

Abstract
Coconut palm (Cocos nucifera,2n = 32), a member of genus Cocos and family Arecaceae (Palmaceae), is an important tropical fruit and oil crop. Currently, coconut palm is cultivated in 93 countries, including Central and South America, East and West Africa, Southeast Asia and the Pacific Islands, with a total growth area of more than 12 million hectares [1]. Coconut palm is generally classified into 2 main categories: “Tall” (flowering 8–10 years after planting) and “Dwarf” (flowering 4–6 years after planting), based on morphological characteristics and breeding habits. This Palmae species has a long growth period before reproductive years, which hinders conventional breeding progress. In spite of initial successes, improvements made by conventional breeding have been very slow. In the present study, we obtained de novo sequences of the Cocos nucifera genome: a major genomic resource that could be used to facilitate molecular breeding in Cocos nucifera and accelerate the breeding process in this important crop. A total of 419.67 gigabases (Gb) of raw reads were generated by the Illumina HiSeq 2000 platform using a series of paired-end and mate-pair libraries, covering the predicted Cocos nucifera genome length (2.42 Gb, variety “Hainan Tall”) to an estimated ×173.32 read depth. A total scaffold length of 2.20 Gb was generated (N50 = 418 Kb), representing 90.91% of the genome. The coconut genome was predicted to harbor 28 039 protein-coding genes, which is less than in Phoenix dactylifera (PDK30: 28 889), Phoenix dactylifera (DPV01: 41 660), and Elaeis guineensis (EG5: 34 802). BUSCO evaluation demonstrated that the obtained scaffold sequences covered 90.8% of the coconut genome and that the genome annotation was 74.1% complete. Genome annotation results revealed that 72.75% of the coconut genome consisted of transposable elements, of which long-terminal repeat retrotransposons elements (LTRs) accounted for the largest proportion (92.23%). Comparative analysis of the antiporter gene family and ion channel gene families between C. nucifera and Arabidopsis thaliana indicated that significant gene expansion may have occurred in the coconut involving Na+/H+ antiporter, carnitine/acylcarnitine translocase, potassium-dependent sodium-calcium exchanger, and potassium channel genes. Despite its agronomic importance, C. nucifera is still under-studied. In this report, we present a draft genome of C. nucifera and provide genomic information that will facilitate future functional genomics and molecular-assisted breeding in this crop species.
Keywords: coconut palm, genome, assembly, annotation
Data Description
Background
Coconut palm (Cocos nucifera, 2n = 32), the only species in the genus Cocos in the family Arecaceae, is a tropical oil crop and widely cultivated in tropical regions due to its extensive application in agriculture and industry. Coconut palm is thought to have originated from the Southwest and Western Pacific region (including the Malay Peninsula and Archipelago, New Guinea, and the Bismarck Archipelago). At present, this tropical tree crop is distributed across 93 tropical countries [2], including Central and South America, East and West Africa, Southeast Asia, and the Pacific Islands, and is grown over 12 million hectares of land [1].
In China, coconut palm grows in the subtropical regions—Hainan and Yunnan provinces—as an economic and ornamental plant. Coconut palm is cultivated over approximately 43 000 hectares in Hainan, with the “Hainan Tall” (HAT) variety covering 36 000 hectares [3]. The HAT coconut needs 8–10 years to enter its reproductive stage and has a height of 20–30 meters, with a medium to large sized nut. The HAT cultivar is highly tolerant to salt and drought stress, but sensitive to temperatures below 10°C. Coconut palm can disseminate through ocean currents: floating nuts sprout and grow naturally upon washing up on beaches. The ability to adapt to a high-salt environment is closely related to this dissemination feature and to these natural growth conditions. The morphological characteristics of the HAT cultivar are shown in Fig. 1. Here, we present the genome sequence of the Hainan Tall coconut and an analysis of the antiporter and ion channel gene families, relevant to salinity tolerance. As draft genome sequences of coconut relatives (e.g., Elaeis guineensis [4] and Phoenix dactylifera [5, 6]) have previously been reported, we also performed a comparative analysis between the coconut and these relative species for genome assembly and annotation characteristics.
Figure 1:

Morphological characteristic of the coconut tree (A), spica (B), female flower (C), male flower (D), coconut nut (E), coconut nut without skin (F), and vertical section of coconut nut (G).
Data Description
Sample collection and sequencing strategy
The genomic DNA was extracted from the spear leaf of an individual of the variety “Hainan Tall” coconut (Cocos nucifera L. Taxonomy ID: 13 894; 19°33’3”N, 110°47’25”E) from the coconut garden of the Coconut Research Institute (Wenchang, Hainan Province, China) by using the CTAB extraction method [7]. Subsequently, 4 paired-end (PE) libraries with insert sizes of 170 bp, 500 bp, 450 bp, and 800 bp and 5 mate-pair (MP) libraries with insert sizes of 2 Kb, 5 Kb, 10 Kb, 20 Kb, and 40 Kb were constructed using the standard procedure provided by Illumina (San Diego, CA, USA). After library preparation and quality control of the DNA samples, template DNA fragments were hybridized to the surface of the flow cells on an Illumina HiSeq2000 sequencer, amplified to form clusters, and then sequenced by following the standard Illumina manual. Finally, we generated 714.67 Gb of raw reads from all constructed libraries. The raw outputs for each sequenced library are summarized in Table 1. Before assembly, the raw reads were pretreated using the following stringent filtering processes via SOAPfilter (v2.2) [8] software: (1) removed reads with 25% low-quality bases (quality scores ≤7); (2) removed reads with N bases more than 1%; (3) discarded reads with adapter contamination and/or polymerase chain reaction duplicates; (4) removed reads with undersized insert sizes. Finally, 419.08 Gb (estimated 173.17 × read depth) of high-quality sequences were obtained for genome assembly.
Table 1:
Data outputs produced by sequencing different insert size libraries.
| Library type | Lane | Reads length, bp | Insert size, bp | Raw data, Gb | Clean data, Gb |
|---|---|---|---|---|---|
| PE101 | 3 | 100 | 170 | 128.75 (53.20) | 111.32 (46) |
| PE251 | 2 | 250 | 450 | 73.86 (30.52) | 56.42 (23.31) |
| PE101 | 2 | 100 | 500 | 64 (26.45) | 65.11 (26.90) |
| PE101 | 2 | 100 | 800 | 78.16 (32.30) | 64.90 (26.82) |
| MP50 | 3 | 49 | 2000 | 128.6 (53.14) | 60.70 (25.08) |
| MP50 | 2 | 49 | 5000 | 71.75 (29.65) | 18.62 (7.69) |
| MP50 | 2 | 49 | 10 000 | 74.65 (30.85) | 18.53 (7.66) |
| MP50 | 2 | 49 | 20 000 | 70.7 (29.21) | 19.35 (7.99) |
| MP50 | 1 | 49 | 40 000 | 24.2 (10.08) | 4.13 (1.71) |
| Total | 19 | 714.67 (295.32) | 419.08 (173.17) |
The sequencing depth is shown in parentheses, calculated based on a genome size of 2.42 G. Clean data were obtained by filtering raw data with low-quality and duplicate reads.
De novo assembly of short reads of Cocos nucifera
We used 209.38 Gb of clean reads of the short-insert libraries (excluding the 450-bp library) to estimate the coconut genome size by k-mer frequency distribution analysis [8]. The genome size (G) of Cocos nucifera could be estimated by the following formula:
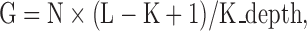 |
where N represents the total of number of reads, L represents the read length, K represents the k-mer value used in the analysis, and K_depth refers to the main peak in the k-mer distribution curve. In our calculations, N was 2 049 520 223, L was 100, and K_depth was 71 for K = 17. As a result, the Cocos nucifera genome was estimated to be 2.42 gigabases (Gb). K-mer size distribution analysis (Fig. 2) indicated that Cocos nucifera was a diploid species with low heterozygosity and a high proportion of repetitive sequences.
Figure 2:
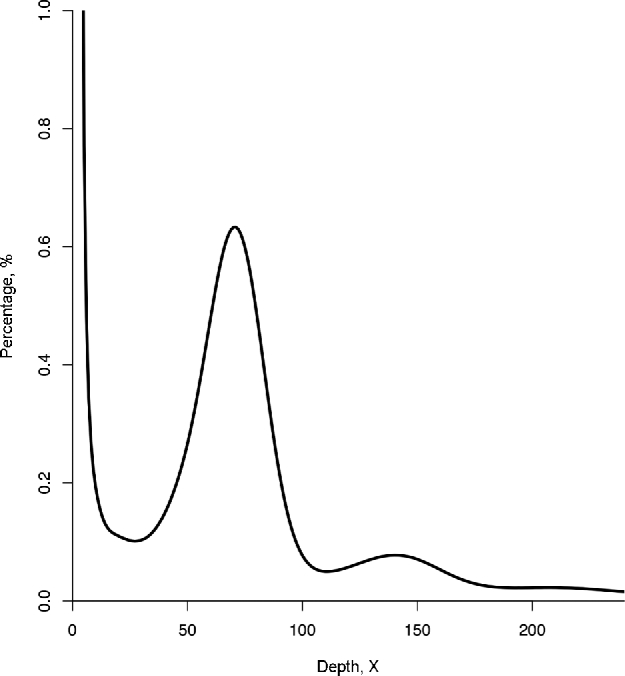
K-mer analysis of the coconut genome.
We then assembled the Cocos nucifera genome using the software SOAPdenovo2 (SOAPdenovo2, RRID:SCR_014986) in 3 steps: contig construction, scaffold construction, and gap filling. In the contig construction step, the SOAPdenovo2 was run with the parameters “pregraph -K 63 -R -d 1” to construct de Bruijn graphs from paired-end libraries with insert sizes ranging from 170 to 800 bp. The k-mers from the de Bruijn graphs were then used to form contiguous sequences (contigs) with the parameters “contig -R” by clipping tips, merging bubbles, and removing low-coverage links. In the scaffold construction step, the orders of the contigs were determined by using paired-end and mate-pair information with parameters “map -k 43” and “scaff -F -u”. In more detail, SOAPdenovo2 maps the reads from paired-end and mate-pair libraries to contigs based on a hash table (keys are unique k-mers on contigs; values are positions). In such cases, 2 contigs are considered to be linked if the bridging of the contigs is supported by 5 paired-end read pairs or 3 mate-pair read pairs. In the gap filling step, gaps within scaffolds were filled by utilizing KGF [8] v1.06 and GapCloser v1.12-r6 (GapCloser, RRID:SCR_015026) [8] with paired-end libraries (having an insert size from 170 to 800 bp in cases, where 1 end could be mapped to 1 contig and the other end extended into a gap). To optimize the assembled sequence, Rabbit (a Poisson-based k-mer model software [9]) was used to remove the redundant sequences. A final length of 2.20 Gb for the scaffolds was obtained and used for further analysis, accounting for 90.91% of the predicted genome size and larger than the African oil palm and date palm genomes (Table 2). Meanwhile, the N50 of the obtained contigs was 72.64 Kb and 418.06 Kb for the scaffolds, which have excluding scaffolds of less than 100 bp. The comparison of N50 values for the assembled coconut genome and for the 4 previously published palm genomes Elaeis guineensis [4], Elaeis oleifera [4], Phoenix dactylifera (PDK30) [5], and Phoenix dactylifera (DPV01) [6] is listed in Table 2.
Table 2:
Comparison analysis of genome sizes, assembly, and annotation of 4 palmae species, including coconut, Phoenix dacylifera (PDK30 and DPV01, 2 different versions), Elaeis guineensis (EG), and Elaeis oleifera (EO).
| Sequencing | Sequence | Estimated | Assembly | Contig | Scaffold | Gene | TEs, | |
|---|---|---|---|---|---|---|---|---|
| Species | technology | coverage | size, Gb | size, Gb | N50, Kb | N50, Kb | number | % |
| Phoenix dactylifera (PDK30) | Illumina GAIIx | ×53.4 | 0.66 | 0.38 | 6.44 | 30.48 | 28 889 | 23.6 |
| Phoenix dactylifera (DPV01) | 454, SOLiD, ABI3730 | ×139 | 0.67 | 0.56 | 10.81 | 334.08 | 41 660 | 38.87 |
| Elaeis guineensis (African oil palm) | 454 | ×16 | 1.8 | 1.54 | 9.37 | 1045.41 | 34 802 | 43.24 |
| Elaeis oleifera (American oil palm) | 454 | ×16 | 1.8 | 1.40 | 8.45 | 333.11 | – | – |
| Cocos nucifera (Hai nan Tall) | Illumina HiSeq | ×173 | 2.42 | 2.20 | 72.64 | 418.07 | 28 039 | 72.75 |
Coconut: Cocos nucifera (Hainan Tall); PDK30: Phoenix dactylifera (PDK30); DPV01: Phoenix dactylifera (DPV01); EG: Elaeis guineensis (Africanoil palm E5 build); EO: Elaeis oleifera (American oil palm, O8-build). TE results were obtained using the same pipeline as for the coconut genome
Genome evaluation
The 57 304 unigenes (transcript obtained from 3 different tissues, spear leaves, young leaves, and fruit flesh), as previously reported by Fan et al. [10], were aligned to the assembled genome of Cocos nucifera using BLAT (BLAT, RRID:SCR_011919) [11] with default parameters. The alignment results indicated that the assembled genome of Cocos nucifera covered 96.78% of the expressed unigenes, suggesting that a high level of coverage has been reached for the assembled genome (Table 3).
Table 3:
The gene coverage of Cocos nucifera based on transcriptome data.
| Data set | Number | Total length, bp | Base coverage by assembly | Sequence coverage by assembly, % |
|---|---|---|---|---|
| All | 57 304 | 43 090 665 | 96.78 | 99.57 |
| >200 bp | 57 304 | 43 090 665 | 96.78 | 99.57 |
| >500 bp | 25 713 | 33 470 388 | 96.36 | 99.85 |
| >1000 bp | 13 796 | 25 004 919 | 95.99 | 99.94 |
We also evaluated the level of genome completeness for the assembled sequences by using BUSCO v2.0 (BUSCO, RRID:SCR_015008) [12], which quantitatively assesses genome completeness using evolutionarily informed expectations of gene content from near-universal single-copy orthologs selected from OrthoDB v9 (OrthoDB, RRID:SCR_011980; plant set) [13]. BUSCO analysis showed that 90.8% and 3.4% of the 1440 expected plant genes were identified as complete and fragmented genes, respectively, while 5.8% of genes were considered to be missing from the assembled coconut genome sequence. The comparative results of the BUSCO estimation in the coconut and in the 4 other palm genome sequences indicates that the smallest fraction of missing genes as predicted by BUSCO was found in the coconut genome assembly (Table 4).
Table 4:
The comparative analysis of assembly results of 5 palm species with BUSCO software, including coconut, Phoenix dacylifera (PDK30 and DPV01, 2 varieties), Elaeis guineensis (EG), and Elaeis oleifera (EO).
| Coconut | PDK30 | DPV01 | EG | EO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BUSCOs | No. | P, % | No. | P, % | No. | P, % | No. | P, % | No. | P, % |
| Total | 1440 | 1440 | 1440 | 1440 | 1440 | |||||
| Complete single-copy | 1192 | 82.8 | 1042 | 72.4 | 1160 | 80.6 | 1100 | 76.4 | 1004 | 69.7 |
| Complete duplicated | 115 | 8.0 | 81 | 5.6 | 134 | 9.3 | 116 | 8.1 | 63 | 4.4 |
| Fragment | 49 | 3.4 | 98 | 6.8 | 42 | 2.9 | 60 | 4.2 | 84 | 5.8 |
| Missing | 84 | 5.8 | 219 | 15.2 | 104 | 7.2 | 164 | 11.3 | 289 | 20.1 |
Coconut: Cocos nucifera (the Hainan Tall); PDK30: Phoenix dactylifera (PDK30); DPV01: Phoenix dactylifera (DPV01); EG: Elaeis guineensis (African oil palm E5 build); EO: Elaeis oleifera (American oil palm, O8-build).
Repeat annotation
We combined homology-based annotation and a de novo method to identify transposable elements (TEs) and the tandem repeats in the Cocos nucifera genome. In the homology-based annotation step, TEs were identified by searching against the Repbase library (v20.04) [14] with RepeatMasker (v4.0.5; RepeatMasker, RRID:SCR_012954) [15] and RepeatProteinMasker (v4.0.5) [15]. In the de novo step, de novo libraries were constructed based on the genome sequences using the de novo prediction program RepeatModeler (RepeatModeler, RRID:SCR_015027) and LTR_FINDER (LTR_FINDER, RRID:SCR_015247) [16] by removing contaminant and multi-copy genes. Subsequently, novel transposable elements were identified and classified using RepeatMasker. Tandem repeat sequences were identified by Tandem Repeat Finder (TRF) software [17] with the following parameters “Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 50 and MaxPerid = 2000”. The total length of the tandem repeat sequences predicted by the software was 151 229 585 bp, comprising 6.86% of the coconut genome. Finally, 1.6 Gb of non-redundant repetitive elements were identified, accounting for 74.48% of the coconut genome. Transposable elements took up 72.75% of the total 1.6 Gb of repetitive elements, with the long-terminal repeat retrotransposon (LTR) class accounting for 92.23% of all TEs and 67.1% of the coconut genome (Table 5).
Table 5:
Classification of predicted transposable elements in the coconut genome.
| Repabse TEs | Protein TEs | De novo TEs | Combined TEs | ||
|---|---|---|---|---|---|
| Length | Length | Length | Length | Percentage | |
| DNA | 20 936 158 | 24 655 089 | 35 131 002 | 58 119 982 | 2.64 |
| LINE | 4 251 185 | 9 631 472 | 7 610 172 | 19 197 064 | 0.87 |
| SINE | 85 717 | 0.00 | 186 364 | 270 055 | 0.012 |
| LTR | 361 968 154 | 512 700 933 | 1 419 281 798 | 1 478 182 089 | 67.10 |
| Other | 8145 | 0.00 | 0.00 | 8145 | 0.0004 |
| Unknown | 0.00 | 12 360 | 139 084 335 | 139 096 695 | 6.31 |
| Total | 385 037 442 | 546 965 774 | 1 552 582 881 | 1 602 630 396 | 72.75 |
Note: Repabse TEs means RepeatMask against Repbase; Protein TEs means RepeatProteinMask result against Repbase protein; De novo TEs means RepeatMask against the de novo library; Combined TEs: the combined results of these 3 steps.
Gene prediction
We combined 3 strategies to predict genes in the Cocos nucifera genome: homology-based, de novo, and transcript alignment. For homology-based annotation, the protein sequences of Arabidopsis thaliana [18], Oryza sativa [19], Sorghum bicolor [20], Zea mays [21], Elaeis guineensis, and Phoenix dactylifera (DPV01) were downloaded from each corresponding source (see “Availability of data sources”). The coconut genome was aligned against these downloaded databases using TBLASTN [22] with parameter “-e 1e-5 -F -m 8” and BLAST results were processed by solar (v0.9) with parameter “-aprot 2 genome2 -z” to determine the candidate gene loci. Next, we extracted the genomic sequences of candidate gene loci, along with 1 kb of flanking sequences, and applied GeneWise 2.2.0 (GeneWise, RRID:SCR_015054) [23] to define the intron—exon boundaries. The genes with pre-stop codon or frame-shifts were excluded from further analysis.
For de novo prediction, we randomly selected 1000 full-length genes (GeneWise score equal to 100, intact structure: start codon, stop codon, perfect intron-exon boundary) from gene models predicted by homology-based methods to train the model parameters for AUGUSTUS 2.5 (Augustus: Gene Prediction, RRID:SCR_008417) [24]. Two software programs, AUGUSTUS 2.5 and GENSCAN (GENSCAN, RRID:SCR_012902) 1.0 [25], were used to do de novo prediction on the repeat-masked genome of Cocos nucifera. Genes with incomplete structure or a protein coding length of less than 150 bp were filtered out.
Subsequently, genes from both homology-based and de novo methods were combined to obtain non-redundant gene sets by using GLEAN [26] with the following parameters: minimum coding sequence length of 150 bp and maximum intron length of 50 kb. Genes were filtered with the same thresholds as were used for homology-based annotation.
For transcriptome-based prediction, RNA-seq data (SRR606452), as previously reported by Fan et al. [10], were mapped onto the coconut genome to identify the splice junctions using the software TopHat v2.1.1 (TopHat, RRID:SCR_013035) [27]. The software Cufflinks v2.2.1 (Cufflinks, RRID:SCR_014597) [28] was then used to assemble transcripts with the aligned reads. The coding potential of these transcripts was identified using a fifth-order Hidden Markov Model, which was estimated with the same gene sets used in AUGUSTUS training by train GlimmerHMM, an application in the GlimmerHMM package (GlimmerHMM, RRID:SCR_002654) [29]. The transcripts with intact open reading frames (ORFs) were extracted, and the longest transcript was retrieved as a representative of a gene from multiple transcripts on the same locus.
Finally, we merged the GLEAN and the transcriptome result to form a comprehensive gene set using an in-house annotation pipeline with the following steps: first, all-to-all BLASTP analysis of protein sequences was performed between GLEAN results and transcript assemblies, with an E-value cutoff of 1e-10. These transcript assemblies were added to the GLEAN result to form untranslated region (UTRs) or alternative splicing products, depending on whether the coverage and identity of the alignment results reached 0.9 or not. If the transcript assemblies had no BLAST hit with the GLEAN results, these transcript assemblies were added to the final gene set as a novel gene. The protocol for integrating GLEAN and transcriptome data is shown in Fig. 3.
Figure 3:
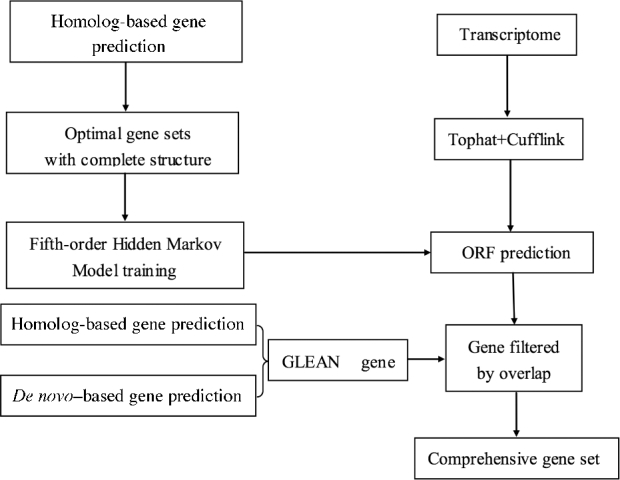
The protocol for integrating GLEAN and transcriptome data.
Gene evaluation
The annotation processes identified 28 039 protein-coding genes (Table 2), which is less than the predicted gene numbers of Phoenix dactylifera (PDK30, 28 889), Phoenix dactylifera (DPV01, 41 660), and Elaeis guineensis (34 802). Meanwhile, the BUSCO evaluation showed that 74.1% and 11.2% of 1440 expected plant genes were identified as complete and fragmented, with 14.7% of genes considered missing in the gene sets. The BUSCO results showed that our gene prediction was more complete than that of Phoenix dactylifera (PDK30) and Elaeis guineensis, but less complete than that of Phoenix dactylifera (DPV01) (Table 6).
Table 6:
The comparative analysis of gene prediction results of 4 palm species with BUSCO software.
| Coconut | PDK30 | DPV01 | EG | |||||
|---|---|---|---|---|---|---|---|---|
| BUSCOs | No. | P, % | No. | P, % | No. | P, % | No. | P, % |
| Total | 1440 | 1440 | 1440 | 1440 | ||||
| Complete single-copy | 965 | 74.1 | 748 | 51.9 | 1195 | 83.0 | 555 | 38.5 |
| Complete duplicated | 102 | 7.1 | 81 | 5.6 | 159 | 11.0 | 53 | 3.7 |
| Fragment | 162 | 11.2 | 255 | 17.7 | 44 | 3.1 | 270 | 18.8 |
| Missing | 211 | 14.7 | 356 | 24.8 | 42 | 2.9 | 562 | 39.0 |
Note: Coconut: Cocos nucifera (the Hainan Tall); PDK30: Phoenix dactylifera (PDK30); DPV01: Phoenix dactylifera (DPV01); EG: Elaeis guineensis (African oil palm E5 build). The gene of Elaeis oleifera (American oil palm, O8-build) was missing, not attained from the public database.
Gene function
Gene function annotation was done based on sequence similarity and domains conservation. First, the coconut protein coding genes were aligned against the KEGG (KEGG, RRID:SCR_012773) protein database [30], SwissProt, and TrEMBL [31], using BLASTP at a cut-off E-value threshold of 10−5. Subsequently, the best match from the alignment was used to represent the gene function. We obtained 18 445 KEGG, 18 867 Swissprot, and 24 882 Tremble annotated genes. Second, InterProScan (InterProScan, RRID:SCR_005829) 5.11–51.0 software [32] was employed to identify the motif and domain based on the public databases Pfam (Pfam, RRID:SCR_004726) [33], PRINTS (PRINTS, RRID:SCR_003412) [34], ProDom (ProDom, RRID:SCR_006969) [35], SMART (SMART, RRID:SCR_005026) [36], PANTHER (PANTHER, RRID:SCR_004869) [37], TIGRFAM (JCVI TIGRFAMS, RRID:SCR_005493) [38], and SUPERFAMILY (SUPERFAMILY, RRID:SCR_007952) [39]. The gene function annotation demonstrated that 21 087 of the coconut proteins had conserved motifs, and 1622 gene ontology (GO) terms were assigned to 15 705 coconut proteins from the corresponding InterPro (InterPro, RRID:SCR_006695) entry [40]. In total, approximately 89.41% of these genes were functionally annotated using the above methods.
Gene family construction
Protein sequences of 13 angiosperms, including Elaeis guineensis, Phoenix dactylifera (DPV01), Sorghum bicolor, Prunus persica, Solanum tuberosum, Glycine max, Arabidopsis thaliana, Theobroma cacao, Vitis vinifera, Musa acuminata, Carica papaya, Populus trichocarpa, and Amborella trichopoda, were downloaded from each corresponding ftp site (see “Availability of data sources”). For genes with alternative splicing variants, the longest transcripts were selected to represent the gene. The gene numbers of Elaeis guineensis and Phoenix dactylifera (DPV01) were greatly different from the research paper published in 2013 [4, 6], because genes of these 2 species were re-predicted using the NCBI Prokaryotic Genome Annotation Pipeline, which seemed to be more reasonable. Similarities between paired sequences were calculated using BLASTP with an E-value threshold of 1e-5. OrthoMCL (OrthoMCL DB: Ortholog Groups of Protein Sequences, RRID:SCR_007839) [41] was used to identify gene family based on the similarities of the genes and a Markov Chain Clustering (MCL) with default parameters. About 79.80% of Cocos nucifera genes were assigned to 14 411 families, of which 282 families only existed in Cocos nucifera (coconut specific families) (Table 7). Fig. 4 shows the shared gene families for orthologous genes. There are 544 orthologous families shared by 5 monocot species and 7706 orthologous families shared by all monocot and dicot species, suggesting 544 monocot unique functions shared by 5 monocot species and 7706 ancestral functions in the most recent common ancestor of the angiosperms.
Table 7:
Statistical analysis of gene families of different species.
| Species | Genes number | Genes in families | Unclustered genes | Family number | Unique families | Average genes per family |
|---|---|---|---|---|---|---|
| C. nucifera | 28 039 | 22 376 | 5663 | 14 411 | 282 | 1.55 |
| E. guineensis | 30 430 | 22 021 | 8409 | 13 415 | 262 | 1.64 |
| P. dactylifera | 24 908 | 22 193 | 2715 | 14 074 | 112 | 1.58 |
| S. bicolor | 27 159 | 22 016 | 5143 | 12 992 | 916 | 1.69 |
| P. persica | 27 792 | 24 276 | 3516 | 14 443 | 497 | 1.68 |
| S. tuberosum | 34 879 | 28 288 | 6591 | 13 206 | 1119 | 2.14 |
| G. max | 42 859 | 38 104 | 4755 | 14 589 | 1145 | 2.61 |
| A. thaliana | 26 637 | 22 990 | 3647 | 13 292 | 674 | 1.73 |
| T. cacao | 28 624 | 23 776 | 4848 | 14 928 | 625 | 1.59 |
| V. vinifera | 25 329 | 19 122 | 6207 | 13 309 | 599 | 1.44 |
| M. acuminata | 36 538 | 24 354 | 12 184 | 13 089 | 620 | 1.86 |
Figure 4:

Groups of orthologues shared among the angiosperms Cocos nucifera (Coconut), Elaeis guineensis (Oil palm), Phoenix dactylifera (Date palm), Sorghum bicolor (Sorghum), Musa acuminate (Banana) and Arabidopsis thaliana (Arabidopsis). Venn diagram generated by http://www.interactivenn.net/.
Phylogenetic analysis
We extracted 247 single-copy orthologous genes derived from the gene family analysis step, and then aligned the protein sequences of each family with MUSCLE (v3.8.31; MUSCLE, RRID:SCR_011812) [42]. Next, the protein alignments were converted to corresponding coding sequences (CDS) using an in-house Perl script. These coding sequences of each single-copy gene family were concatenated to form 1 super gene for each species. The nucleotides at positions 2 (phase 1 site) and 3 (4 degenerate sites) of codon were extracted separately to construct the phylogenetic tree by PhyML 3.0 (PhyML, RRID:SCR_014629) [43] using a HKY85 substitution model and a gamma distribution across sites. The tree constructed by phase 1 sites was consistent with the tree constructed by 4 degenerate sites.
Divergence time
The Bayesian relaxed molecular clock approach was used to estimate species divergence time using MCMCTREE in PAML (PAML, RRID:SCR_014932) [44], based on the 4 degenerate sites and the data set used in phylogenetic analysis, with previously published calibration times (divergence between Arabidopsis thaliana and Carica papaya was 54–90 Mya, divergence between Arabidopsis thaliana and Populus trichocarpa was 100–120 Mya) [45]. The divergence time between coconut and oil palm is about 46.0 Mya (25.4–83.3 Mya) (Fig. 5), which is less than the divergence time between coconut and date palm.
Figure 5:
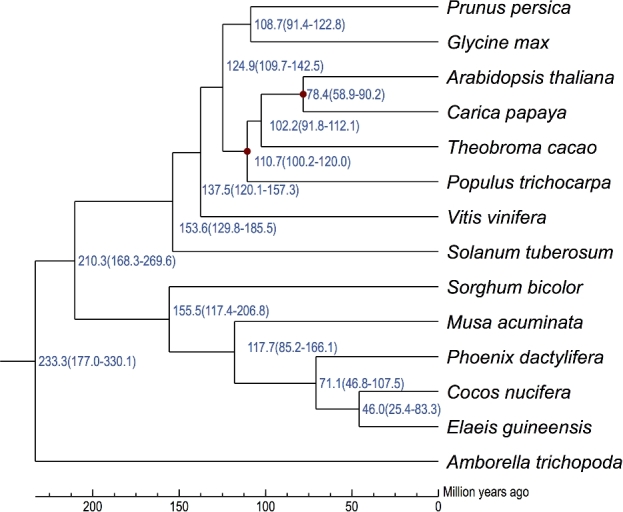
Estimation of divergence time. The blue numbers on the nodes are the divergence time from present (million years ago); the red nodes indicate the previously published calibration times.
Identification of antiporter genes in coconut genome
Antiporters are transmembrane proteins involved in the exchange of substances within and outside the membrane. In Arabidopsis, the functions of antiporter genes have been well characterized experimentally, and this gene family was subdivided into 13 different functional groups. Among them, 3 functional clusters were involved in Na+/H+ antiporters, some of which were documented to be associated with salt tolerance [46, 47].
The amino acid sequences of 70 antiporter genes of Arabidopsis were downloaded from the Arabidopsis Information Resource TAIR website (TAIR, RRID:SCR_004618) [48] and used as queries for BLASTP against the predicted proteins in the Cocos nucifera genome with a cut-off E-value of 1e-10. A total of 126 antiporter genes were identified in coconut genome. Using local Hidden Markov Model–based HMMER (v3.0) searches and the Pfam database, 7 antiporter genes were excluded from further analysis because of the lack of conserved domain. The detailed information of the 119 antiporter genes is listed in Additional file 1.
In order to elucidate the evolutionary relationship and potential functions of the antiporters identified in the study, we applied phylogenetic analysis of Arabidopsis and C. nucifera antiporter proteins using the neighbor joining method (Fig. 6). Phylogenetic analysis showed that the 119 antiporter genes from C. nucifera can be subdivided into 12 groups and that almost all antiporter genes were clustered together with the functional groups in Arabidopsis thaliana.
Figure 6:

Phylogenetic tree of antiporter genes from C. nucifera and Arabidopsis thaliana. Every cluster is indicated with a different colored arc line arc. The potential function of every cluster is indicated with the function groups found in Arabidopsis thaliana. Colored stars indicate antiporter genes of C. nucifera.
Phylogenetic analysis showed that the number of antiporter genes was equal between Arabidopsis thaliana and C. nucifera for most groups, except for G1 (1 of 3 Na+/H+ antiporter family), G3 (carnitine/acylcarnitine translocase family), and G12 (potassium-dependent sodium-calcium exchanger). The G1 group (1 of 3 Na+/H+ antiporter families) contained only 1 Arabidopsis antiporter gene and but 14 C. nucifera antiporters (1-At/14-Cn), whereas G3 (carnitine/acylcarnitine translocase family) contained 1-At/29-Cn, and G13 (potassium-dependent sodium-calcium exchanger) contained 3-At/11-Cn. The Na+/H+ antiporter family had been reported to be associated with salt stress. The expansion of the Na+/H+ antiporter gene family in the coconut palm may be associated with the high salt tolerance of coconut. Meanwhile, carnitine/acylcarnitine translocase is involved in fatty acid transport across the mitochondrial membranes. This gene family expansion may be associated with accumulation of fatty acid in coconut pulp. Moreover, coconut water contains a high density of potassium ion, approximately 312 mg potassium ion per 100 g of coconut water [49]. In this study, the gene number of potassium-dependent sodium-calcium exchangers was also detected to be significantly increased compared to Arabidopsis.
Identification of ion channel genes in coconut genome
A total of 67 ion channel genes were identified in the coconut genome (Additional file 2). The amino acid sequences of 67 C. nucifera and 60 Arabidopsis ion channel genes were used to analyze their evolutionary relationship (Fig. 7). Almost all ion channel genes from C. nucifera can be clustered into the function groups found in Arabidopsis thaliana. The number of ion channel genes was equal between Arabidopsis thaliana and Cocos nucifera in most groups except for G5 (potassium channel). Many more genes (21) from C. nucifera than from Arabidopsis thaliana (9 genes) were present in group 5 (potassium channels). The gene family expansion may be associated with the accumulation of potassium ions in coconut water.
Figure 7:

Phylogenetic tree of ion channel genes from C. nucifera and Arabidopsis thaliana. Every cluster was indicated with different colored arc line arc. The potential function of every cluster was indicated with the function groups found in Arabidopsis thaliana. Colored stars indicate ion channel genes of C. nucifera.
Conclusion
Cocos nucifera (2n = 32) is an important tropical crop, and it is also used as an ornamental plant in the tropics. In the present study, we sequenced and de novo assembled the coconut genome. A total scaffold length of 2.2 Gb was generated, with scaffold N50 of 418 Kb. The divergence time of Cocos nucifera and Elaeis guineensis is more recent than that of Cocos nucifera and Phoenix dactylifera, suggesting a closer relationship between C. nucifera and E. guineensis. Comparative analysis of antiporter and ion channels between C. nucifera and Arabidopsis thaliana showed significant gene family expansions, maybe involving Na+/H+ antiporters, carnitine/acylcarnitine translocases, potassium-dependent sodium-calcium exchangers, and potassium channels. The expansion of these gene families may be associated with adaptation to salt stress, accumulation of fatty acid in coconut pulp, and potassium ions in coconut water. The data output of the coconut genome will provide a valuable resource and reference information for the development of high-density molecular makers, construction of high-density linkage maps, detection of quantitative trait loci, genome-wide association mapping, and molecular breeding.
Availability of supporting data
Supporting data are available in the GigaDB database (GigaDB, RRID:SCR_004002) [50]. Raw data were deposited in the Sequence Read Archive (SRA539146) with the project accession code PRJNA374600 for the Cocos nucifera genome. Previously published RNA-seq data used for transcriptome-based prediction are available under accession number SRR606452.
Availability of other angiosperms data sources
Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, Zea mays, Sorghum bicolor, Solanum tuberosum, Prunus persica, Theobroma cacao, Vitis vinifera, Musa acuminata, Carica papaya, Populus trichocarpa, Amborella trichopoda: https://phytozome.jgi.doe.gov/pz/portal.html (phytozomev9.1)
Elaeis guineensis: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/442/705/GCF_000442705.1_EG5/
Phoenix dactylifera (DPV01): ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/413/155/GCF_000413155.1_DPV01/
Phoenix dactylifera (PDK30): http://qatar-weill.cornell.edu/research/datepalmGenome/download.html
Additional files
Additional file 1: Identification and characterization of antiporter genes in the genome of Cocos nucifera.
Additional file 2: Identification and characterization of ion channel genes in the genome of Cocos nucifera.
Abbreviations
bp: base pair; CDS: coding sequence; CTAB: Cetyl trimethylammonium bromide; EG: Elaeis guineensis; Gb: giga base; HAT: Hainan Tall; Kb: kilo base; KEGG: Kyoto Encyclopedia of Genes and Genomes; LTRs: long-terminal repeat retrotransposon; Mb: mega base; MCL: Markov Chain Clustering; MP mate-pair; PE: paired-end; SRA: Seqeunce Read Archive; TE: transposable elements; UTRs: untranslated region.
Competing interests
The authors declare that they have no competing interests.
Funding
This study was supported by International Science and Technology Cooperation projects of Hainan Province (No. KJHZ2014–24), Hainan Natural Science Foundation (No. 313 058), the major Technology Project of Hainan (No. ZDZX2013023–1), the fundamental Scientific Research Funds for the Chinese Academy of Tropical Agriculture Sciences (CATAS-No. 1 630 032 012 044, 1 630 052 014 002, 1 630 052 015 050, 1 630 152 017 019, and 1 630 152 016 006), and the Central Public-interest Scientific Institution Basal Research Fund for Innovative Research Team Program of CATAS (No.17CXTD-28).
Author contributions
Y.X., H.F., Y.Y., M.P., Q.L., and A.G. designed the study and contributed to the project coordination. X.Y., P.X., and W.X. wrote the paper. L.Z., J.L., and Y.W. collected the samples and extracted the genomic DNA. Y.X., B.L., B.S., J.X., A.A., E.I., and N.L. conducted the genome analyses.
Supplementary Material
Acknowledgements
Annaliese S. Mason is gratefully acknowledged for assistance with language editing and manuscript revisions.
References
- 1. www.fao.org/faostat/en/. Accessed May 2, 2017.
- 2. Batugal P, Ramanatha Rao V, Oliver J, eds. Coconut Genetic Resources. International Plant Genetic Resources Institute – Regional Office for Asia, the Pacific and Oceania (IPGRI-APO). Serdang, Selangor DE, Malaysia; 2005. [Google Scholar]
- 3. Tang B, Tang M, Chen C et al. . Characteristics of soil fauna community in the Dongjiao coconut plantation ecosystem in Hainan, China. Acta Ecologica Sinica 2006;26(1):26–32. [Google Scholar]
- 4. Singh R, Ong-Abdullah M, Low ET et al. . Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 2013;500(7462):335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al-Dous EK, George B, Al-Mahmoud ME et al. . De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol 2011;29(6):521–7. [DOI] [PubMed] [Google Scholar]
- 6. Al-Mssallem IS, Hu S, Zhang X et al. . Genome sequence of the date palm Phoenix dactylifera L. Nat Commun 2013;4:2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 1980;8(19):4321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo R, Liu B, Xie Y et al. . SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 2012;1(1):18 doi:10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhan D. Rabbit Genome Assembler. 2017. https://github.com/gigascience/rabbit-genome-assembler.
- 10. Fan H, Xiao Y, Yang Y et al. . RNA-Seq analysis of Cocos nucifera: transcriptome sequencing and de novo assembly for subsequent functional genomics approaches. PLoS One 2013;8(3):e59997 doi:10.1371/journal.pone.0059997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simao FA, Waterhouse RM, Ioannidis P et al. . BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 13. http://busco.ezlab.org/.
- 14. Jurka J, Kapitonov VV, Pavlicek A et al. . Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005;110(1-4):462–7. [DOI] [PubMed] [Google Scholar]
- 15. Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 2009;Chapter 4:Unit 4.10. doi:10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 16. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 2007;35(Web Server issue):W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27(2):573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000;408(6814):796–815. [DOI] [PubMed] [Google Scholar]
- 19. Goff SA, Ricke D, Lan TH et al. . A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002;296(5565):92–100. [DOI] [PubMed] [Google Scholar]
- 20. Paterson AH, Bowers JE, Bruggmann R et al. . The Sorghum bicolor genome and the diversification of grasses. Nature 2009;457(7229):551–6. [DOI] [PubMed] [Google Scholar]
- 21. Schnable PS, Ware D, Fulton RS et al. . The B73 maize genome: complexity, diversity, and dynamics. Science 2009;326(5956):1112–5. [DOI] [PubMed] [Google Scholar]
- 22. Altschul SF, Madden TL, Schaffer AA et al. . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res 2004;14(5):988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stanke M, Keller O, Gunduz I et al. . AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 2006;34(Web Server issue):W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997;268(1):78–94. [DOI] [PubMed] [Google Scholar]
- 26. Elsik CG, Mackey AJ, Reese JT et al. . Creating a honey bee consensus gene set. Genome Biol 2007;8(1):R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25(9):1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trapnell C, Williams BA, Pertea G et al. . Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28(5):511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2004;20(16):2878–9. [DOI] [PubMed] [Google Scholar]
- 30. Ogata H, Goto S, Sato K et al. . KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 1999;27(1):29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 2000;28(1):45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones P, Binns D, Chang HY et al. . InterProScan 5: genome-scale protein function classification. Bioinformatics 2014;30(9):1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bateman A, Birney E, Durbin R et al. . The Pfam Protein Families Database. Nucleic Acids Res 2000;28(1):263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Attwood TK, Croning MDR, Flower DR et al. . PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Res 2000;28(1):225–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corpet F, Gouzy J, Kahn D. Recent improvements of the ProDom database of protein domain families. Nucleic Acids Res 1999;27(1):263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schultz J, Copley RR, Doerks T et al. . SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res 2000;28(1):231–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mi H, Huang X, Muruganujan A et al. . PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45(Database issue):D183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Selengut JD, Haft DH, Davidsen T et al. . TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res 2007;35(Database issue):D260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson D, Pethica R, Zhou Y et al. . SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res 2009;37(Database issue):D380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burge S, Kelly E, Lonsdale D et al. . Manual GO annotation of predictive protein signatures: the InterPro approach to GO curation. Database (Oxford) 2012;2012:bar068 doi:10.1093/database/bar068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004;32(5):1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guindon S, Dufayard JF, Lefort V et al. . New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010;59(3):307–21. [DOI] [PubMed] [Google Scholar]
- 44. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 45. Tuskan GA, Difazio S, Jansson S et al. . The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006;313(5793):1596–604. [DOI] [PubMed] [Google Scholar]
- 46. Shi H, Lee BH, Wu SJ et al. . Overexpression of a plasma membrane Na+/H+ antiporter gene improves salt tolerance in Arabidopsis thaliana. Nat Biotechnol 2003;21(1):81–5. [DOI] [PubMed] [Google Scholar]
- 47. Brini F, Hanin M, Mezghani I et al. . Overexpression of wheat Na+/H+ antiporter TNHX1 and H+-pyrophosphatase TVP1 improve salt- and drought-stress tolerance in Arabidopsis thaliana plants. J Exp Bot 2007;58(2):301–8. [DOI] [PubMed] [Google Scholar]
- 48. http://www.arabidopsis.org/.
- 49. Yong JW, Ge L, Ng YF et al. . The chemical composition and biological properties of coconut (Cocos nucifera L.) water. Molecules 2009;14(12):5144–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiao Y, Xu P, Fan H et al. . Supporting data for “The genome draft of coconut (Cocos nucifera).” Gigascience Database 2017. http://dx.doi.org/10.5524/100347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.