solvent is composed of H2O and 0.1 % trifluoroacetic acid and the nonpolar solvent is comprised of 90 % acetonitrile, 10 % H2O, and 0.1 % trifluoroacetic acid.
3 Methods
3.1 Design of α3D Derivatives Using PyMOL
Run PyMOL and open the α3DIV (PDB 2MTQ) or α3D structure (PDB 2A3D).
Show structure as carton.
To model a new metal-binding site in the layers described in the introduction (Fig. 1), in the Menu tab, choose Wizard and then Mutagenesis.
In Mutagenesis option, select backbone- dependent rotamers and show residues as sticks.
Pick a residue to mutate and then the desired residue that can provide a metal-binding ligand such as S(Cys), N(His), or O(Asp or Glu).
Notice that several rotamers are possible. Choose the rotamer that is conducive to metal binding, that is, where the ligand is oriented toward the hydrophobic core.
Repeat according to the number of desired ligands (see Note 5).
Under the Wizard tab, use the Measurement option to determine the distances between the ligands. To obtain a qualitative sense of a suitable metal binding, these distances should be between 3.5 and 4.5 Å.
3.2 Transformation
Prior to the transformation experiment, prepare the amino acid sequence with the desired mutations. The gene for this sequence is placed between restriction sites BamHI and NcoI in the pET-15b vector (see Note 6).
Add 4–5 μL of 1 ng/μL of DNA to a tube of one-shot (50 μL) BL21(DE3) chemically competent E. coli cells thawed on ice for 10 min. Let stand for 10 min.
Heat shock in a 42 °C water bath for 30 s.
Cool on ice for 2 min.
Add 200 μL SOC and shake at 200 rpm in 37 °C for 30–50 min.
Prepare a diluted culture solution by adding 10 μL of BL21(DE3) cells into 90 μL fresh SOC.
Plate 100 μL culture on LB agar amp plate and incubate upside down overnight in 37 °C.
Save unused cells in 4 °C, which can be re-platted if the overnight plate does not show single colonies or is overgrown with no distinguishable single colonies.
3.3 Protein Expression Using Autoinduction Media
Pick single colonies from the overnight plate and inoculate 20 mL LB broth containing 20 μL of 100 mg/mL amp. Grow cultures overnight at 200 rpm and 37 °C.
Add 2 mL of 100 mg/mL amp to a 2 L autoinduction media. Inoculate with a 20 mL overnight culture.
Incubate overnight, 16–20 h, at 180 rpm and 25–30 °C.
Harvest cells by spinning down in 1 L centrifuge tubes at 8,000 × g and 4 °C. Re-suspend pelleted cells with 15–25 mL of lysis buffer pre-chilled on ice.
Transfer re-suspended cells in a steel cup chilled on an ice bucket.
Insert sonicator tip in steel cup, ~80 % submerged in resuspended cells. Keep steel cup on ice.
Sonicate for total of 5 min, at 30 s on and 30 s off intervals. Repeat three times or until the solution has turned translucent.
Transfer to centrifuge tubes (50 mL) and spin-down at 17,000 × g and 4 °C for 30 min.
Transfer supernatant to 50 mL conical tubes and heat denature at 55 °C for 20–30 min. Transfer to the appropriate centrifuge tubes and spin-down at 17,000 × g and 4 °C for 30 min.
Pour supernatant in a beaker and acidify to pH 1.9 to precipitate salts and cellular debris. Transfer to the appropriate centrifuge tubes and spin-down at 17,000 × g and 4 °C for 30 min.
Place supernatant in 50 mL conical tubes and flash-freeze in liquid nitrogen for 10–15 min or until completely frozen. Lyophilize frozen protein for 2–3 days or until dry.
3.4 Protein Purification
Redissolve dry protein powder in H2O (15–20 mL) and check pH (see Note 7).
Purify on a reversed-phase C18 HPLC using a flow rate of 20 mL/min and a linear gradient of polar solvent (0.1 % TFA in water) to nonpolar solvent (0.1 % TFA in 9:1 CH3CN/H2O) over 45 min.
Retention time of α3D constructs is between 26 and 30 min.
The molecular weight is determined using an electrospray mode on a Micromass LCT Time-of-Flight mass ionization spectrometer. The MW accounts for 72 of the 73 amino acids as Met1 is cleaved posttranslation (see Note 8).
Acknowledgments
J.S.P. and V.L.P. would like to thank the National Institutes of Health (NIH) for financial support for this research (ES012236). J.S.P. thanks the Rackham Graduate School at the University of Michigan for a research fellowship.
Footnotes
The sequence of α3DH3 was extended with a GSGA tail to increase the overall protein stability [14]. This extension also improved protein expression yield to ~100 mg/L compared from 50 mg/L compared to α3DIV.
Autoinduction media and induction via IPTG work by the same mechanism, which involves the induction of gene expression by relieving the repression of the lac promoter. Using the latter induction technique, repression is relieved by the binding of IPTG (allolactose analog). However, in the case of autoinduction media, protein overexpression is controlled by the availability of sugar source instead of the addition of IPTG. Cell density relies on sugar source, as well as the expression of the designed gene without monitoring the cell density at 600 nm (OD600). After exhausting the more metabolically available sugars, glucose and glycerol, the cells will use lactose as the sugar source. This natural switch will turn on the components of the lac operon, including our gene of interest that is downstream of the lac promoter region in pET- 15b. Overall, compared to IPTG induction in LB media, the autoinduction technique eliminates 3–5 h waiting for the cell density to reach the proper OD600 and has also improved our protein yield by 20–50 mg/mL.
For the sugar solution, autoclave using a short liquid program to avoid sugar oxidation, which can make them less bioavailable. If this sugar oxidation is suspected, the solution can be prepared by dissolving all the components in autoclaved water and vacuum filter through a sterile 0.22 μm filter.
Prepare the autoreduction media several hours before inoculation to avoid contamination. Contamination was often observed when the media was prepared one to two days prior to expression.
Depending on your design goals, each metal-binding residue can be positioned on separate helices to form a triangular pocket. Or the two ligands can be placed on the same helix, spaced by two to three residues, to replicate a chelate-like motif and the third on a second helix, requiring only two of the three helices to form a metal-binding site.
Carefully indicate where to place the stop codon in the sequence. If a 73 amino acid sequence is desired, place the stop codon (TAA, TAG, or TGA) after Asn73. If a GSGA or GSGC tail is desired, place the stop codon after Ala/Cys77 (see Note 1). As described in the introduction and Note 1, the addition of a GSGA tail had a significant effect on the expression yield (100 mg/L) of α3DH3 and improved the ∆GU of α3DH3 by 0.6 kcal/mol compared to α3DIV. The latter effect, which demonstrates an increase in stability, showed that the addition of these tails does not change or perturb the overall framework of α3D. Moreover, an A77C mutation to generate a GSGC tail also had the same effect on the expression yield of α3DH3. Therefore, we expect that the addition of a GSGA or GSGC tail is essential in stabilizing α3D derivatives that aim to modify layers 2 or 3 (see Fig. 1b).
The pH of the polar solvent is about 1.9. It is important to make sure that the crude protein solution matches this pH condition. When the pH conditions do not match, we have observed precipitation after the crude solution is mixed with the polar solvent. This precipitate can clog the HPLC tubings and lines and ultimately damage the solvent pump system.
Met1-containing species is observed in the mass spectrum of α3DIV and α3DH3 but at a low amount compared to the Met1- cleaved species. Met1-containing species make up about <5 % of the total protein.
References
- 1.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. De novo design and structural characterization of proteins and metalloproteins. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Berry SM, Pfister TD. Engineering novel metalloproteins: design of metal-binding sites into native protein scaffolds. Chem Rev. 2001;101(10):3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 3.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Protein design: toward functional metalloenzymes. Chem Rev. 2014;114(7):3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460(7257):855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peacock AF, Iranzo O, Pecoraro VL. Harnessing nature’s ability to control metal ion coordination geometry using de novo designed peptides. Dalton Trans. 2009;13:2271–2280. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Identifying important structural characteristics of arsenic resistance proteins by using designed three-stranded coiled coils. Proc Natl Acad Sci U S A. 2007;104(29):11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Designing a functional type 2 copper center that has nitrite reductase activity within alpha-helical coiled coils. Proc Natl Acad Sci U S A. 2012;109(52):21234–21239. doi: 10.1073/pnas.1212893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu F, Penner-Hahn JE, Pecoraro VL. De novo -designed metallopeptides with type 2 copper centers: modulation of reduction potentials and nitrite reductase activities. J Am Chem Soc. 2013;135(48):18096–18107. doi: 10.1021/ja406648n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zastrow ML, Peacock AF, Stuckey JA, Pecoraro VL. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat Chem. 2012;4(2):118–123. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zastrow ML, Pecoraro VL. Influence of active site location on catalytic activity in de novo -designed zinc metalloenzymes. J Am Chem Soc. 2013;135(15):5895–5903. doi: 10.1021/ja401537t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh ST, Cheng H, Bryson JW, Roder H, DeGrado WF. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc Natl Acad Sci U S A. 1999;96(10):5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Design of a three-helix bundle capable of binding heavy metals in a triscysteine environment. Angew Chem Int Ed Engl. 2011;50(9):2049–2053. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plegaria JS, Pecoraro VL. Sculpting metal- binding environments in de novo designed three-helix bundles. Isr J Chem. 2015;55(1):85–95. doi: 10.1002/ijch.201400146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cangelosi VM, Deb A, Penner-Hahn JE, Pecoraro VL. A de novo designed metalloenzyme for the hydration of CO 2. Angew Chem Int Ed Engl. 2014;53(30):7900–7903. doi: 10.1002/anie.201404925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plegaria JS, Duca M, Tard C, Friedlander TJ, Deb A, Penner-Hahn JE, Pecoraro VL. De novo design and characterization of copper metallopeptides inspired by native cupredoxins. Inorg Chem. 2015 doi: 10.1021/acs.inorgchem.5b01330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 17.Studier FW. Protein production by auto- induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]