

Kang et al. confirm PRC1–Br140 and PRC1–Fs(1)h interactions and identify their genomic binding sites. PRC1–Br140 bind developmental genes in fly embryos, with analogous co-occupancy of PRC1 and BRD1 at bivalent loci in human ES cells.
Keywords: Polycomb, Enok, Br140, Fs(1)h, bivalent chromatin, BioTAP-XL
Abstract
Regulatory decisions in Drosophila require Polycomb group (PcG) proteins to maintain the silent state and Trithorax group (TrxG) proteins to oppose silencing. Since PcG and TrxG are ubiquitous and lack apparent sequence specificity, a long-standing model is that targeting occurs via protein interactions; for instance, between repressors and PcG proteins. Instead, we found that Pc-repressive complex 1 (PRC1) purifies with coactivators Fs(1)h [female sterile (1) homeotic] and Enok/Br140 during embryogenesis. Fs(1)h is a TrxG member and the ortholog of BRD4, a bromodomain protein that binds to acetylated histones and is a key transcriptional coactivator in mammals. Enok and Br140, another bromodomain protein, are orthologous to subunits of a mammalian MOZ/MORF acetyltransferase complex. Here we confirm PRC1–Br140 and PRC1–Fs(1)h interactions and identify their genomic binding sites. PRC1–Br140 bind developmental genes in fly embryos, with analogous co-occupancy of PRC1 and a Br140 ortholog, BRD1, at bivalent loci in human embryonic stem (ES) cells. We propose that identification of PRC1–Br140 “bivalent complexes” in fly embryos supports and extends the bivalency model posited in mammalian cells, in which the coexistence of H3K4me3 and H3K27me3 at developmental promoters represents a poised transcriptional state. We further speculate that local competition between acetylation and deacetylation may play a critical role in the resolution of bivalent protein complexes during development.
The Polycomb group (PcG) of transcriptional silencing factors is a key player in gene regulation during development (Lewis 1978; Struhl 1981; Simon et al. 1992; Nègre et al. 2006; Schwartz et al. 2006; Tolhuis et al. 2006), and many of the activators that oppose silencing, including the Trithorax group (TrxG), have also been identified (for review, see Geisler and Paro 2015). These two groups of chromatin regulatory factors are essential for normal development and are often mutated in cancer (Sparmann and van Lohuizen 2006; Kingston and Tamkun 2014; Geisler and Paro 2015; Koppens and van Lohuizen 2016). Pc complexes are highly conserved between flies and mammals, but mechanisms for their targeting to genomic binding sites, while still poorly understood, are thought to differ. Furthermore, developmental genes in mammalian embryonic stem (ES) cells are frequently marked with both H3K27me3, the PcG silencing mark, and H3K4me3, a defining mark of active promoters. This “bivalent” state is postulated to precede resolution into full transcriptional activation or silencing during cell type specification (Bernstein et al. 2006), but analogous bivalency has not been identified during Drosophila embryogenesis (Schuettengruber et al. 2009).
We approached the analysis of targeting and function of Pc-repressive complexes (PRC1 and PRC2) by taking a chromatin-based approach in Drosophila. Despite their important functions in pattern formation, PcG and TrxG factors are ubiquitously expressed during development and lack apparent DNA sequence specificity (Bienz and Müller 1995; Grimaud et al. 2006; Geisler and Paro 2015; Bauer et al. 2016). Therefore, sequence-specific transcription factors are thought to attract the relevant chromatin complexes to their appropriate context-dependent targets through protein–protein interactions (Kassis and Brown 2013).
We wished to test this model by using BioTAP-XL, an approach combining cross-linking with tandem affinity purification to facilitate identification of critical protein interactions that might be disrupted by the salt or detergent required for the removal of DNA-binding factors from chromatin (Alekseyenko et al. 2015). We performed BioTAP-XL pull-downs of Pc (PRC1) and E(z) (PRC2) from embryos with the aim of testing a new approach to the recovery of relevant DNA-binding repressor proteins (Fig. 1; Kang et al. 2015). Contrary to our expectation, our analysis in Drosophila embryos revealed that PRC1 strongly interacts with transcriptional coactivators Fs(1)h [female sterile (1) homeotic] and Enok/Br140 during embryogenesis. Fs(1)h is the Drosophila ortholog of BRD4, a double-bromodomain protein that binds to acetylated histone marks on active chromatin and is a key transcriptional coactivator in mammals (Kanno et al. 2004; Kockmann et al. 2013). BRD4 is known to positively affect gene expression through Mediator and P-TEFb interactions and reinforce gene activation through its interactions with acetylated histones (Kanno et al. 2004; Jang et al. 2005; Di Micco et al. 2014). Enok and Br140 are orthologous to subunits of a well-defined mammalian MOZ/MORF histone acetyltransferase complex, which is strongly implicated in hematopoietic stem cell function and human leukemias (Yang and Ullah 2007; Yang 2015).
Figure 1.
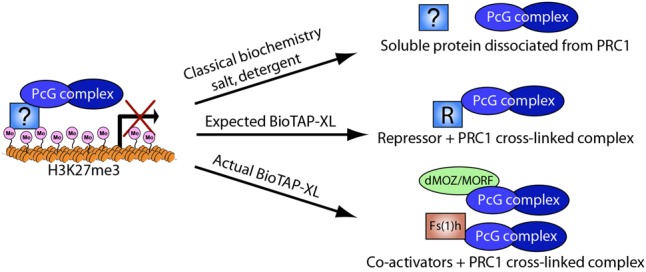
Overview of hypothetical and observed PRC1 interactors. BioTAP-XL is a cross-linking, affinity purification, and mass spectrometric approach for the discovery of protein interactions that may be labile to removal from DNA in the absence of cross-linking (Alekseyenko et al. 2015).
Here, we explore the implications of our discovery of the strong and unexpected PRC1 association with these specific coactivators during embryonic development in Drosophila. As mentioned above, classical models for transcriptional regulation generally posit that sequence-specific transcription factors drive developmental regulation by choosing the locations for de novo activation or repression. Here we propose an alternative, potentially unifying model for flies and mammals—that bivalent regulatory complexes respond to transcription factor occupancy rather than being set up by such factors. The response is likely to involve tipping the balance in the local acetylation/deacetylation cycle. While speculative, our model is consistent with the strong protein interactions and genomic co-occupancy seen in our fly data, analogous mapping data in human ES cells, and the bivalency model in mammals.
Results
Reciprocal BioTAP-XL analysis confirms the specificity of PRC1 interaction with Br140 and Fs(1)h
The original aim of our cross-linking and affinity purification of Drosophila PRC1 and PRC2 was to identify interactions between specific transcription factors/repressors and PcG proteins as predicted by classical models for targeting of Pc-dependent silencing (Fig. 1). However, even with cross-linking, we did not capture strong candidates for such interactions and instead found enrichment of coactivators Fs(1)h (dBRD4) and Enok and Br140 (subunits of dMOZ/MORF) in Pc BioTAP-XL purifications (Kang et al. 2015).
To probe the specificity of those unexpected interactions, we sought to validate the proteomic data by performing reciprocal affinity purifications. We selected Br140 for affinity purification of the Drosophila MOZ/MORF complex because BRPF1, one of the mammalian orthologs of BR140, acts as the scaffold for MOZ/MORF complex formation (Ullah et al. 2008). We incorporated an N-terminal BioTAP tag into a Br140 transgene containing the genomic region of Br140, including the endogenous promoter and flanking upstream and downstream regions. Expression of the BioTAP-tagged Br140 fusion protein in both transgenic embryos and S2 cells was detected by Western blot (Supplemental Fig. S1A). We found that the fusion protein was fully functional by transgenic rescue of Br140 mutant recessive lethality (Supplemental Fig. S1C). As the Br140 transgene was expressed from its own promoter from a unique insertion site, the level of Br140 expression and its regulation should be comparable with the endogenous protein.
To test whether the strong Pc–Br140 interaction that we detected previously in Drosophila embryos would be replicated in a reciprocal manner using Br140 as bait, we used BioTAP-XL affinity purification coupled with mass spectrometry in embryos carrying epitope-tagged Br140 and lacking any endogenous untagged Br140 protein. We recovered all dMOZ/MORF complex components (Enok, Eaf6, and Ing5) as well as Elg1, the PCNA unloader previously shown to interact with Br140 (Huang et al. 2016), and Ash1, a TrxG protein (Fig. 2; Supplemental Fig. S2). To investigate whether PRC1 components are reciprocally and specifically copurified by Br140 pull-down, we compared the embryonic Br140 interactome with our previous embryonic Pc (PRC1) and E(z) (PRC2) interactomes (Fig. 2A,B). Although recovery of peptides beyond the dMOZ/MORF complex itself was low, PRC1 components Psc and Su(z)2 and redundant components Ph-p + Ph-d fell within the top one percentile of enrichment in both the Br140 and the Pc pull-downs from embryos (Fig. 2A), while PRC2 components were not enriched with Br140 (Fig. 2B), confirming the specificity of the PRC1/Br140 interaction.
Figure 2.

Proteomic analysis of Br140 and Fs(1)h complexes. (A,B) Scatter plots of Br140 and Pc (A) or E(z) (B) pull-down enrichment over total embryonic chromatin proteins. Each point represents an individual protein, with coordinates corresponding to its log enrichment in Br140 pull-downs (X-axis) and Pc (A) or E(z) (B) pull-downs (Y-axis). Dashed lines represent the 99th percentile of Br140 enrichment (vertical line) and Pc (A) or E(z) (B) enrichment (horizontal line). PRC1 (A) and PRC2 (B) components are highlighted in blue and pink, respectively. Known dMOZ/MORF components are highlighted in green. (C) Scatter plot showing Fs(1)h enrichment over total S2 cell chromatin input. Each point represents an individual protein, with coordinates corresponding to its enrichment in BioTAP-N Fs(1)h pull-downs (X-axis) and Fs(1)h C-BioTAP pull-downs (Y-axis). “L + S” indicates that both long and short Fs(1)h isoforms carried the BioTAP tag, while “L” indicates that only the long Fs(1)h isoform was tagged. Dashed lines represent the 98th percentile of BioTAP-N Fs(1)h enrichment (vertical line) and Fs(1)h C-BioTAP enrichment (horizontal line). See Supplemental Data File S1 for full results. (D) The zoomed-in view of the top right quadrant of the scatter plot in C shows selected protein identities above the 98th percentile. Mediator and P-TEFb complex proteins are highlighted in red, and PcG proteins are highlighted in blue.
We also wished to test whether the Pc–Fs(1)h interaction would be replicated using Fs(1)h as bait. The fs(1)h gene produces two conserved isoforms of Fs(1)h protein via alternative splicing, so we inserted the BioTAP sequence at the common 5′ ATG to tag both short and long isoforms and at the furthest downstream 3′ end to tag only the long form (Supplemental Fig. S1B). We were unable to obtain transgenic fly lines expressing either N-BioTAP-fused or C-BioTAP-fused Fs(1)h protein after multiple attempts. Therefore, we sought to validate the PRC1 and Fs(1)h interaction in stably transformed S2 tissue culture cells. Our proteomic analyses of these cell lines revealed consistent results independent of the position of the BioTAP tag (Fig. 2C,D; Supplemental Fig. S2). Like Br140, Fs(1)h recovered low amounts of PRC1 but not PRC2 (Fig. 2D). Interestingly, although they are both coactivators, Br140 and Fs(1)h did not exhibit a strong pairwise interaction, suggesting that while they both interact with PRC1, they are unlikely to function in direct physical contact with each other. From these data, we cannot determine whether they are physically distant components that can simultaneously interact with PRC1 or whether they participate in completely separate PRC1–Br140 or PRC1–Fs(1)h configurations.
Recombinant Fs(1)h copurifies with PRC1 upon coexpression in Sf9 insect cells
In parallel, we also tested for an interaction between recombinant Fs(1)h and PRC1 components using baculoviral expression and affinity pull-downs from soluble nuclear extracts. We expressed the Fs(1)h short form alone, with the four PRC1 core components, or with the PRC2 core components. Affinity purifications were performed using Flag epitope tags attached to the Psc and Esc subunits of PRC1 and PRC2, respectively (Fig. 3A). The reconstituted PRC1 and PRC2 complexes were validated through silver staining of SDS-PAGE gels (Fig. 3B). We observed increased intensity of a putative Fs(1)h band in the PRC1 + Fs(1)h elution sample, although a band of slightly faster mobility detected in the PRC2 + Fs(1)h elution sample obfuscated this result. To unambiguously identify Fs(1)h, we performed a Western blot with an anti-Fs(1)h antibody. Figure 3C shows that while Fs(1)h was detected in input at similar levels in the PRC1 and PRC2 coexpressions and more highly in the Fs(1)h-only extract, strong Fs(1)h enrichment was detected only in the PRC1 pull-down and elution. As the affinity purifications were subject to very stringent salt washes (stepwise to 2 M KCl) and since interaction was not observed with PRC2 coexpression, our results suggest that a very strong and specific interaction occurs between recombinant Fs(1)h and PRC1.
Figure 3.

Recombinant Fs(1)h interacts with the recombinant PRC1 complex. (A) The purification scheme using M2 anti-Flag antibody. Nuclear extracts were generated from Sf9 cells that were infected with baculovirus expressing recombinant Fs(1)h alone or coinfected with PRC1 (Psc-Flag, Pc, Ph, and Sce) or PRC2 [Esc-Flag, E(z), Su(z)12, and Caf1-55] complex components. Entire elution fractions were boiled in SDS-PAGE loading buffer and resolved on an 8% Tris-glycine gel. (B) The presence of the Fs(1)h along with the intact PRC1 or PRC2 complexes was verified by silver staining. (C) The specificity of the Fs(1)h–PRC1 interaction was confirmed by Western blotting using anti-Fs(1)h antibodies. Nuclear extract lanes show Fs(1)h expression levels in input samples (0.5% input loaded).
If PRC1 has such specific interactions with coactivators Fs(1)h and dMOZ/MORF during Drosophila embryogenesis, why were these not recovered in previous purifications? We believe that the BioTAP-XL approach enables the retention of interactions on chromatin that may be sensitive to the salt and detergent treatments typically used to extract soluble chromatin complexes for biochemical study. Consistent with this possibility, both Fs(1)h and Enok/Br140 were recovered in substoichiometric amounts from a previous affinity purification of Pc in the absence of cross-linking (Strubbe et al. 2011). Therefore, BioTAP-XL may allow enhanced recognition of important interactions simply lost during release from DNA, as we propose above. However, it is also important to note that when we describe complexes going forward, these may differ from classically defined soluble complexes. For example, some of these interactions may be dependent on physical linkage to DNA or nucleosomes and thus would not be referred to as complexes using a classical biochemical definition.
Embryonic ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) of Pc-Br140 reveals potential bivalency and genes in the active state
Our original Pc pull-downs and the reciprocal BioTAP-XL analyses in embryos and S2 cells presented here confirm specific interactions between PRC1 and the Fs(1)h and dMOZ/MORF coactivators. We next asked where these interactions were occurring. As the BioTAP-XL approach is designed to perform ChIP-seq for binding sites in parallel with the mass spectrometric analyses (Alekseyenko et al. 2014, 2015), we mapped genomic localization for dMOZ/MORF from the same cross-linked embryonic chromatin that yielded our Br140 proteomic results. We also performed anti-Fs(1)h ChIP in parallel. We compared our Br140 and Fs(1)h binding results in the context of several additional data sets, including Pc-BioTAP from our previous work (Kang et al. 2015) and histone modifications H3K27me3, H3K27ac, and H3K4me3 from the modENCODE project (Ho et al. 2014). In general, trimethylation at H3K27 correlates with PcG silencing, while acetylation at the same position indicates transcriptional activation. H3K4me3 typically marks active promoters. We noted that the majority of strong binding for Pc, Br140, and Fs(1)h overlaps with genes, so, instead of focusing on individual peaks, we focused on shared signals at the gene level.
It should be noted that a confounding issue when performing genomic studies on embryos is that even a single embryo is a collection of cells with differing positional information and developmental fates. Furthermore, a collection of embryos represents a spectrum of developmental ages. In essence, ChIP-seq occupancy can represent a broadly similar result over a majority of cells or, alternatively, an average over a very diverse mixture of cells. However, an important insight that we gained using BioTAP-XL was physical evidence for protein–protein interactions that are cross-linked within nuclei. Therefore, while we cannot say that any two proteins interact at all locations where they show overlapping ChIP-seq binding, depending on the apparent strength of their coaffinity purification, we can say that they are very likely to overlap at many or most places. Particularly in the case of Pc pull-down of Fs(1)h and dMOZ/MORF in embryos, the abundant recovery of the two coactivators (Supplemental Fig. S2; Kang et al. 2015) suggests potential co-occupancy at the majority of Pc binding sites.
Interestingly, we found >2000 genes as common targets of Pc and Br140 in embryos (Fig. 4A), and, with more stringent peak calling, practically all Pc peaks are also bound by Br140 (447 of 483) (Supplemental Fig. S3B,C). That these results largely represent Pc–Br140 complex formation within nuclei—as opposed to heterogeneity within a mixture of cells—is supported by their clearly skewed average H3K27 modification status. The Pc/Br140-binding sites appear to segregate into either strong K27me3 (cluster 1; n = 406) or strong K27ac (cluster 2; n = 1621) without the ambiguity that would be expected if a sizeable fraction of cells displayed opposite fates in different parts of the embryo (Fig. 4B). Interestingly, in both clusters, the transcription start sites (TSSs) of genes are marked with H3K4me3. In the case of cluster 1, our results are suggestive of a bivalent state, as these genes (listed in Supplemental Data File S2) are also marked with H3K27me3. However, as discussed above, we cannot formally exclude the possibility that the chromatin marks are not coincident but occur in separate cells. Cluster 2 genes are instead enriched for H3K27ac, indicative of an active state. Thus, our results are consistent with binding of Pc–Br140 at both active and bivalent genes. Interestingly, Pc and Br140 appear stronger in cluster 1, marked by H3K27me3, while Fs(1)h correlates instead with H3K27ac enrichment. This ChIP-seq result is consistent with our proteomic results, as the lack of a strong pairwise interaction between Br140 and Fs(1)h (Fig. 2) suggests that PRC1 may interact closely with the two coactivators, whereas they can be at least partially independent of each other. Screenshots of representative genes from the two clusters are shown in Figure 4C. Note that H3K27me3 or H3K27ac often display broad binding over whole genes, while H3K4me3 is typically focused on the 5′ TSS. This focused H3K4me3 signal differs from the strikingly broad H3K4me3 seen over bivalent genes in mammalian ES cells and may have contributed to a consensus view that bivalency does not occur in flies.
Figure 4.

Co-occupancy of Pc and Br140 in embryos. (A) Venn diagram of the overlap between Pc- and Br140-bound genes in embryos. (B) Heat maps of embryo ChIP-seq enrichment patterns of BioTAP-tagged Pc, Br140, Fs(1)h, and indicated histone H3 modifications within ±10 kb of TSSs of genes cobound by Pc and Br140. Heat maps are centered at TSSs and ordered by H3K27me3 intensity. Signals were clustered into two groups by k-means clustering based on the distributions of the proteins and histone modifications surrounding the cobound promoters. (C) Genome browser views from embryo ChIP-seq data showing binding profiles of the indicated proteins in representative regions with high H3K27me3 and low H3K27ac (cluster 1; top) or low H3K27me3 and high H3K27ac (cluster 2; bottom). (D) Gene ontology (GO) term enrichment for genes with promoters cobound by Br140 + Pc or bound only by Br140 or Pc as indicated. The numbers in parentheses indicate the number of genes from our data sets that corresponded to each category. The size of each dot indicates that each GO term was found exclusively (100%) with only one of the three sets of genes (Rb140 only, cobound, or Pc only) rather than divided between them.
To assess bivalent complex formation of Pc and Br140 with an alternative method, we analyzed the post-translational marks on histones recovered separately from Br140- and Pc-BioTAP-XL affinity pull-downs (Supplemental Fig. S4A). Marks such as H4K20 methylation were recovered in relative abundances similar to those found in bulk genomic histones (Supplemental Fig. S4B). However, in addition to association with histone acetylation marks (Supplemental Fig. S4C–E), the relative levels of H3K27me3 were also enriched by Br140 over the genomic input (Supplemental Fig. S4F). If Br140 only bound sites in an active configuration, we would not expect this enrichment for H3K27me3-modified histones. Similarly, Pc enriches for acetylated histones as well as for H3K27me3. Together, these results provide additional evidence that Pc and Br140 do form bivalent complexes in regions enriched for H3K27me3 and active marks.
While most Pc sites are coincident with Br140 binding, there are many genes where Br140 and Fs(1)h occur independently of Pc (Fig. 4A; Supplemental Fig. 3B) and potentially independently of each other. Based on the H3K4me3 and H3K27ac signals and additional marks at those sites (data not shown), these regions represent active promoters depleted for Pc and H3K27me3. We conclude that dMOZ/MORF and Fs(1)h occupy a broad class of active genes that may not be subject to coregulation by PRC1, at least in embryos. This occupancy is consistent with our proteomic data, where PRC1 subunits were recovered in both Br140 and Fs(1)h pull-downs but were not their top interactors (Fig. 2; Supplemental Fig S2; Supplemental Data File S1). Interestingly, the gene ontology (GO) terms for genes with PRC1-independent binding were enriched for typical cellular functions and differed significantly from potentially cobound genes, which are enriched for developmental terms (Fig. 4D). Cobound genes also display expression profiles consistent with regulation during development (Supplemental Fig. S3D). Overall, our ChIP-seq analyses together with our cross-linking and affinity purification results strongly suggest that PRC1 and dMOZ/MORF form composite complexes at a set of bivalent and active developmental genes in embryos, with Fs(1)h enriched as genes transition toward a more active configuration (Fig. 4B).
If bivalent, does resolution occur as cells differentiate?
If the bivalent PRC1/H3K27me3/coactivator state in embryos is a dynamic precursor to gene activation or repression, then we might expect to see more stable states in established cells in culture, such as Drosophila S2 cells. Therefore, we proceeded with BioTAP-XL analyses of stably transformed S2 cell lines. When analyzing the ChIP-seq data, we again used heat maps to depict enrichment or depletion of chromatin proteins and histone marks centered on the TSSs of bound genes (Fig. 5).
Figure 5.

Genome-wide localization of Br140 and Fs(1)h in S2 cells. (A) Venn diagram of the overlap between Pc- and Br140-bound genes in S2 cells. (B,C) Heat maps of S2 cell ChIP-seq enrichment patterns of BioTAP-tagged Pc, Br140, Fs(1)h, and the indicated histone H3 modifications within ±10 kb of TSSs of genes bound by only Pc or Br140 (B) or cobound by Pc and Br140 (C). Heat maps are centered at TSSs and ordered by H3K27me3 intensity. Signals in C were clustered into two groups by k-means clustering based on the distributions of the proteins and histone modifications surrounding the cobound promoters. (D) Genome browser view of H3K27me3, Pc, and Br140 binding across the Bithorax complex (BX-C) in embryos and S2 cells.
Interestingly, we found that the major categories of S2-binding sites differed dramatically from our embryonic results. For example, only a subset of Pc sites overlaps with Br140 sites that are also enriched for H3K27me3 (n = 73) (Fig. 5C, cluster 1). Instead, PRC1 is colocalized with either Br140 and H3K27ac on active genes (n = 1086) (Fig. 5C, cluster 2) or H3K27me3 on silent regulatory regions (n = 3157) (Fig. 5B, Pc-only). We speculate that these potentially more stable classes may have arisen from a previous bivalent state.
The existence of cluster 2, with Pc-Br140 bound to sites carrying marks of active transcription such as H3K4me3 and H3K27ac, is consistent with recent data from both flies and mammals that PRC1 is often associated with active genes (Frangini et al. 2013; Schaaf et al. 2013; Gao et al. 2014). These results could be consistent with a bivalent model in which PRC1 might toggle into either active (cluster 2) or silent states (Fig. 5B, Pc-only) dependent on its local protein–protein interactions. While they no longer comprise a major cluster, a small group of potentially bivalent genes (n = 73) marked by PRC1, Br140, and H3K27me3 can still be found in S2 cells (Fig. 5C top; genes listed in Supplemental Data File 2). These results are consistent with bivalency in mammalian cells, which is seen at many genes in ES cells but at smaller and differing subsets of genes in differentiated cells and tissues (Bernstein et al. 2006; Mikkelsen et al. 2007).
Are Fs(1)h and dMOZ/MORF the key activators of the Bithorax complex (BX-C) opposing Pc- and H3K27me3-mediated silencing?
Regulation of the body plan through maintenance of the on/off states of HOX genes within the BX-C is the hallmark of PcG function. PcG mutants exhibit ectopic expression of the BX-C and aberrant segment identity. Therefore, it was originally expected that PcG proteins would bind local Pc response elements (PREs) to promote silencing and, conversely, be absent from PREs to allow activation. However, the work of multiple groups has shown that the occupancy of several PcG proteins at PREs in the BX-C and at additional loci appears similar in both the active (H3K27ac) and silent (H3K27me3) chromatin state (Papp and Müller 2006; Langlais et al. 2012; Bowman et al. 2014). Therefore, it has remained an intriguing mystery how PcG governs the specific patterning required for proper development. We wished to re-examine this question by asking whether differential dMOZ/MORF or Fs(1)h interactions with PRC1 might help explain its functional specificity.
Interestingly, when we compared our ChIP-seq data for Pc and Br140 in embryos and S2 cells, we found that Br140 is strongly bound to the entire BX-C during embryogenesis but strongly depleted from the region in S2 cells (Fig. 5D). This result offers a potential explanation for the paradox of PcG binding in both the active and the silent states. Perhaps the composition of PcG complexes—with or without coactivators—might differ locally in the two conditions. If so, Pc levels would remain relatively constant, while inclusion of Br140 and Fs(1)h in bivalent complexes might change upon differentiation in vivo, with association seen only in places where the BX-C is active.
To test this model, we turned to ChIP analysis in imaginal discs, the precursors to adult tissues that form within Drosophila larvae prior to metamorphosis. The Ubx homeotic gene is expressed in the haltere and third leg discs, while it is repressed in the wing disc (Fig. 6A). When we performed ChIP-seq from these tissues, we found that H3K27me3 covered the whole BX-C in wing discs, as expected. The haltere and third leg discs displayed an overlap of active and silent marks over the bxd region, but the active Ubx gene was selectively associated with H3K27ac instead (Fig. 6B). However, due to the small amount of material recovered from hand dissection, we were unable to obtain reproducible ChIP-seq results for the proteins implicated in bivalency. Therefore, we turned to ChIP-qPCR (ChIP combined with quantitative PCR), assaying potential regulatory regions within the BX-C (Fig. 6C) for Pc, Fs(1)h, and Enok (the Drosophila MOZ/MORF enzyme) (Huang et al. 2014).
Figure 6.

Differential localization of PRC1 and coactivators at various developmental stages. (A) Wing, haltere, and third leg imaginal discs labeled with Hoechst (top panel) and an antibody against Ubx (bottom panel). (B) Genome browser view of H3K27me3 and H3K27ac modifications across the BX-C that correlate with its expression state in haltere and third leg discs and repression in the wing disc. (C) Zoomed-in view of the region in B indicating sites selected for analysis by ChIP-qPCR. (D) ChIP-qPCR for Pc, Enok, and Fs(1)h in wing versus haltere and third leg discs. C1 and C2 correspond to regions selected as negative and positive controls for Pc, respectively. See Supplemental Table S1 for primer sequences. Results are presented as means ± standard error of three independent biological replicates. (*) P < 0.05; (**) P < 0.01, one-tailed Student's t-test.
Consistent with previous reports (Papp and Müller 2006), we found small but significant differences in Pc binding at multiple sites near the Ubx gene, with more Pc correlating with the repressed state (wing > haltere). Contrary to our expectation for the resolution of bivalency, we did not detect significant differences between wing and haltere binding of Enok across the BX-C region except at the Ubx promoter. However, Fs(1)h was skewed toward increased occupancy at the Ubx promoter and other sites when active (haltere > wing), showing an inverse relationship to Pc ChIP (Fig. 6D). We conclude that relative ratios of Fs(1)h and PRC1, rather than the absolute presence or absence of each component, may contribute to complementary transcriptional states at the BX-C. It appears that, in spite of a strong difference in gene expression, bivalency still persists at this stage of development, perhaps to retain the ability for reversible switching of transcriptional states. Revisiting our embryonic data, this result is consistent with surprising levels of apparent bivalency at genes that are in mainly repressed patterns; for example, at the Abd-B locus. While the vast majority of cells represses Abd-B in mid to late embryogenesis, a strong Br140 ChIP signal is retained, suggesting the continued occupancy of bivalent complexes at that stage in development (Fig. 5D).
Conservation of PRC1/MOZ/MORF at bivalent genes in human stem cells
To ask whether Br140 interaction with PRC1 may be conserved in mammalian development, we performed ChIP-seq in HUES64 human ES cells with antibodies against RING1B to probe for human PRC1 and an antibody against BRD1, one of three mammalian orthologs of Br140 (BRPF1, BRPF2/BRD1, and BRPF3). We found that binding sites for RING1B (n = 951) were broad and largely overlapped genes (n = 707 of 951) (Fig. 7A, SIX6 and SIX1). BRD1 exhibited much more expansive binding than PRC1 (n = 6893) but also largely overlapped genes (n = 5953 of 6893) (Fig. 7A, SIX6, SIX1, and SIX4). Consistent with our analyses in Drosophila embryos, most genes bound by RING1B (∼75%) were co-occupied by BRD1 (Fig. 7B). In total, we identified 609 genes that were cobound by RING1B and BRD1 in all replicates (Fig. 7B; listed in Supplemental Data File S2). Bivalent chromatin (marked by active H3K4me3 and repressive H3K27me3) is prevalent in ES cells, particularly at developmental genes. To understand whether the identified cobound genes are marked by bivalent chromatin, we compared the cobound sites with high-confidence bivalent regions in human ES cells (Court and Arnaud 2017). Strikingly, virtually all cobound genes were identified previously as bivalent genes (566 bivalent out of 609 cobound). To confirm the presence of bivalent chromatin at these sites in HUES64 cells, we visualized publicly available ChIP-seq data for H3K4me3 and H3K27me3 (Fig. 7A,C; Gifford et al. 2013). The cobound genes exhibit strong H3K27me3 signal and moderate H3K4me3 signal, indicating that these genes are indeed bivalent in HUES64 cells. Similar to the Br140 results from Drosophila embryos, there is also a large group of additional BRD1 sites that do not overlap RING1B-binding sites (n = 6219 of 6893) and are largely found at active gene promoters that exhibit strong H3K4me3 (Fig. 7A, SIX4).
Figure 7.

Conserved PRC1/MOZ/MORF co-occupancy in human ES cells. (A) Genome browser screenshot of HUES64 ChIP-seq enrichment (immunoprecipitation/input) profiles for RING1B, BRD1, H3K4me3, and H3K27me3 across a genomic region containing two cobound genes (SIX6 and SIX1) and one BRD1-only gene (SIX4). (B) Venn diagram of the overlap between BRD1-bound and RING1B-bound genes. (C) Heat maps of HUES64 ChIP-seq enrichment patterns of BRD1, RING2, and the indicated histone H3 modifications within ±20 kb of TSSs of cobound genes. n = 609. Heat maps are centered at TSSs and ordered by RING1B intensity.
To gain insight into the types of genes that are cobound by BRD1 and RING1B, we performed a GO analysis of genes overlapping (1) cobound regions and (2) BRD1-only regions. The cobound regions, which essentially include all binding sites for RING1B, are significantly enriched for developmental processes and transcription, consistent with known roles for PRC1 (Supplemental Fig. S5, cobound). BRD1 sites without RING1B are enriched for a variety of processes, including immunity, metabolism, and transcription (Supplemental Fig. S5, BRD1-only).
Discussion
Inappropriate activation and/or repression of gene expression underlies many human diseases, yet the mechanisms that execute transitions in developmental gene expression remain poorly defined. How are genes chosen to be initially active or repressed, and how are transitions in gene activity managed with fidelity? Transcription factors clearly regulate these changes, but how can this regulation occur with such specificity when their consensus binding sites and genomic occupancy appear so promiscuous? Together, our proteomic and ChIP-seq analyses suggest a model in which PRC1 and MOZ/MORF function to create a poised regulatory state during development (Fig. 8). As cells differentiate, bivalent protein complexes may eventually be diminished locally, as most loci resolve into either an active or silent state. We speculate that the choice of activation may occur via increased acetylation, influenced by nearby transcription factors (Poux et al. 2002), and subsequent enrichment of Fs(1)h and TrxG proteins such as Ash1 (Papp and Müller 2006), which we specifically recovered in our Br140 pull-down (Fig. 2; Supplemental Fig. S2). A transition toward silencing may involve deacetylation and a decrease in TrxG.
Figure 8.

Model for the role of bivalent complexes in developmental transitions of transcriptional state. During embryonic development, PRC1 and dMOZ/MORF form bivalent complexes at poised bivalent genes (shown in the center). The choice between a transcriptionally active or silent state may be triggered by specific transcriptional factors that alter the acetylation state of the chromatin environment, favoring the association of increased Fs(1)h or PRC2, respectively.
The retention of some bivalency after initial transcriptional choices are made in embryogenesis is likely to allow critical reversibility for subsequent gene expression programming. However, if transcriptional state is not dictated strictly by the occupancy of bivalent components, how are these states manifested? We speculate that local post-translational modifications (PTMs) may be critical for the specification of transcriptional state and for reversibility. For example, the Enok subunit of dMOZ/MORF is known to acetylate H3K23, while this mark is incompatible with Pc chromodomain binding to H3K27me3 on the same histone tail (Fischle et al. 2003; Huang et al. 2014). Interestingly, we did not observe enrichment of H3K23ac from modENCODE data sets on our set of potentially bivalent genes (Supplemental Fig. S3A), but further analysis will be required to investigate the significance of this finding. Competition between the cognate enzymatic activities within bivalent complexes and their interactors may be central to their ability to act as reversible switches of transcriptional state. Future studies to address this hypothesis will require improved approaches to comprehensive PTM detection as well as in vitro reconstitution of key interactions and biochemical activities of bivalent complexes containing the appropriately modified subunits.
Our results are consistent with recent studies in which PRC1 is found on active genes in many systems (Schaaf et al. 2013; Pemberton et al. 2014; van den Boom et al. 2016), and PRC1 targeting is largely independent of PRC2 (Kahn et al. 2016). Most exciting is the likely conservation in zebrafish (Laue et al. 2008) and mice (Sheikh et al. 2015), based on the opposing genetic activities of PRC1 and MOZ/MORF complexes in regulation of the Hox genes. The reliance on a universal transducer of transcription factor activity in developmental decisions would be an elegant solution to the problem of widespread binding of sequence-specific regulators, as, in our model, only local interactions with preset bivalency will result in functional consequences.
Key fundamental questions remain. In particular, how are PRC1 and MOZ/MORF targeted in the first place? PREs are cis-acting regulatory elements that can recruit PRC1 and PRC2 to target genes in Drosophila. PREs lack universal consensus sequences but contain combinations of motifs for many DNA-binding proteins (Kassis and Brown 2013). Therefore, diverse protein–protein interactions with the PcG could be critical for initial binding, as postulated from classical genetics. A speculative alternative is that the 5′ TSSs of developmentally regulated genes may remain epigenetically marked throughout the life cycle of the organism to specify the initial association of bivalent complexes. Both BRD4 and BRPF1 have been identified as “bookmarking proteins” that may retain vital information throughout the cell cycle, based on their ability to remain at their chromosomal binding sites through mitosis (Dey et al. 2003; Laue et al. 2008). Furthermore, Fs(1)h and Enok are essential for oogenesis, and genic acetylation is detected very early in embryogenesis (Li et al. 2014). Finally, the importance of maternal E(z) suggests that H3K27me3 could be at least part of such an inherited mark for developmental genes (Zenk et al. 2017).
In summary, our results provide evidence for bivalent protein complexes that may correspond to a bivalent transcriptional state in Drosophila embryos and mammalian stem cells. Beyond identification of these intriguing protein interactions in flies, we speculate that their identity reveals a likely role for acetylation in the resolution of bivalency. We envision that the choice toward activation may be triggered and maintained by cell-specific transcription factors that drive the acetylated state, favoring MOZ/MORF and BRD4 bromodomain-dependent association with chromatin. Cell type decisions may be governed by a constant assessment of the amount of acetylation at each TSS, consistent with the enrichment of deacetylases on even very active genes (Delcuve et al. 2012). Deacetylation would favor loss of bromodomain–acetyl interactions and ultimately the loss of coactivators, leading toward the establishment of a stably silenced state. The ability to regulate genes while only partially resolving bivalent complexes is likely to be critical for reversibility in response to changes in cell type-specific transcription factor expression and binding. We propose that regulatory elements possess the intrinsic ability to switch fate dependent on this local balance, with de novo targeting rarely required.
Materials and methods
Cell culture
S2 cells were cultured at 25°C in Schneider's Drosophila medium (Gibco) supplemented with 1× antibiotic–anti-mycotic (Gibco). HUES64 cells were cultured at 37°C in mTeSR 1 medium (Stem Cell Technologies) on plates coated with Matrigel (Corning). The medium was replaced daily, and cells were passaged with 0.05 M EDTA/PBS.
BioTAP-tagged transgene constructs and transgenesis
All transgenes containing N-terminal or C-terminal BioTAP tags were constructed by the Red/ET recombination system. The N-terminal BioTAP tag was introduced into BACR26B18 using the counterselection BAC modification kit (GeneBridges, K002). The 10-kb genomic fragment containing BioTAP-N-Br140 and its upstream and downstream regions was recombineered into the pFly backbone vector (Wang et al. 2013). The BioTAP-N-Br140 transgene was injected into ΦC31 flies (strain 9732) for site-specific integration at the 76A2 cytosite on chromosome 3 (Miller et al. 2002). For Fs(1)h, the N-terminal and C-terminal BioTAP tags were introduced into BAC CH321-60P23, and the 27-kb genomic regions of Fs(1)h tagged with N-terminal or C-terminal BioTAP were transferred to the pFly vector. N-terminal or C-terminal BioTAP-Fs(1)h and BioTAP-N-Br140 transgenes were cotransfected with a hygromycin-resistant vector at a 10:1 mass ratio into S2 cells using a calcium phosphate transfection kit (Invitrogen), and stable transgenic S2 cell lines were selected by supplementing Schneider's medium with hygromycin B at a final concentration of 300 µg/mL.
BioTAP-XL
The BioTAP-XL protocol was followed as described in Alekseyenko et al. (2015). Briefly, 12- to 24-h-old embryos from transgenic fly lines expressing BioTAP-N Br140 were collected and stored for up to 3 d at 4°C. Embryos were cross-linked with 3% formaldehyde, and extracts were prepared as described and snap-frozen in liquid nitrogen for storage at −80°C. Twenty grams of embryos was used for each experiment. S2 cell lines stably expressing BioTAP-N Br140, BioTAP-N Fs(1)h, and Fs(1)h C-BioTAP were grown at 26.5°C to a density of ∼1 × 107 cells per milliliter using 500 mL of HyClone CCM3 serum-free medium in 2.8-L Fernbach glass flasks with shaking at 90 rpm. Cross-linked nuclear extracts from S2 cells were prepared from a total of 5 × 109 cells. After sonication, tandem affinity purification was performed to purify the BioTAP-tagged bait along with its protein interaction partners and associated genomic DNA. Genomic localization was determined by high-throughput sequencing of libraries generated from the tandem affinity-purified material using the NEBNext ChIP-seq library preparation master mix set for Illumina (New England Biolabs, catalog no. E6240S) and TruSeq adaptors (Illumina). ChIP libraries were sequenced at the Tufts University Core Facility using an Illumina HiSeq 2500.
For protein identification, streptavidin–agarose complexes were digested with trypsin (Promega) while rotating overnight at 37°C, and peptides were desalted with in-house-constructed C18 (3M) tips as described previously (Zee et al. 2016). Peptides were lyophilized with a vacuum centrifuge and stored in −80°C until liquid chromatography-mass spectrometry (LC-MS) analysis as described previously (Alekseyenko et al. 2015). Mass spectrometric data for protein identification have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaino et al. 2016) with the data set identifier PXD006079.
For histone PTM analysis, streptavidin-bound complexes were derivatized with propionic anhydride (Sigma-Aldrich) prior to trypsin digestion as described previously (Zee et al. 2016). Another round of derivatization was performed following trypsin digestion before desalting and LC-MS analysis. Mass spectrometric data for the histone PTM analysis have been deposited to the Harvard Dataverse (https://dataverse.harvard.edu) with the data set identifier doi:10.7910/DVN/Y0BG1S.
To determine enrichments from the protein identification samples, LC-MS files were first searched using the SEQUEST algorithm with precursor mass tolerance of either 1 amu or 20 ppm, depending on whether the MS1 data were acquired on either an ion trap or Orbitrap, respectively. For both instruments, b and y fragment mass tolerance was set at 0.9 Da, and variable methionine oxidation was specified. Peptide identifications were further filtered with XCorr ≥ 2 and XCorr ≥ 3 for z = 2 and z = 3, respectively, and ΔCorr ≥ 0.1.
Scatter plots were generated as described previously (Kang et al. 2015). Briefly, once peptides were mapped to the proteome, the total spectral counts were divided by the molecular weight (kilodalton) of the respective protein to control for protein size. A pseudocount of 0.5 peptides was substituted for proteins with zero peptides. Weight-normalized counts for each protein were then divided by the sum of weight-normalized counts across all proteins in the entire sample to control for overall peptide amount. After natural log transformation of the weight- and sample-normalized counts, the immunoprecipitations were then divided by the inputs and log10-normalized again to yield enrichments of binding partners for the BioTAP-tagged baits.
Imaginal disc ChIP-seq and ChIP-qPCR
Imaginal disc ChIP-seq was performed as described in Langlais et al. (2012) with some modifications. Fifty to ∼100 wing discs or haltere and third leg discs were collected from dissection of third instar larvae, cross-linked in 2% formaldehyde, and sonicated using a Bioruptor (Diagenode) for 15 cycles (30 sec on/30 sec off) on high power. Five percent of the total volume of sonicated chromatin was saved for input. ChIP was performed with 1:200 dilutions of anti-H3K27me3 (Cell Signaling, catalog no. C36B11) and anti-H3K27ac (Active Motif, catalog no. 39133). DNA was purified from immunoprecipitated samples. Illumina ChIP-seq libraries for H3K27me3 and H3K27ac were prepared using the NEBNext ChIP-seq library preparation kit (New England Biolabs) and sequenced as described above.
For ChIP-qPCR, 200 wing discs or haltere and third leg discs were collected from dissection of third instar larvae, cross-linked in 3% formaldehyde, and sonicated using the Covaris E210 ultrasonicator for 5 min (30 sec on/30 sec off) on 6% duty cycle. ChIP was performed with 1:100 dilutions of anti-Pc (Santa Cruz Biotechnology), anti-Enok (a kind gift from Dr. Jerry L. Workman), and anti-Fs(1)h (a kind gift from Dr. Igor B. Dawid). qPCR was performed using Platinum SYBR Green qPCR Supermix-UDG with ROX (Invitrogen) on the ABI Prism 7000 sequence detection system (Applied Biosystems). Primers used for qPCR are listed in Supplemental Table S1. Primer efficiencies, calculated using standard dilutions, were close to 100%, and specificity of the PCR products was confirmed by dissociation curve analysis.
Fly genetics
The crossing schemes for rescue tests of the BioTAP-N Br140 transgene are described in Supplemental Figure S1C. Rescued adult flies were assessed by the absence of balancer markers and the expression of mini-white from the transgenic construct. In the absence of the BioTAP-tagged transgene, transheterozygous mutants were lethal. Br140S203 and Br140S781 fly stocks were kindly provided by Dr. T. Suzuki.
Immunostaining of imaginal disc tissues
Immunostaining of Drosophila imaginal discs was performed as described previously (Beuchle et al. 2001). Briefly, wing, third leg, and haltere imaginal discs were dissected from third instar larvae in PBS and fixed with 4% formaldehyde in PBS + 0.1% Triton X-100 (PBST). Discs were washed several times and blocked in BBT (1% bovine serum albumin, 0.1% Triton X-100 in PBS) and labeled with mouse anti-Ubx (1:100; Developmental Hybridoma Studies Bank, FP6.87). Donkey anti-mouse Alexa fluor 488 (1:500 dilution in BBT; Invitrogen) was used as a secondary antibody, and imaginal discs were mounted in Prolong Gold (Invitrogen) for imaging.
Western blots
Embryos (12–24 h) from BioTAP-N Br140 transgenic flies were collected and dechorionated by immersion in 50% bleach for 3 min, rinsed with distilled water, and dried. Embryos (0.2 g) were suspended in 150 µL of nuclear extraction buffer (10% sucrose, 20 mM HEPES at pH 7.6, 10 mM NaCl, 3mM MgCl2, 0.1% Triton X-100) supplemented with 0.1 mM PMSF in a 1.5-mL tube and homogenized on ice with a motorized pestle. Nuclear extraction buffer was added up to 1 mL, and the lysate was centrifuged at 2000g for 5 min at 4°C. The nuclear pellet was resuspended in 150 µL of nuclear extraction buffer, and homogenization and centrifugation steps were repeated. The final crude nuclear pellet was resuspended in 600 µL of Novex Tris-glycine SDS sample buffer (Invitrogen), including NuPAGE sample-reducing agent (Invitrogen), and then boiled for 10 min. For S2 cells, ∼5 × 106 cells expressing BioTAP transgenes were harvested; washed with PBS; lysed with 200 μL of Novex Tris-glycine SDS sample buffer (Invitrogen), including NuPAGE sample-reducing agent (Invitrogen); and then boiled for 10 min.
Twenty microliters was used for each Western blot. PAP antibody (1:1000; Sigma, P1291) diluted in TBST + 5% milk was used to detect the Protein A epitope of the BioTAP tag, and Clarity Western ECL substrate (Bio-Rad) was used for detection.
Preparation of baculovirus-infected nuclear extracts and Flag purification
PRC1 and PRC2 component baculovirus constructs and viruses were kind gifts from R. Kingston (Francis et al. 2001; Müller et al. 2002). The Fs(1)h baculovirus was constructed by insertion of a short-form cDNA of Fs(1)h from Drosophila Genomics Resource Center stock LD26482 into pFastBac1. Fs(1)h baculovirus was generated using the Bac-to-Bac baculovirus expression system (Invitrogen). Viral stocks were reamplified individually in T-225 flasks for 5–7 d in Insect-Xpress medium (Lonza) supplemented with 10% FBS (Sigma) prior to use in protein expression experiments. For protein coexpression infections, baculoviruses were mixed at a ratio of 1:1:1:0.5:0.4 for Fs(1)h:E(z):Suz12:Caf1-55:Flag-Esc or 1.25:1:2:0.6:0.7 for Fs(1)h:Flag-Psc:Ph:Pc:Sce and incubated for 72 h at 26.7°C in 50-mL or 500-mL total volume (starting cell density 2 × 106 Sf9 cells per milliliter). Extracts and Flag purifications were made as described previously (Kang et al. 2015). Briefly, soluble nuclear extracts were made using salt extraction of crude nuclear lysate. For copurifications, 50–60 μL of anti-Flag magnetic resin was incubated overnight in 1 mL of the appropriate nuclear extract. Bound complexes were washed extensively with a maximum salt concentration of 2 M KCl. Elution fractions were taken with 0.4 mg/mL Flag peptide, and the protein content of elution fractions was analyzed by silver staining of SDS-PAGE gels. The presence of Fs(1)h was further probed by Western blotting using anti-Fs(1)h (a kind gift from Dr. Igor B. Dawid) and anti-rabbit HRP.
Embryo, S2 cell, and imaginal disc ChIP-seq analyses
To trim adaptor sequences, Cutadapt version 1.2.1 (Martin 2011) was used. For the alignment, the trimmed reads were mapped to the Drosophila genome (dm3 assembly) using Bowtie version 12.0 (Langmead et al. 2009) with the parameter “-m 1.” Only uniquely aligned reads were used for the entire analysis. To obtain input normalized profiles, the callpeak function was used with parameters “-B --nomodel --extsize 75 --SPMR” and the bgdcmp function “-m FE” using MACS version 2 (Zhang et al. 2008). Significantly enriched regions were identified using the “get.broad.enrichment.clusters” function of the SPP R package (Kharchenko et al. 2008) with a 1-kb window size and a z-score threshold of 3, and we called these enriched regions “relaxed peaks.” To determine whether the replicates are consistent, genome-wide correlations with 1-kb bins were calculated. For Pc and Br140 in embryos, enriched regions consistently detected from the two replicates were used for the analysis. For the heat map figures, one representative replicate with a higher quality was used. In S2 analysis, the top 80% Pc and Br140 peaks were used. For the comparison between promoters bound by Pc and/or Br140 in the Venn diagrams, a 500-base-pair (bp) margin around the TSS, as defined from the dm3 annotations, was used. Stringent Pc peaks were defined as peaks whose enrichment around promoters within a 1-kb window were greater than twofold. For the heat map figures, signals were averaged in 200 bins at the promoters with a 10-kb window. Five groups in the heat map figures in embryos (Supplemental Fig. S3A) were determined with the k-means clustering method based on the distributions of Pc, Br140, H3K27me3, and H3K27ac around the promoters with a 5-kb window, and each row was sorted by H3K27me3 intensities for each cluster. For the S2 cell heat map in Figure 5, the four groups around the peaks of promoters were determined by the presence of Pc, Br140, and H3K27me3 (window size of 2 kb) and ordered by the intensity of H3K27me3. For the GO analysis, DAVID (Dennis et al. 2003) was used, and the result was visualized for the top 10 GO categories for each group using the R package clusterProfiler (Yu et al. 2012). Embryo histone modification data files from modENCODE were obtained from Gene Expression Omnibus (GEO) for H3K4me3 (GSE47285), H3K27me3 (GSE47230), and H3K27ac (GSE47237) and processed as above. For S2 cells, log2 M-value profiles were obtained from GEO for H3K4me3 (GSE20787), H3K27me3 (GSE20781), and H3K27ac (GSE20779).
Globally/broadly expressed or constitutive genes were obtained from Ho et al. (2014), and the gene expression variability score was calculated as the ratio of the standard deviation and mean of expression across multiple developmental stages and samples in the modENCODE project. A higher expression variability score indicates that the gene is more likely to be specifically expressed/regulated. For comparison between cobound genes and the control, a number of globally expressed genes equal to the number of genes in the cobound group were chosen at random.
HUES64 ChIP-seq and analyses
ChIP was performed in HUES64 cells using the Myers laboratory ChIP-seq protocol (http://myers.hudsonalpha.org/documents/Myers%20Lab%20ChIP-seq%20Protocol%20v041610.pdf) with the following modifications. Nuclei were sonicated using a Bioruptor (Diagenode) for 20 cycles (30 sec on/30 sec off) on high power. After centrifugation at 20,000g for 15 min at 4°C, 5% of total chromatin was set aside for input, and the remainder was incubated with 10 µg of anti-BRD1 (Abcam, ab71877) or anti-RING1B (Abcam, ab3832) overnight at 4°C. Chromatin/antibody complexes were captured by incubation with Protein A Dynabeads (Invitrogen) for 2 h at 4°C with rotation. After cross-link reversal, DNA was purified by phenol-chloroform extraction and ethanol precipitation. Illumina ChIP-seq libraries were prepared using the NEBNext ChIP-seq library preparation kit (New England Biolabs) and sequenced as described above.
Raw FASTQ files were quality-filtered using the FASTX-Toolkit “fastq_quality_filter” tool (http://hannonlab.cshl.edu/fastx_toolkit) so that only reads containing a quality score of ≥20 for at least 80% of base positions were kept. Filtered reads were aligned to the hg19 human genome annotation using Bowtie2 with default parameters (Langmead et al. 2009). Reads with a MAPQ ≥ 20 were kept and extended to an estimated fragment length of 150 bp using deepTools (Ramirez et al. 2014). Peaks were called for each immunoprecipitation replicate using the HOMER “findPeaks” tool with a matched input as control. Considering the broad nature of peaks upon visual inspection, we set peak parameters so that the minimum peak size was 1000 bp and the minimum allowable distance between peaks was 2500 bp. Common peaks from two biological replicates were identified using DiffBind (R Bioconductor) for each immunoprecipitation, and these common peak sets were used for downstream analyses. HUES64 ChIP-seq data files for H3K4me3 and H3K27me3 were obtained from GEO (GSE19465) and processed as above.
Genomic binding profiles for all HUES64 ChIP-seq replicates were generated using the deepTools “bamCompare” tool with a bin size of 20 and “ratio” normalized to a matched input. The resulting bigWig files were visualized in the Integrative Genome Viewer (IGV). Heat maps were generated using the deepTools “computeMatrix” and “plotHeatmap” functions on the generated genomic binding profiles.
GO analyses were performed on the indicated gene sets using the PANTHER database (Mi et al. 2017). Significantly enriched terms after Bonferroni correction, were plotted as a bubble plot in R, where fill represents the corrected P-value, size represents gene percentage, and terms were sorted by decreasing fold enrichment.
Accession numbers for ChIP-seq data files
ChIP-seq data have been submitted to the NCBI GEO public repository under the accession number GSE104059.
Supplementary Material
Acknowledgments
We are grateful to Dr. R. Kingston, Dr. I. Dawid, Dr. J. Workman, Dr. R. Jones, Dr. T. Suzuki, and Dr. W. Bender for generous sharing of reagents, and R. Tomaino for help with protein samples (Taplin Mass Spectrometry Facility, Harvard Medical School). We thank A. Alekseyenko for numerous helpful discussions and critical reading of the manuscript. We thank the Bloomington Stock Center (National Institutes of Health [NIH] P40OD018537) for providing fly stocks and/or DNA constructs used in this study. This work was supported by NIH grant GM101958 to M.I.K. K.A.M. was supported by the Joint Training Program in Molecules, Cells, and Organisms (T32GM007598 from the NIH). B.M.Z was supported by a fellowship from the Jane Coffin Childs Memorial Fund.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.305987.117.
References
- Alekseyenko AA, Gorchakov AA, Kharchenko PV, Kuroda MI. 2014. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci 111: 2488–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, McElroy KA, Kang H, Zee BM, Kharchenko PV, Kuroda MI. 2015. BioTAP-XL: cross-linking/tandem affinity purification to study DNA targets, RNA, and protein components of chromatin-associated complexes. Curr Protoc Mol Biol 109: 21.30.1–21.30.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Trupke J, Ringrose L. 2016. The quest for mammalian Polycomb response elements: are we there yet? Chromosoma 125: 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. [DOI] [PubMed] [Google Scholar]
- Beuchle D, Struhl G, Müller J. 2001. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development 128: 993–1004. [DOI] [PubMed] [Google Scholar]
- Bienz M, Müller J. 1995. Transcriptional silencing of homeotic genes in Drosophila. Bioessays 17: 775–784. [DOI] [PubMed] [Google Scholar]
- Bowman SK, Deaton AM, Domingues H, Wang PI, Sadreyev RI, Kingston RE, Bender W. 2014. H3K27 modifications define segmental regulatory domains in the Drosophila Bithorax complex. Elife 3: e02833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F, Arnaud P. 2017. An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research. Oncotarget 8: 4110–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcuve GP, Khan DH, Davie JR. 2012. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics 4: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4: P3. [PubMed] [Google Scholar]
- Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci 100: 8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fontanals-Cirera B, Low V, Ntziachristos P, Yuen SK, Lovell CD, Dolgalev I, Yonekubo Y, Zhang G, Rusinova E, et al. 2014. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep 9: 234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Saurin AJ, Shao Z, Kingston RE. 2001. Reconstitution of a functional core polycomb repressive complex. Mol Cell 8: 545–556. [DOI] [PubMed] [Google Scholar]
- Frangini A, Sjöberg M, Roman-Trufero M, Dharmalingam G, Haberle V, Bartke T, Lenhard B, Malumbres M, Vidal M, Dillon N. 2013. The aurora B kinase and the polycomb protein ring1B combine to regulate active promoters in quiescent lymphocytes. Mol Cell 51: 647–661. [DOI] [PubMed] [Google Scholar]
- Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, Reinberg D. 2014. An AUTS2–Polycomb complex activates gene expression in the CNS. Nature 516: 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler SJ, Paro R. 2015. Trithorax and Polycomb group-dependent regulation: a tale of opposing activities. Development 142: 2876–2887. [DOI] [PubMed] [Google Scholar]
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. 2013. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 153: 1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud C, Nègre N, Cavalli G. 2006. From genetics to epigenetics: the tale of Polycomb group and Trithorax group genes. Chromosome Res 14: 363–375. [DOI] [PubMed] [Google Scholar]
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512: 449–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Paulson A, Dutta A, Venkatesh S, Smolle M, Abmayr SM, Workman JL. 2014. Histone acetyltransferase Enok regulates oocyte polarization by promoting expression of the actin nucleation factor spire. Genes Dev 28: 2750–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Saraf A, Florens L, Kusch T, Swanson SK, Szerszen LT, Li G, Dutta A, Washburn MP, Abmayr SM, et al. 2016. The Enok acetyltransferase complex interacts with Elg1 and negatively regulates PCNA unloading to promote the G1/S transition. Genes Dev 30: 1198–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19: 523–534. [DOI] [PubMed] [Google Scholar]
- Kahn TG, Dorafshan E, Schultheis D, Zare A, Stenberg P, Reim I, Pirrotta V, Schwartz YB. 2016. Interdependence of PRC1 and PRC2 for recruitment to Polycomb response elements. Nucleic Acids Res 44: 10132–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, McElroy KA, Jung YL, Alekseyenko AA, Zee BM, Park PJ, Kuroda MI. 2015. Sex comb on midleg (Scm) is a functional link between PcG-repressive complexes in Drosophila. Genes Dev 29: 1136–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K. 2004. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol Cell 13: 33–43. [DOI] [PubMed] [Google Scholar]
- Kassis JA, Brown JL. 2013. Polycomb group response elements in Drosophila and vertebrates. Adv Genet 81: 83–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ. 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston RE, Tamkun JW. 2014. Transcriptional regulation by trithorax-group proteins. Cold Spring Harb Perspect Biol 6: a019349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockmann T, Gerstung M, Schlumpf T, Xhinzhou Z, Hess D, Beerenwinkel N, Beisel C, Paro R. 2013. The BET protein FSH functionally interacts with ASH1 to orchestrate global gene activity in Drosophila. Genome Biol 14: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppens M, van Lohuizen M. 2016. Context-dependent actions of Polycomb repressors in cancer. Oncogene 35: 1341–1352. [DOI] [PubMed] [Google Scholar]
- Langlais KK, Brown JL, Kassis JA. 2012. Polycomb group proteins bind an engrailed PRE in both the ‘on’ and ‘off’ transcriptional states of engrailed. PLoS One 7: e48765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue K, Daujat S, Crump JG, Plaster N, Roehl HH, Tubingen Screen C, Kimmel CB, Schneider R, Hammerschmidt M. 2008. The multidomain protein Brpf1 binds histones and is required for Hox gene expression and segmental identity. Development 135: 1935–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276: 565–570. [DOI] [PubMed] [Google Scholar]
- Li XY, Harrison MM, Villalta JE, Kaplan T, Eisen MB. 2014. Establishment of regions of genomic activity during the Drosophila maternal to zygotic transition. Elife 3: e03737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10. [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. 2017. PANTHER version 11: expanded annotation data from gene ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 45: D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DF, Holtzman SL, Kaufman TC. 2002. Customized microinjection glass capillary needles for P-element transformations in Drosophila melanogaster. Biotechniques 33: 366–367. [DOI] [PubMed] [Google Scholar]
- Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197–208. [DOI] [PubMed] [Google Scholar]
- Nègre N, Hennetin J, Sun LV, Lavrov S, Bellis M, White KP, Cavalli G. 2006. Chromosomal distribution of PcG proteins during Drosophila development. PLoS Biol 4: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Müller J. 2006. Histone trimethylation and the maintenance of transcriptional on and off states by trxG and PcG proteins. Genes Dev 20: 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton H, Anderton E, Patel H, Brookes S, Chandler H, Palermo R, Stock J, Rodriguez-Niedenführ M, Racek T, de Breed L, et al. 2014. Genome-wide co-localization of Polycomb orthologs and their effects on gene expression in human fibroblasts. Genome Biol 15: R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poux S, Horard B, Sigrist CJ, Pirrotta V. 2002. The Drosophila trithorax protein is a coactivator required to prevent re-establishment of polycomb silencing. Development 129: 2483–2493. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42: W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CA, Misulovin Z, Gause M, Koenig A, Gohara DW, Watson A, Dorsett D. 2013. Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLoS Genet 9: e1003560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Ganapathi M, Leblanc B, Portoso M, Jaschek R, Tolhuis B, van Lohuizen M, Tanay A, Cavalli G. 2009. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol 7: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, Pirrotta V. 2006. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet 38: 700–705. [DOI] [PubMed] [Google Scholar]
- Sheikh BN, Downer NL, Phipson B, Vanyai HK, Kueh AJ, McCarthy DJ, Smyth GK, Thomas T, Voss AK. 2015. MOZ and BMI1 play opposing roles during Hox gene activation in ES cells and in body segment identity specification in vivo. Proc Natl Acad Sci 112: 5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J, Chiang A, Bender W. 1992. Ten different Polycomb group genes are required for spatial control of the abdA and AbdB homeotic products. Development 114: 493–505. [DOI] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. 2006. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 6: 846–856. [DOI] [PubMed] [Google Scholar]
- Strubbe G, Popp C, Schmidt A, Pauli A, Ringrose L, Beisel C, Paro R. 2011. Polycomb purification by in vivo biotinylation tagging reveals cohesin and Trithorax group proteins as interaction partners. Proc Natl Acad Sci 108: 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G. 1981. A gene product required for correct initiation of segmental determination in Drosophila. Nature 293: 36–41. [DOI] [PubMed] [Google Scholar]
- Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, van Lohuizen M. 2006. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat Genet 38: 694–699. [DOI] [PubMed] [Google Scholar]
- Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, Tahmasebi S, Cayrou C, Doyon Y, Goh SL, et al. 2008. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol Cell Biol 28: 6828–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom V, Maat H, Geugien M, Rodríguez López A, Sotoca AM, Jaques J, Brouwers-Vos AZ, Fusetti F, Groen RW, Yuan H, et al. 2016. Non-canonical PRC1.1 targets active genes independent of H3K27me3 and is essential for leukemogenesis. Cell Rep 14: 332–346. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, Kuroda MI. 2013. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat Struct Mol Biol 20: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ. 2015. MOZ and MORF acetyltransferases: molecular interaction, animal development and human disease. Biochim Biophys Acta 1853: 1818–1826. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ullah M. 2007. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 26: 5408–5419. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Alekseyenko AA, McElroy KA, Kuroda MI. 2016. Streamlined discovery of cross-linked chromatin complexes and associated histone modifications by mass spectrometry. Proc Natl Acad Sci 113: 1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenk F, Loeser E, Schiavo R, Kilpert F, Bogdanović O, Iovino N. 2017. Germ line-inherited H3K27me3 restricts enhancer function during maternal-to-zygotic transition. Science 357: 212–216. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.