


Abstract
Translation rate per mRNA molecule correlates positively with mRNA abundance. As a result, protein levels do not scale linearly with mRNA levels, but instead scale with the abundance of mRNA raised to the power of an ‘amplification exponent’. Here we show that to quantitate translational control, the translation rate must be decomposed into two components. One, TRmD, depends on the mRNA level and defines the amplification exponent. The other, TRmIND, is independent of mRNA amount and impacts the correlation coefficient between protein and mRNA levels. We show that in Saccharomyces cerevisiae TRmD represents ∼20% of the variance in translation and directs an amplification exponent of 1.20 with a 95% confidence interval [1.14, 1.26]. TRmIND constitutes the remaining ∼80% of the variance in translation and explains ∼5% of the variance in protein expression. We also find that TRmD and TRmIND are preferentially determined by different mRNA sequence features: TRmIND by the length of the open reading frame and TRmD both by a ∼60 nucleotide element that spans the initiating AUG and by codon and amino acid frequency. Our work provides more appropriate estimates of translational control and implies that TRmIND is under different evolutionary selective pressures than TRmD.
INTRODUCTION
The relative contributions of transcriptional and post-transcriptional control to protein expression levels in eukaryotes are the topic of ongoing debate (1–3). One view suggests that translation and protein degradation together play the dominant role because protein and mRNA abundance data correlate poorly (coefficient of determination for log10 transformed values R2prot–RNA = 0.2–0.45) (4–9). Other work, though, has shown that the correlation is much higher when measurement error is considered (R2prot–RNA = 0.66–0.83), implying that transcription dominates (10–12). In addition, the variance in translation rates affects not only the correlation coefficient between protein and mRNA, but also the slope of the relationship because translation rates increase with mRNA abundance (12). Whereas most studies assumed that protein abundances scale linearly with mRNA levels, Csardi et al. demonstrate that protein abundances scale with mRNA levels raised to the power of an ‘amplification exponent’ (bprot–RNA). Presumably the mRNAs of genes that are expressed at high levels, such as those for ribosomal proteins and glycolytic enzymes, contain nucleotide sequence signals that promote faster rates of translation per message than observed for less abundant mRNAs (12).
In this article, we argue that because translation affects both R2prot–RNA and bprot–RNA, the approaches used previously to quantify the contribution of translation to protein expression are improper. Prior approaches have sought to provide a single metric to estimate translational control: the impact of translation on R2prot–RNA. We propose that, instead, proper quantification requires that translation rates (TR) be decomposed mathematically into two components: one that is dependent on mRNA abundance (TRmD) and one that is not (TRmIND). For a given gene i
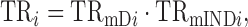 |
where TR is the number of protein molecules translated per mRNA molecule; TRmD determines bprot–RNA; and TRmIND only contributes to R2prot–RNA (not bprot–RNA).
The traditional view of the steady-state relationship between protein and mRNA for gene i can be expressed as
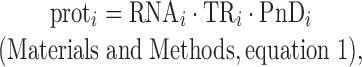 |
where prot and RNA are the number of protein molecules and mRNA molecules per cell, respectively, and PnD is the fraction of protein that is not degraded per cell cycle (0 ≤ PnD ≤ 1). Once TR is decomposed, this equation can be reformulated as
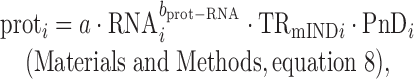 |
where a and bprot–RNA are positive constants for all genes. This reformulated equation has the advantage that it explicitly describes the non-linear relationship between protein and mRNA levels as well as permitting correct quantitation of translation's contribution to protein levels.
Three idealized scenarios explain the complex dependency of protein abundances on mRNA levels and the two components of translation. Plots of log10-transformed data are employed because the amplification exponent bprot–RNA is simply the linear slope of the relationship in logarithmic space (Figure 1), i.e.,
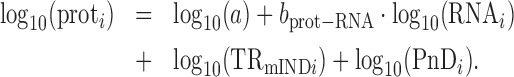 |
Figure 1.

Three scenarios explain the relationships between the steps in protein expression. (A) Translation rates for all expressed genes are equal, as are protein degradation rates. (B) Translation rates vary between genes but correlate perfectly with the amount of mRNA. Degradation rates for all proteins are constant. (C) Translation and protein degradation rates vary but are uncorrelated with mRNA abundance. Upper panels show the relationship between protein and mRNA levels; lower panels show the relationship between translation rates and mRNA levels. The coefficients of determination (R2) and slopes (b) are indicated.
In the first scenario, translation rates are equal for all genes (i.e. TRi = constant) as are protein degradation rates. Therefore, R2prot–RNA = 1 and bprot–RNA = 1 (Figure 1A). In the second scenario, translation rates correlate perfectly with mRNA levels (i.e., TRi = TRmDi), while the protein degradation rate is constant for all genes. Thus, R2prot–RNA = 1 and bprot–RNA > 1. (Figure 1B). In the third scenario, translation and protein degradation rates are both uncorrelated with mRNA (i.e. TRi = TRmINDi). Therefore, R2prot–RNA < 1 and bprot–RNA = 1 (Figure 1C).
The third scenario is the one most widely considered in the literature. Csardi et al. argue, though, that the truth is a hybrid of this scenario and the second scenario because translation is partially, but not fully, correlated with mRNA abundance. A Bayesian model was employed to estimate protein and mRNA abundances for 5,854 annotated protein-coding genes in S. cerevisiae, including 842 genes for which either protein or mRNA abundance data was lacking. From the modeled abundances, it was estimated that bprot–RNA = 1.69 and R2prot–RNA = 0.85 (12).
Csardi et al.’s basic premise is important. Their Bayesian model, however, did not take into account which methods provide accurately scaled abundance data, and they did not decompose TR. Bayesian models are, in addition, inherently subjective because priors are chosen by the researcher. Therefore, we adopted a non-modeling approach that considers empirically determined abundance measurements that have been scaled using internal concentration standards, and we decomposed TR. We find that in S. cerevisiae TRmD represents ∼20% of the variance in translation and results in an amplification exponent of 1.20 with a 95% confidence interval [1.14, 1.26] and that TRmIND constitutes the remaining ∼80% of the variance in translation and explains ∼5% of the variance in protein expression. By taking into account protein degradation data and measurement error, we also show that the expected correlation between the abundances of protein and mRNA is R2prot–RNA ∼0.94. This value is markedly higher than the R2prot–RNA = 0.80 obtained between the Bayesian model's abundance estimates for the 5,012 genes for which empirical data are available. Finally, we examined which mRNA sequence elements explain the variance in TRmD and TRmIND using a model that predicts 80% of the variance in TR from mRNA sequence data alone. We find that TRmD is most strongly determined both by RNA secondary structure within a ∼60 nucleotide element that spans the initiating AUG and by the fact that the amino acid and codon frequencies encoded in highly expressed mRNAs more closely correlate with the abundances of their cognate tRNAs than is the case for mRNAs expressed at lower levels. TRmIND, by contrast, is chiefly determined by the length of the protein coding region. TRmIND is thus likely under different evolutionary selective pressures than TRmD and predominantly controlled by different mechanisms. Our work establishes more accurate estimates of translational control than earlier research. In addition, our analysis illustrates that decomposing translation rates allows insights into the mRNA sequence dependence of translation that would not otherwise be apparent.
MATERIALS AND METHODS
Data and code
All of the data used are provided in Datasets S1–S9. The mRNA and protein abundance datasets used by Csardi et al. as input to their Bayesian model (Dataset S1) are from their file ‘scer-mrna-protein-raw.txt’ (12). The estimates for the true abundances of mRNA and protein generated by Csardi et al.’s Bayesian model (Dataset S2) are from their file ‘scer-mrna-protein-absolute-estimate-sample.txt’ and are for a single sample from their ‘SCM’ values (12). The scaling-standard mRNA data are from NanoString (13,14), qPCR (15) and competitive PCR (16) studies (Dataset S3). Three scaling-standard protein datasets were measured by western blot (17), flow cytometry (18) and selected reaction monitoring mass spectrometry (19) (Dataset S3). A fourth scaling-standard protein dataset was compiled as an extension of one by von der Haar (20) to which additional data were added (21–26) (Dataset S3). The ribosome profiling data comprise median values from several studies provided by Csardi et al. (12) and, separately, the translation-initiation efficiency values from Weinberg et al. (27) (Dataset S4). Protein degradation data is from Christiano et al. (28) (Dataset S5). The mRNA sequence feature information was from Weinberg et al. and Subtelny et al. or was calculated as described in Supplementary Methods S4 (27,29) (Datasets S6–S9). The fraction of RNA not degraded was calculated from Presnyak et al. as described in Supplementary Methods S4 (30) (Dataset S6). Dataset S2 also includes our corrected versions of the Csardi et al. protein and mRNA abundance data. Dataset S4 includes corrected versions of Weinberg et al. mRNA abundance and ribosome density data as well as calculated values of TRmD and TRmIND. For the details on our correction of the Csardi et al. protein and mRNA abundance data and the Weinberg et al. mRNA abundance and ribosome density data, please refer to Supplementary Method S1.
The R code used in the analyses are provided in Dataset S10. Both an R Markdown file and its output Word file are provided.
The relationship between the steps in protein production
For simplicity we consider the ideal case where there is no measurement error, i.e. where the true values are measured. It is assumed that the system is at steady state. We denote
RNA = the abundance of a particular mRNA (molecules per cell)
prot = the abundance of a particular protein (molecules per cell)
TR = the translation rate of a particular mRNA (the number of protein molecules translated per mRNA molecule)
TRmIND = the mRNA abundance-independent component of TR
TRmD = the mRNA abundance-dependent component of TR
PnD = a scaling factor that gives the fraction of a particular protein that remains undegraded per cell cycle, i.e. (1 – the fraction of the protein degraded per cell cycle); 0 ≤ PnD ≤ 1.
a = a constant for all genes
bTR–RNA = a constant for all genes that is the slope of the relationship between log10-transformed translation rates and log-transformed mRNA levels. It thus measures the amplification of translation rates due to mRNA abundance
bprot–RNA = a constant for all genes that is the slope of the relationship between log10-transformed protein abundance and log-transformed mRNA levels. It is thus also the amplification exponent for the relationship between the unlogged abundances.
We assume that PnD is not correlated with mRNA abundance and, thus, has no impact on bprot–RNA. This appears to be a reasonable assumption because the correlation between measured values for PnD and mRNA abundance is very low (R2PnD–RNA < 0.005; Supplementary Table S2).
The abundance of a chosen protein is given by
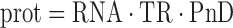 |
(1) |
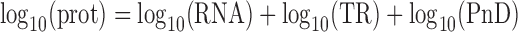 |
(2) |
In an idealized situation where log10-transformed translation rates correlate perfectly with log10-transformed mRNA levels (i.e. R2TR–RNA = 1; and TRmIND = 1)
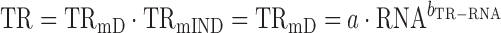 |
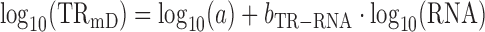 |
(3) |
When log10-transformed translation rates only partially correlate with log10-transformed mRNA levels then
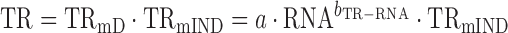 |
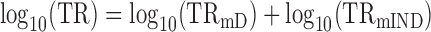 |
(4) |
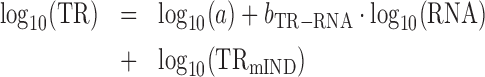 |
(5) |
Combining (2) and (5)
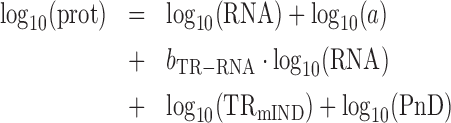 |
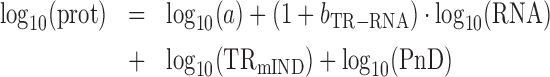 |
(6) |
From (6), the slope of the relationship between log10(prot) and log10(RNA) is (1 + bTR–RNA), i.e.
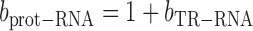 |
(7) |
Combining (6) and (7)
 |
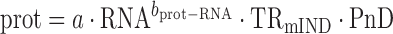 |
(8) |
Estimating the slope bTR–RNA and the contributions of TRmIND and TRmD to TR
Having defined the basic relationships between steps in protein expression, we now estimate the value for bTR–RNA and the contributions of TRmIND and TRmD to TR.
From (4) and the fact that log(TRmD) and log(TRmIND) are uncorrelated by definition
 |
(9) |
where var is the variance.
From (3) and given that var(log10 (a)) = 0
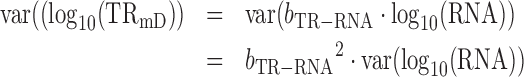 |
(10) |
Combining (9) and (10)
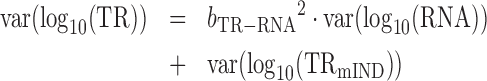 |
From (10)
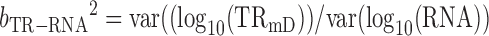 |
Therefore true slope
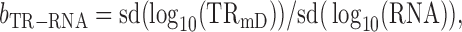 |
(11) |
where sd is the standard deviation.
We considered three different regressions with log10(TR) as the response variable and log10(RNA) as the explanatory variable for estimating the value of the true slope bTR–RNA, finding that the Ordinary Least Squares (OLS) regression described is the most appropriate (Supplementary Methods S3).
Estimating the contributions of mRNA, TRmIND and PnD to prot
Motivated by the model for true abundances (on log10 scale) in Equation (6), the following statistical model was used to quantitate the contribution of mRNA, TRmIND and PnD to protein expression:
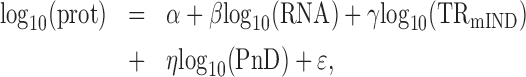 |
(12) |
where ϵ denotes the error term. The OLS regression is used to estimate the intercept α and slopes β, γ and η. Given these estimates, the variance of log10-transformed protein abundance was decomposed into the variances explained by log10-transformed RNA abundance, log10-transformed TRmIND, log10-transformed PnD, and unexplained variance (i.e. error).
RESULTS
Estimates for bprot–RNA from protein and mRNA abundances
Csardi et al.’s estimate for bprot–RNA was derived using a Bayesian model to determine the true levels of mRNAs and proteins based on multiple abundance datasets from the literature and imputed values when data were lacking (12). However, the methods used to produce most of the empirical data input to this model (e.g. mRNA microarray, RNA-seq, and label-free mass spectrometry) do not employ internal concentration standards. As a result, the standard deviations of the data can be—depending on the method—either systematically compressed or systematically expanded relative to the true values (10,12,31–33). There is no guarantee that such reproducible biases can be corrected by a Bayesian model. The slope of any relationship depends on the standard deviations of the x and y values, so improperly scaled data is likely to exhibit an inaccurate slope.
We therefore re-estimated bprot–RNA by correcting abundances of protein and mRNA using datasets that had been derived by methods employing internal concentration standards. The internal standards are used to account for any linear or non-linear scaling bias in the raw data, and thus the final data produced by these methods should be reasonably scaled. Data for individual genes will still include some gene specific error, but the standard deviation of the whole dataset will not be much impacted by such error. We refer to these datasets as ‘scaling-standards’. NanoString (13,14), qPCR (15) and competitive PCR (16) studies provided four independent mRNA scaling-standards (Dataset S3). Western blot (17); flow cytometry (18); selected reaction monitoring mass spectrometry (19) and a compilation of assorted methods (20–26) each provided one of four protein scaling-standards (Dataset S3). Plots of these scaling-standards against the corresponding abundance values from the Bayesian model reveal the relative scaling of each dataset: scaling-mRNA versus Bayesian mRNA and scaling-protein versus Bayesian protein (Figure 2 and Supplementary Figure S1). The slope and intercept of a linear regression fit to the log10-transformed data for each of the eight pairwise comparisons was then used to correct the scaling of the Bayesian abundance estimates (i.e., re-centering and re-scaling them; Supplementary Methods S1 and Dataset S2). The Reduced Major Axis (RuMA) regression was used as it is the only one that allows the scaling of a dataset to be adjusted such that its standard deviation becomes equal to that of a scaling-standard (34) (Supplementary Methods S1).
Figure 2.

The slopes between Bayesian-model abundance data and scaling-standards. (A) The four protein scaling-standards are compared to the Bayesian protein abundance data. (B) The four mRNA scaling-standards are compared to the Bayesian mRNA data. The colored lines are RuMA regressions that demonstrate slope 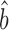 . The lines have been shifted to give the same value at the origin, allowing ready comparison of the slopes. The dashed black lines show slope b = 1, the case where the standard deviations of the x and y values are equal and thus what would be seen if the data from the Bayesian model were scaled identically to a scaling-standard.
. The lines have been shifted to give the same value at the origin, allowing ready comparison of the slopes. The dashed black lines show slope b = 1, the case where the standard deviations of the x and y values are equal and thus what would be seen if the data from the Bayesian model were scaled identically to a scaling-standard.
The standard deviation of the uncorrected Bayesian protein dataset approximates those of the scaling-protein datasets (RuMA slope 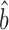 sprot–prot = 0.87–1.11), while the standard deviation of the uncorrected Bayesian mRNA data is less than those of the scaling-mRNA sets (RuMA slope
sprot–prot = 0.87–1.11), while the standard deviation of the uncorrected Bayesian mRNA data is less than those of the scaling-mRNA sets (RuMA slope 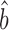 sRNA–RNA = 1.34–1.54) (Figure 2 and Supplementary Figure S1). After correcting the scaling bias of the Bayesian data by our linear transformation, we bootstrapped the corrected versions of the data to obtain a mean RuMA estimate of
sRNA–RNA = 1.34–1.54) (Figure 2 and Supplementary Figure S1). After correcting the scaling bias of the Bayesian data by our linear transformation, we bootstrapped the corrected versions of the data to obtain a mean RuMA estimate of 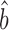 prot–RNA = 1.17 with a 95% quantile confidence interval [1.10, 1.26] (Figure 3B).
prot–RNA = 1.17 with a 95% quantile confidence interval [1.10, 1.26] (Figure 3B).
Figure 3.

Different predictions for slope bprot–RNA. Red lines show the regression of protein on mRNA amount; dashed red lines show the 95% quantile confidence limits. Dashed black lines illustrate a slope of one. (A) The RuMA regression between the uncorrected values for protein and mRNA amounts from the Bayesian model. (B) The mean slope of the RuMA regressions for 16 pair wise comparisons between our corrected versions of the Bayesian protein and mRNA abundance data. (C) The true slope predicted by the relationship between the corrected Weinberg translation rate and mRNA abundance data. The predicted slope shown is 1 + the mean of the slopes of the OLS regressions for our corrected versions of the Weinberg data. The intercept in this panel was derived such that the total number of expected protein molecules per cell is the same as in panel B.
Csardi et al. used the Ranged Major Axis (RgMA) to estimate the slope bprot–RNA. This other type of regression yields a slope that is nearly identical to that of the RuMA regression for our corrected versions of the protein and mRNA abundance data, 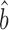 prot–RNA = 1.16 with a 95% quantile confidence interval [1.09, 1.25]. We also considered two additional, though more approximate, approaches to determine
prot–RNA = 1.16 with a 95% quantile confidence interval [1.09, 1.25]. We also considered two additional, though more approximate, approaches to determine 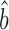 prot–RNA. These two methods estimate that
prot–RNA. These two methods estimate that 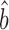 prot–RNA = 1.08 or 1.10 (see Supplementary Methods S2 and Table S1). Thus, the evidence strongly suggests that the amplification exponent bprot–RNA is much smaller than the previously reported value of 1.69 (Figure 3).
prot–RNA = 1.08 or 1.10 (see Supplementary Methods S2 and Table S1). Thus, the evidence strongly suggests that the amplification exponent bprot–RNA is much smaller than the previously reported value of 1.69 (Figure 3).
To investigate the basis for Csardi et al.’s higher estimate of bprot–RNA, we compared the standard deviations of the datasets input to their Bayesian model with those of our scaling-standards and with the abundances output by the Bayesian model. While the standard deviations of the 20 input protein datasets range above and below that of the protein scaling-standards, their mean scalings are similar (i.e. mean RuMA 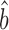 sprot–prot 0.96) and agree closely with that of the Bayesian model (Figure 4A). By contrast, while most of the 38 input mRNA datasets are scaled similarly to the mRNA scaling-standards, 33 out of the 38 input mRNA datasets have a larger standard deviation than the Bayesian model's abundance estimates (Figure 4B). The model has, in effect, given greater weight to the small minority of the input mRNA data that have the most compressed scaling. This minority is dominated by mRNA microarray data (Figure 4B), which is known to give compressed abundance estimates relative to the true values (32,33). The Bayesian model's strong weighting on biased microarray data thus appears to explain its high estimate for bprot–RNA.
sprot–prot 0.96) and agree closely with that of the Bayesian model (Figure 4A). By contrast, while most of the 38 input mRNA datasets are scaled similarly to the mRNA scaling-standards, 33 out of the 38 input mRNA datasets have a larger standard deviation than the Bayesian model's abundance estimates (Figure 4B). The model has, in effect, given greater weight to the small minority of the input mRNA data that have the most compressed scaling. This minority is dominated by mRNA microarray data (Figure 4B), which is known to give compressed abundance estimates relative to the true values (32,33). The Bayesian model's strong weighting on biased microarray data thus appears to explain its high estimate for bprot–RNA.
Figure 4.

The RuMA slope between scaling-standards and the datasets input to the Bayesian model. (A) Protein data. (B) mRNA data. Each data point is the mean of the RuMA slopes between a single input dataset versus each of the corresponding scaling-standards, where RuMA slope = standard deviation of a scaling-standard / standard deviation of an input dataset. The results are grouped by the method used to produce the input dataset, and the numbers of datasets in each group are indicated (N). An RuMA slope of 1 is shown by the dashed black line, the case where the standard deviation of the input dataset is equal to the mean of the standard deviation of the scaling-standards. The mean RuMA slope between the scaling-standards and the abundance estimates from the Bayesian model is shown by the solid black line.
Estimates for bprot–RNA from ribosome profiling data
The previous study by Csardi et al. used a ‘toy’ model to independently determine bprot–RNA from the slope and correlation between translation rates and mRNA abundances (12). Using averaged measurements of translation rates and mRNA abundances from several ribosome profiling studies (29,35–37), it was suggested that the toy model was consistent with bprot–RNA = 1.69 (12). Since our results are inconsistent with this estimate for bprot–RNA, we have independently explored the relationship between bprot–RNA and ribosome profiling data. Again we adopted a non-modeling approach that defines the appropriate mathematical equations and employs the most accurate datasets available.
The correlation between measured protein degradation data and mRNA abundance data is negligible (R2PnD–RNA < 0.005; Materials and Methods and Supplementary Table S2) (28). Thus, we can assume that protein degradation has no impact on bprot–RNA. Hence, the relationship between bprot–RNA and bTR–RNA is
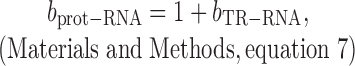 |
where bTR–RNA is the true slope between log10-transformed translation rates versus log10-transformed mRNA levels.
To estimate bTR–RNA we employed two available ribosome profiling datasets: one used by Csardi et al. (12), which we refer to as ‘Csardi–median’, and another from Weinberg et al. (27) (Dataset S4). The Weinberg data eliminates a poly-A mRNA selection bias and has been corrected to reduce two additional sources of bias (27). As a result, these data show a higher correlation between translation rates and mRNA levels than previously observed (27) and appear to be more accurate than the Csardi-median data because they correlate more highly with both the mRNA and the protein scaling-standards (Supplementary Table S3). The standard deviations of the Weinberg ribosome-density and mRNA data differ modestly from that of their respective scaling-standards (mean RuMA 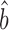 sprot–RD = 0.98; RuMA
sprot–RD = 0.98; RuMA 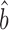 sRNA–RNA = 1.07). We corrected this miss-scaling in the Weinberg data using the scaling-standards (Dataset S4) and then used the Ordinary Least Squares (OLS) regression to estimate bTR–RNA on the corrected data. The result suggests that the amplification exponent bprot–RNA = 1 + 0.22 = 1.22 with a 95% bootstrap quantile confidence interval [1.13, 1.29] (Figure 3C; Supplementary Table S4).
sRNA–RNA = 1.07). We corrected this miss-scaling in the Weinberg data using the scaling-standards (Dataset S4) and then used the Ordinary Least Squares (OLS) regression to estimate bTR–RNA on the corrected data. The result suggests that the amplification exponent bprot–RNA = 1 + 0.22 = 1.22 with a 95% bootstrap quantile confidence interval [1.13, 1.29] (Figure 3C; Supplementary Table S4).
Rather than correcting the Csardi-median data, we analyzed it in its original form so that we could compare analysis strategies on the same data. The result suggests that bprot–RNA = 1+ 0.28 = 1.28 with a 95% bootstrap quantile confidence interval [1.26, 1.31] (Supplementary Table S4). Csardi et al.’s claim that ribosome profiling data were consistent with an amplification exponent of 1.69 must therefore be largely due to differences between our analysis methods and those that they employed, not the data used.
Csardi et al. estimated bTR–RNA using the RgMA regression rather than OLS. For the corrected Weinberg data, RgMA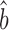 TR–RNA predicts bprot–RNA = 1 + 0.31 = 1.31; for the Csardi–median dataset, RgMA
TR–RNA predicts bprot–RNA = 1 + 0.31 = 1.31; for the Csardi–median dataset, RgMA 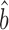 TR–RNA predicts bprot–RNA = 1 + 0.55 = 1.55 (Supplementary Table S4). The RgMA slope, however, is insensitive to the correlation coefficient (Supplementary Table S5 and Methods S3). In effect, this regression assumes that the true translation rates and true mRNA levels correlate perfectly and that the poor correlations observed between the data (R2TR–RNA ≤ 0.28; Supplementary Table S4) are due only to measurement errors that are somewhat evenly split between the TR and mRNA data. The OLS regression, by contrast, down-weights the slope as the correlation decreases (34) (Supplementary Table S5 and Methods S3). It effectively assumes that the poor correlation between translation and mRNA abundance is largely due to a genuine biological phenomenon rather than measurement error. OLS-based estimates better match current thinking that translational control includes a substantial component that is unrelated to the abundance of each mRNA. In addition, OLS
TR–RNA predicts bprot–RNA = 1 + 0.55 = 1.55 (Supplementary Table S4). The RgMA slope, however, is insensitive to the correlation coefficient (Supplementary Table S5 and Methods S3). In effect, this regression assumes that the true translation rates and true mRNA levels correlate perfectly and that the poor correlations observed between the data (R2TR–RNA ≤ 0.28; Supplementary Table S4) are due only to measurement errors that are somewhat evenly split between the TR and mRNA data. The OLS regression, by contrast, down-weights the slope as the correlation decreases (34) (Supplementary Table S5 and Methods S3). It effectively assumes that the poor correlation between translation and mRNA abundance is largely due to a genuine biological phenomenon rather than measurement error. OLS-based estimates better match current thinking that translational control includes a substantial component that is unrelated to the abundance of each mRNA. In addition, OLS 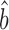 TR–RNA predicts a value for the amplification exponent that is more similar to that we obtained from scaling-standard-rescaled protein and mRNA abundances (1.22 versus 1.17 respectively) than to RgMA
TR–RNA predicts a value for the amplification exponent that is more similar to that we obtained from scaling-standard-rescaled protein and mRNA abundances (1.22 versus 1.17 respectively) than to RgMA 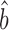 TR–RNA (1.31 or 1.55 versus 1.17). Thus, OLS
TR–RNA (1.31 or 1.55 versus 1.17). Thus, OLS 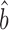 TR–RNA should give a more accurate estimate of bprot–RNA (see Supplementary Methods S3 for further justification). Averaging our estimate from ribosome profiling data with that from corrected protein and mRNA abundances (i.e. the estimates in Figure 3B and C) provides our most accurate estimate for bprot–RNA as 1.20 with a 95% confidence interval [1.14, 1.26].
TR–RNA should give a more accurate estimate of bprot–RNA (see Supplementary Methods S3 for further justification). Averaging our estimate from ribosome profiling data with that from corrected protein and mRNA abundances (i.e. the estimates in Figure 3B and C) provides our most accurate estimate for bprot–RNA as 1.20 with a 95% confidence interval [1.14, 1.26].
Estimating mRNA abundance-dependent and independent translational control
The variance in protein levels is caused by gene-specific differences in mRNA abundances, translation rates, and protein degradation rates. Because translation rates correlate with mRNA levels, it has been suggested that the percent of the variance in true protein amounts that is explained by the true individual contributions of mRNA, translation, and protein degradation sum to more than 100% (12). This argument is, however, misleading. The correlation coefficient between translation and protein abundance is not a legitimate measure of the contribution of translation to protein expression because it breaches one of the essential requirements for analysis of variance (ANOVA). ANOVA is only valid when the true explanatory variables (in this case mRNA abundance, translation and protein degradation) are fully uncorrelated with each other (i.e. when they are not collinear) and, as a result, when their marginal contributions sum to exactly 100%. Therefore, as briefly explained in the Introduction, to determine the contribution of translation rates (TR) to protein expression it is essential to decompose TR into two components: one that is dependent on mRNA abundance (TRmD) and a second that is independent of mRNA abundance (TRmIND), where TR = TRmD · TRmIND. TRmIND determines the variance in protein levels that is not explained by mRNA or protein degradation; it has no impact on bprot–RNA. TRmD, by contrast, determines the amplification exponent bprot–RNA. The abundance of any protein i is then given by the following
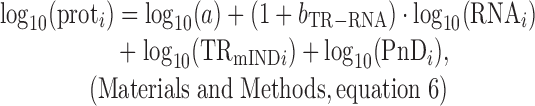 |
 |
where sd is the standard deviation; a is positive constant for all genes; PnD is the fraction of protein not degraded; and (1+bTR–RNA) = bprot–RNA. As one consequence of this 100% of the variance in true protein expression is explained by the sum of the contributions of the variances of true RNA, TRmIND and PnD values.
To quantitate the contribution of translation to protein expression, we first calculated gene-specific values of TRmIND and TRmD from OLS regressions of translation efficiency on mRNA abundance for both the Csardi-median and the Weinberg datasets (Figure 5 and Dataset S4). In addition, from these same regressions we determined the percent of the variance in TR that is explained by the variances in TRmD and TRmIND. Assuming no measurement error, these values are 19%–21% and 79%–81% respectively (Supplementary Table S4).
Figure 5.
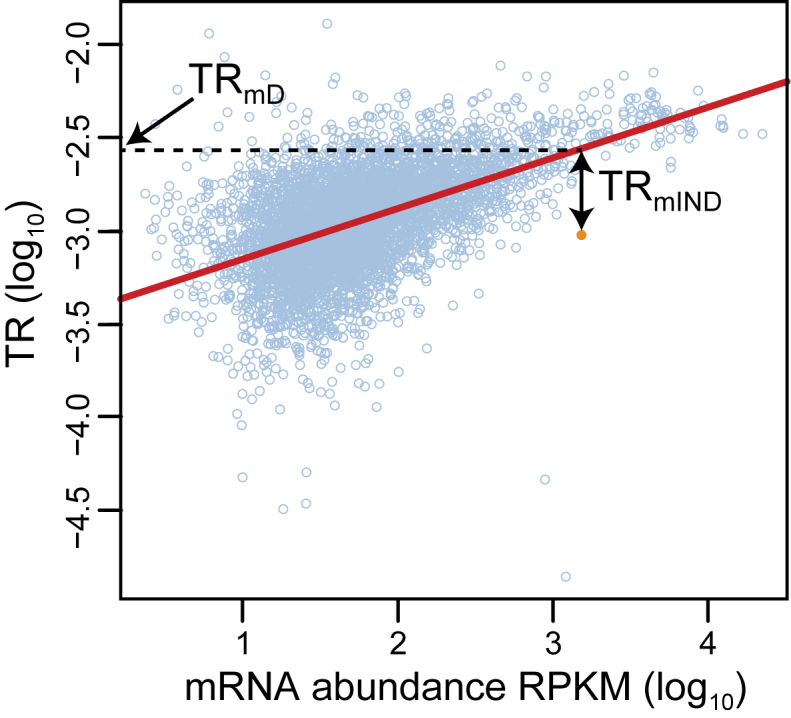
The estimation of TRmD and TRmIND for a single gene. The linear regression between log10 translation rate data (y-axis) and log10 mRNA abundance data (x-axis) is shown by the red line (data from (27)). The data point for an example gene is highlighted in orange, whereas those for all remaining genes are shown in light blue circles. The gene specific values for log10(TRmD) and log10(TRmIND) are shown for the highlighted gene. The value for log10(TRmD) is the intercept on the y-axis of a horizontal line that intercepts the regression at the mRNA abundance of the gene. The value for log10(TRmIND) = log10(TR) – log10(TRmD). Values are determined likewise for the remaining genes. Values for log10(TRmIND) thus have both positive and negative values depending on if the data point lies above or below the regression. Values for log10(TRmD) fall within the range of values for log10(TR), all of which are negative.
The amplification effect of TRmD on the contribution of mRNA to protein abundance is given by the amplification exponent bprot–RNA, which we have estimated earlier as 1.20 with a 95% confidence interval [1.14, 1.26]. The contribution of TRmIND to protein abundance was derived from OLS linear regressions of the gene specific values of protein data on TRmIND using a statistical model (Equation 12) based on Equation (6) (Materials and Methods). TRmIND only accounted for 1%–3% of the variance in the protein abundance estimates from the Bayesian model (Supplementary Table S4). Because these percentages were surprisingly low, we recalculated the contribution of TRmIND by regressing the protein scaling-standards against it to test for an unknown bias in the output of the Bayesian model. The mean contributions of TRmIND to the variance in the scaling-standard protein datasets were also low: 4% (Supplementary Table S6). We also re-estimated TRmIND by regressing translation efficiencies against the Bayesian mRNA abundances to avoid any potential bias in the mRNA data from the ribosome profiling studies. These re-calculated values for TRmIND, though, still only explain <1% of the variance in the Bayesian protein data.
To compare our new metrics to one derived from undecomposed TR, we determined the R2 coefficient of determination between undecomposed TR and protein abundance data. R2prot–TR = 0.24–0.28 (Supplementary Table S4). This relatively high value helps expose why R2prot–TR cannot be used as measure of the contribution of translation to protein abundance. TRmIND represents ∼80% of the variance in TR, yet R2prot –TRmIND is dramatically lower than R2prot–TR (0.01–0.04 versus 0.24–0.28). TRmD accounts for only ∼20% of the variance in TR and yet is chiefly responsible for the fact that R2prot–TR >> R2prot –TRmIND (Supplementary Table S4). It is counter-intuitive that a ∼20% minority of the variance in TR should have much the dominant contribution to protein expression. In effect, R2prot–TR is a hybrid measure of the correlation of TRmIND with protein abundances combined with some part of the correlation between mRNA abundance and protein levels. Only by decomposing TR can the impact of translation be properly quantitated and provide metrics consistent with the requirement of ANOVA that explanatory variables be completely uncorrelated.
Estimating post-transcriptional control
The contribution of protein degradation to the variance of protein abundance in actively dividing yeast cells is very low because the median half-life of proteins is 3.5 times longer than the cell division rate (28). By our estimate, this contribution is ∼1% (Supplementary Table S2; Materials and Methods). As explained above, the percentage contributions of the variances in the true values of mRNA, TRmIND, and PnD should sum to explain exactly 100% of the variance in true protein levels (Materials and Methods, Equation 5). For measured data, though, the sum of the contributions is no more than 77% (mRNA) + 4% (TRmIND) + 1% (PnD) = 82% (Figure 6A and Supplementary Tables S2, S4 and S6; Materials and Methods). This discrepancy reveals another advantage of our framework. The ∼18% of the variance in protein data that is unexplained (Figure 6A) should be due to measurement error. Our approach thus provides an assessment of the magnitude of error, whereas error cannot be estimated if TR is left undecomposed.
Figure 6.

Contributions to the control protein expression. (A) The maximum percentage contributions of estimates of mRNA abundance, protein degradation (PnD), and TRmIND to the variance in measured levels of protein expression as well as the percent of the variance unexplained (Supplementary Tables S4 and S6). The contributions were calculated by using the OLS regression to fit a statistical model (Materials and Methods, Equation 12). (B) Left, the presumed percentage contributions of true mRNA abundance, protein degradation (PnD) and TRmIND if the unexplained component in A is due to similar proportions of measurement error in each data class. Right, the mean of our estimates for the contribution of TRmD to the amplification exponent bprot–RNA. The dashed black line shows a slope of 1, the shaded area shows the increase in slope due to TRmD.
Further, if we assume that the proportion of measurement error is similar in each data class, we can estimate the contribution of the true values of each step to true protein expression. When we do this, the variance in the true values of TRmIND + PnD explain ∼6% of the variance in true protein levels, while TRmD makes an additional contribution by increasing slope bprot–RNA from its ground state of 1 to more like 1.20 (Figure 6B). The expected correlation between true protein and true mRNA abundances is thus R2prot–RNA ∼0.94 (Figure 6B).
The mRNA sequence determinants of TRmD and TRmIND
The fact that translation rates correlate with mRNA abundances suggests that highly expressed mRNAs contain features in their nucleic acid sequences that specify faster rates of translation than mRNAs present at low levels (12). Such mRNA sequence features would thus correlate with TRmD. TRmIND, on the other hand, is by definition fully uncorrelated with mRNA abundance and with TRmD. It is plausible then that the two components of translation may be specified by different sequence elements and controlled by separate mechanisms. We therefore sought to determine if there are mRNA sequence features that specify TRmD and to assess if these differ from those that define TRmIND.
Detailed prior work has identified several mRNA sequence features that correlate with, and in some cases have been directly shown to affect, rates of translation (27,29,30,38–59). Extending this earlier work, we defined nine sequence features that predict between 5% – 60% of the variance in the rates of translation when tested in pairwise regressions in which only one feature is present. When all nine features are combined in a multivariate model, 80% of translation is explained (Figure 7; Supplementary Table S7 and Methods S4; Datasets S6–S9). Of note, a Translation Initiation Control Region (TICE) that flanks the AUG codon alone explains 33% of the variance in translation rates (Figures 7 and 8). The extent of the TICE was determined by testing Position Weight Matrices (PWMs) of differing lengths, which showed that the TICE is largely encoded by nucleotides –35 to +28 (Figure 8C). The –35 to –1 region is strikingly more A rich and G poor in highly translated mRNAs than in less well translated genes, while the +4 to +28 region shows more complex position specific differences with translation rate (Figure 8A and Supplementary Figure S2). Further analysis revealed that the frequencies of a subset of dinucleotides and trinucleotides within the –35/–1 and the +4/+28 regions allow more complete prediction of translation rates when combined with PWMs (Figure 8D and Dataset S8). Consistent with earlier observations (39–41,44,51), the TICE is much less likely to adopt a folded RNA structure in highly translated mRNAs than it is in poorly translated mRNAs (Figure 8B), suggesting that it functions at least in part by specifying structure.
Figure 7.

mRNA sequences that explain translation rates. The R2 coefficients of determination between nine mRNA sequence features and TR are shown (Supplementary Table S7 and Methods S4). A cartoon below shows to which mRNA region each feature maps. The TICE, CDS amino acid frequency and CDS codon frequency features are multi feature sets comprised of 14, 20 and 61 individual features respectively (Datasets S6 and S8). The other six are single features (Dataset S6).
Figure 8.

The –35 to + 28 Translation Initiation Control Element (TICE). (A) Position weight matrices (PWMs) for the 10% of mRNAs with the highest TR scores (top) and the 10% of mRNAs with the lowest TR scores (bottom). Sequence logos show the frequency of each nucleotide at each position relative to the first nucleotide of the protein coding sequence (CDS) (Dataset S7). (B) The mean predicted RNA folding energy (ΔG, kcal/mol) of 35 nucleotide windows (y-axis). The x-axis shows the position of the 5′ most nucleotide of each window. Windows representing every one nucleotide offset were calculated. (C) The R2 coefficient of determination between translation rates (TR) and PWM scores. PWMs of varying lengths were built from the sequences of the 10% of mRNAs with the highest TRs, and then log odds scores were calculated for all mRNAs that completely contained a given PWM. PWMs extending 5′ from –1 in 5 nucleotide increments were tested (x-axis, right to left) and these were also extended 3′ from +4 in 5 or 10 nucleotide increments (grey to black scale). (D) The R2 coefficients of determination between TICE mRNA sequence features and TR. PWMs corresponding to the three specified TICE mRNA regions (–35/–1, +4/+28 and –35/+28) were used to score each gene (PWM only). Alternatively, PWMs and the frequencies of a small subset of dinucleotides and/or trinucleotides were scored for each gene (PWM + di/tri nuc. freq.) (Datasets S6 and S8).
Using the nine features, the percent of the variances in TRmD and TRmIND that are explained by each in pairwise regressions were determined (Figure 9 and Supplementary Table S7). While TRmD and TRmIND both correlate with multiple features, there are significant differences in the degree to which some features explain TRmD versus TRmIND. CDS length has a much larger impact on TRmIND than on TRmD (Bonferroni corrected P < 0.001). On the other hand, the TICE, the frequencies of amino acids or codons encoded by the CDS, RNA folding of the CDS, and poly A tail length each explains more of TRmD than TRmIND (Bonferroni corrected P < 0.001). The remaining three features—length of the 5′ untranslated region (UTR), number of open reading frames (ORFs) in the 5′ UTR, and RNA folding in the 5′ UTR—show no compelling discrimination in the degree to which they explain TRmIND and TRmD (Bonferroni corrected P > 0.074).
Figure 9.

TRmD and TRmIND are differentially determined by mRNA sequences. The R2 coefficients of determination between mRNA sequence features and TRmD and TRmIND are shown (Supplementary Table S7 and Methods S4). The Bonferroni corrected P-value testing if the correlations with TRmD and TRmIND are equal are given, with significant p-values shown in red. A cartoon below shows to which mRNA region each feature maps.
The mRNA sequence features that correlate with TRmIND are likely to be mechanistic determinants of translation rates, see Discussion. The features that correlate with TRmD, however, could in principle directly affect translation or they could instead only impact mRNA stability. Their correlation with TRmD might not reflect a direct mechanistic role in translation but instead a fortuitous consequence of their impact on mRNA abundance. We therefore determined if measured mRNA degradation rate data could explain the correlation of each feature with TRmD by calculating revised TRmD values (TRmD*) where the expected impact of RNA degradation has been removed (Supplementary Methods S4 and Table S8) (30). Only poly-A length and CDS RNA folding showed a significant reduction in their correlation with TRmD* (Bonferroni corrected P < 0.001). The remaining features showed similar correlations with TRmD and TRmD* (Bonferroni corrected P > 0.072) (Supplementary Table S8). Thus, poly-A tail length and CDS RNA folding likely act at least in part by impacting mRNA stability. The correlation of the other seven features with translation rates appears to reflect direct control of protein synthesis.
The frequencies of codons in different mRNAs correlate with the abundance of the encoded proteins (27,30,45–49,52). Codon frequencies within highly translated mRNAs more closely match the abundances of their cognate tRNAs than is the case for poorly translated messages, resulting in higher rates of translation elongation (27,30,45–49,52). The substantial correlation between amino acid or codon frequencies with TRmD and with TRmIND (Figure 9) therefore likely reflects control of elongation. To directly test this, we first determined the frequencies of amino acids and codons in the 10% of genes with the highest values of TRmD or TRmIND (top cohorts) and separately the frequencies of amino acids and codons in the 10% of genes with the lowest values (bottom cohorts) (Dataset S9). We then correlated these frequencies with tRNA abundance. All cohorts show a positive correlation (Figure 10 A and B; Supplementary Table S9). Top cohorts, however, consistently show a higher correlation than bottom cohorts, though this differences is only statistically significant for codon frequencies not for amino acid frequencies (Figure 10A and B; Supplementary Table S9). Notably, there is a larger difference between the top and bottom TRmD cohorts than seen between the top and bottom TRmIND cohorts.
Figure 10.

Amino acid and codon frequencies correlate with tRNA abundances. The frequencies of amino acid (AA) or codons in the CDS were determined separately for the 10% of genes with the highest scores for TRmD or TRmIND (top TRmD or top TRmIND) and for the 10% of genes with the lowest scores (bottom TRmD or bottom TRmIND) (Dataset S9). (A and B) The coefficient of determination (R2) for top and bottom amino acid or codon frequencies vs their cognate tRNA abundances (Supplementary Table S9). For amino acids, the frequencies of all cognate tRNAs for each amino acid were summed to give a combined tRNA abundance. The Bonferroni corrected P-value testing if the correlation of tRNA abundance with the top cohort is greater than that with the bottom cohort are given, with significant p-values shown in red. (C–F) The ratios between the frequencies of amino acids or codons in the top cohort divided by those in the bottom cohort were determined (Dataset S9). Ratios > 1 thus indicate a higher frequency in the top TRmD or top TRmIND cohorts. Scatter plots are shown between top/bottom frequency ratios and tRNA abundance along with the Pearson correlation coefficients (r). Bonferroni corrected P-values testing if the correlations are significant are also given, with significant P-values shown in red. Dashed vertical lines indicate a ratio of 1.
We also calculated the ratio of amino acid or codon frequencies between top cohorts divided by that of bottom cohorts (Dataset S9). For a given amino acid or codon, a ratio of greater than one thus indicates that it is more abundant in highly translated mRNAs than in poorly translated messages. Scatter plots comparing these ratios to tRNA abundance show positive correlations, with only the TRmIND amino acid ratios not showing a significant correlation (Figure 10C–F). The correlations are stronger for TRmD ratios than for TRmIND ratios. The range of ratios is also markedly larger for TRmD than for TRmIND. In particular, TRmD codon ratios lie between 0.02 to 3.60 while TRmIND codon ratios lie between 0.61 to 1.53, an over 50-fold difference (Figure 10E and F; Dataset S9). We conclude that both amino acid and codon frequencies control TRmD more strongly than they impact TRmIND and do so by their effect on the rate of elongation by the ribosome. The differences in amino acid composition between highly abundant and less abundant proteins have a significant impact on translation rates, but the additional larger variation in the frequencies of individual codons plays a bigger role.
DISCUSSION
We have presented a revised framework for determining the contribution of translation rates to the differences in protein expression between genes. Because translation rates partially correlate with mRNA abundance, it is not possible to provide a single metric to capture system-wide translational control. The R2 coefficient of determination between translation rates and protein expression cannot measure translation's contribution because it mixes the contribution of translation with that of mRNA. Instead, to be consistent with the requirements of ANOVA the contributions of translation to the amplification exponent bprot–RNA and to R2prot–RNA must be estimated separately. To achieve this, translation rates are decomposed into mRNA-abundance dependent and independent components, TRmD and TRmIND respectively. TRmD determines bprot–RNA, whereas TRmIND and protein degradation together determine R2prot–RNA.
We find that in S. cerevisiae TRmD represents ∼20% of the variance in translation and results in an the amplification exponent bprot–RNA of 1.20 with a 95% confidence interval [1.14, 1.26], while TRmIND constitutes the remaining ∼80% of the variance in translation and explains ∼5% of the variance in protein expression (Figure 6B). To overcome the difficulty of comparing the magnitude of contributions that are expressed by different, incommensurable metrics, we suggest that the percent of the variance in translation that each explains be used. In other words, TRmIND could be said to contribute 80/20 = 4-fold more to the control of protein levels than does TRmD.
Our estimates for bprot–RNA are lower than that of the only previous study to assume mRNA-abundance dependent translational amplification (1.20 [1.14, 1.26] versus 1.69) (12). Because bprot–RNA is an amplification exponent for non-logged abundance data, this disagreement between estimates is large. bprot–RNA = 1.20 implies a range of mRNA abundances in the cell that is fifty fold larger than that implied by bprot–RNA = 1.69 (Dataset S2 and Figure 3). One of the two approaches that we used to estimate bprot–RNA is based on multiple protein and mRNA abundance scaling-standard datasets that were each produced using methods that employed internal concentration standards and should thus be properly scaled. Broad agreement is observed between scaling-standards from separate studies that used different methods (Figure 2). Our other estimate of bprot–RNA is based on the correlation between measured translation rate and mRNA abundance data. Our two independent estimates are similar (means 1.17 versus 1.22; Figure 3B and C), implying that they are reasonable. The prior estimate of bprot–RNA = 1.69, by contrast, used a Bayesian model to infer the scaling of true protein and true mRNA abundances from datasets that in some cases were produced by methods that yield biased scalings (Figure 4). The model had no guide for which data input was correctly scaled, and thus it had no way to determine a correct scaling. It was also previously claimed that the correlation between ribosome profiling data and mRNA abundances was consistent with bprot–RNA = 1.69 (12). Our analysis, however, indicates that this claim in effect assumes that true translation rates and true mRNA abundances correlate perfectly (see Results), an idea that is inconsistent with the available evidence.
Given estimates for TRmIND, protein degradation and measurement error, we showed that it is possible to estimate R2prot–RNA for the true abundances of proteins and mRNA. This approach suggests that R2prot–RNA ∼0.94 (Figure 6B). The highest previous estimate for the correlation between protein and mRNA levels was R2prot–RNA = 0.86 (12). This estimate was based on modeled abundances for 5,854 protein-coding genes in S. cerevisiae. For 842 of these genes, however, either protein or mRNA abundance data was lacking; instead, values were imputed using a Bayesian model. When we limit the protein and mRNA abundances produced by the Bayesian model to the 5,012 genes for which empirically measured data is available, R2prot–RNA = 0.80.
Our decomposition of translation rates, thus, provides an estimate for the combined contributions of translation and protein degradation that is ∼3-fold lower than the smallest previous estimates based on measured protein and mRNA abundance data. Results from other approaches, though, support our estimate that R2prot–RNA ∼0.94. For example, ribosome profiling studies have found almost as strong a correlation between mRNA levels and the total number of protein molecules synthesized per gene (R2 = 0.90) (27). In addition, translational regulation of specific transcripts in response to stress in S. cerevisiae is generally less than threefold and limited to a minority of genes (37,60). Finally, unlike animals, plants and other fungi, S. cerevisiae lacks micro RNAs (61). The degree of transcript-specific translational regulation may be limited in this species, and so a particularly high correspondence between protein and mRNA abundances should be unsurprising.
These results should not be taken to suggest that translational control is unimportant, however. Translation and other steps, such as protein degradation, that do not strongly determine protein abundances, contribute to responsivity (3,11). For example, the response to environmental stimuli that change levels of specific mRNAs will be more rapid for those mRNAs that are inherently translated more quickly. Several metrics for control must be considered to properly appreciate the contribution of each step in regulating gene expression.
Quantifying the mechanisms that control translation
By considering which mRNA sequence features determine TRmD and TRmIND, we have also been able to provide insights into the mechanisms governing translation and the degree to which each exerts control. Extending detailed prior studies (27,29,30,38–59), we showed that nine sequence features can explain 80% of the variance in translation rates (Figure 7 and Supplementary Table S7). Importantly, the nine features do not all affect TRmD and TRmIND equally (Figure 9). TRmD—and therefore the amplification exponent—is most strongly determined both by a Translation Initiation Control element (TICE) that spans nucleotides –35 to +28 and by the frequencies of amino acid and codons encoded in the protein coding sequence (CDS). TRmIND, by contrast, is chiefly determined by the length of the CDS. These differences indicate that these two components of translation are under different selective pressures.
Translation initiation in eukaryotes has been proposed to be enhanced by a circularization event that brings the 5′ and 3′ ends of mRNAs into close proximity (55,62). The negative impact that longer CDSs have on translation rates results because this circularization appears less efficient for longer mRNAs than for shorter mRNAs ((27,55,63) Fernandes et al. (2017) Biorxiv doi 10.1101/105296). Given this, it can be readily understood why there might be dramatic differences in the degree to which CDS length specifies TRmD versus TRmIND. CDS length and mRNA abundance are under strong selective pressures that are unrelated to the control of translation rates. The relatively weak negative correlation of CDS length with TRmD should thus be mostly determined by these other strong selective forces. In contrast, TRmIND has no correlation with mRNA abundance, and thus the degree to which circularization efficiency affects translation will be fully reflected in the strong anti-correlation we observe between CDS length and TRmIND.
Previous work indicates that A-rich sequences in the region –10 to –1 result in higher rates of translation initiation and that nucleotides between either +4 to +6 or +10 to +20 also play a role (38,40–42,64). Our analysis defining the TICE is consistent with this evidence, though suggests that the A-rich element is more extensive, stretching from nucleotides –35 to –1, and that all of the region from +4 to +28 is involved (Figure 8 and Supplementary Figure S2). The –35 to –1 region in highly translated mRNAs has a less folded RNA structure than in mRNAs translated at lower rates (Figure 8B) (39–41,44,51). Thus it is possible that the A-rich sequences act only by specifying unfoldedness and perhaps other aspects of structure, such as the degree of base stacking or chain flexibility. It has also been speculated, however, that A-rich sequences might stabilize the interaction of poly-A binding protein with the 5′ UTR and thus enhance translation by mRNA circularization (65). The –35 to –1 portion of the TICE could thus act by two means. The TICE from +4 to +28 is not especially A-rich and instead shows a variety of location specific preferences for different bases between highly translated and poorly translated mRNAs (Figure 8A). Some of these sequence preferences may reflect evolutionary selection for protein function that are unrelated to the control of translation. Unfolded mRNA structure in the +4 to +28 region also correlates positively with translation rates, however, raising the intriguing possibility that the N-terminal nine or so amino acids could in part be selected because of the mRNA structures produced by their codons, rather than for their function within the protein (Figure 8B).
It has long been recognized that rates of translation elongation are higher for mRNAs whose frequency of codons more closely matches the relative abundance of tRNAs (27,30,45–49,52). Our analysis shows that both amino acid and codon frequencies are principally used to determine differences in translation elongation rates between differently abundant mRNAs (i.e. TRmD) (Figures 9 and 10). These two features play a lesser role in modulating the mRNA independent variation in translation rates (i.e. TRmIND) (Figures 9 and 10).
The length of poly-A tails and the degree of RNA folding in the CDS also show strong discrimination in their correlation with TRmD versus with TRmIND (Figure 9). The correlation of these features with TRmD, however, unlike those of our other seven features, may not reflect a direct effect on translation but an impact on mRNA stability and hence mRNA abundance (Supplementary Table S8). The correlation of poly-A length and CDS RNA folding with TR may be entirely fortuitous. On the other hand, codon usage does have dramatic effects on both translation rate and mRNA stability in S. cerevisiae, with mRNAs that have codon frequencies optimized for rapid translation being the most stable (30,52,57–59). Our results confirm that our codon frequency feature correlates with RNA degradation (R2 = 0.21, Supplementary Table S8). These results explain why codon usage is a strong determinant of TRmD and why it has a less strong effect on TRmIND. The control of both translation and mRNA turnover by this one sequence feature will inevitably lead to a correlation of TR and mRNA abundance and—as a consequence—an amplification exponent bprot–RNA > 1. Any feature that impacts mRNA abundance will tend to explain TRmD more so than TRmIND, which is what we observe (Figure 9).
The three remaining features—5′ UTR length, number of 5′ UTR ORFs and 5′ UTR folding—do not show significant differences in their correlation with TRmD and TRmIND. They contribute to both, establishing that TRmD and TRmIND are each specified by multiple features.
Finally, because our model explains the bulk of the variance in translation, we can estimate the relative contributions of control at initiation versus control during elongation. 5′ UTR length, number of 5′ UTR ORFs, 5′ UTR folding, and the TICE all likely affect initiation, not elongation, and collectively explain 42% of the variance in translation. Assuming that the length of the protein coding region also effects initiation rates, 58% of the variance in translation is controlled prior to elongation by the ribosome (Figure 11). Codon frequency controls elongation rate and determines 60% of the variance in translation (Figures 7 and 11). When these six features are combined in a model, 80% of the variance in translation is explained (Figure 11). Initiation and elongation thus appear to share an equal role in controlling translation and to act in a substantially correlated manner. Slightly more than 60% of the control of initiation is collinear with elongation and vice versa (Figure 11): % initiation correlated with elongation = 66% = (58% + 60% - 80%) / 58% × 100% elongation correlated with initiation = 63% = (58% + 60% – 80%)/60% × 100%. Initiation and elongation control features appear to act in tandem, tending to amplify the effect of each other either to both up regulate or to both down regulate rates.
Figure 11.

Sequence information that specifies initiation and elongation rates are highly correlated. We assume that mRNA sequences in the 5′ UTR, the TICE, and the length of the protein coding sequence together control the rate of translation initiation and that codon frequency determines the rate of elongation by the ribosome (top, center). Scatter plots compare measured TR data to the results of three multi-variate models that predict TR based on features controlling initiation only (middle, left), elongation only (middle, right) or initiation or elongation (bottom, center). The R2 coefficients of determination are shown in each case. The results show that initiation and elongation signals each explain >57% of the variance in TR and that a model including both signals explains 80% of TR. Thus, 66% the control of initiation is collinear with the control of elongation (bottom, left) and 63% of the control of elongation is collinear with initiation (bottom, right).
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to David Weinberg, Premal Shah, Joshua Plotkin, and David Bartel for generously providing their ribosome profiling data prior to publication. Likewise, we are grateful to Craig Lawless, Robert Beynon, and Simon Hubbard for early access to their SRM MS protein abundance data. We acknowledge Kevin Weeks for pointing out that A rich RNA is likely unfolded. We thank Peter Bickel, Soile Keranen, Alisyn Nedoma and Han Chen for thoughtful critiques of earlier drafts of this manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Department of Statistics at University of California, Los Angeles [to J.J.L.]; Hellman Fellowship from the Hellman Foundation; PhRMA Foundation Research Starter Grant in Informatics; NIH [R01GM120507]; Lawrence Berkeley National Laboratory was conducted under U.S. Department of Energy Contract [DE-AC02-05CH11231]. Funding for open access charge: NIH [P01GM099655].
Conflict of interest statement. None declared.
REFERENCES
- 1. Li J.J., Biggin M.D.. Gene expression. Statistics requantitates the central dogma. Science. 2015; 347:1066–1067. [DOI] [PubMed] [Google Scholar]
- 2. Albert F.W., Kruglyak L.. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015; 16:197–212. [DOI] [PubMed] [Google Scholar]
- 3. Liu Y., Beyer A., Aebersold R.. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016; 165:535–550. [DOI] [PubMed] [Google Scholar]
- 4. de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C.. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009; 5:1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghazalpour A., Bennett B., Petyuk V.A., Orozco L., Hagopian R., Mungrue I.N., Farber C.R., Sinsheimer J., Kang H.M., Furlotte N. et al. . Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011; 7:e1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M.. Global quantification of mammalian gene expression control. Nature. 2011; 473:337–342. [DOI] [PubMed] [Google Scholar]
- 7. Kristensen A.R., Gsponer J., Foster L.J.. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol. 2013; 9:689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M.C., Zimmerman L.J., Shaddox K.F., Kim S. et al. . Proteogenomic characterization of human colon and rectal cancer. Nature. 2014; 513:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ly T., Ahmad Y., Shlien A., Soroka D., Mills A., Emanuele M.J., Stratton M.R., Lamond A.I.. A proteomic chronology of gene expression through the cell cycle in human myeloid leukemia cells. Elife. 2014; 3:e01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J.J., Bickel P.J., Biggin M.D.. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ. 2014; 2:e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jovanovic M., Rooney M.S., Mertins P., Przybylski D., Chevrier N., Satija R., Rodriguez E.H., Fields A.P., Schwartz S., Raychowdhury R. et al. . Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015; 347:1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Csardi G., Franks A., Choi D.S., Airoldi E.M., Drummond D.A.. Accounting for experimental noise reveals that mRNA levels, amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast. PLoS Genet. 2015; 11:e1005206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yassour M., Pfiffner J., Levin J.Z., Adiconis X., Gnirke A., Nusbaum C., Thompson D.A., Friedman N., Regev A.. Strand-specific RNA sequencing reveals extensive regulated long antisense transcripts that are conserved across yeast species. Genome Biol. 2010; 11:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weiner A., Chen H.V., Liu C.L., Rahat A., Klien A., Soares L., Gudipati M., Pfeffner J., Regev A., Buratowski S. et al. . Systematic dissection of roles for chromatin regulators in a yeast stress response. PLoS Biol. 2012; 10:e1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lipson D., Raz T., Kieu A., Jones D.R., Giladi E., Thayer E., Thompson J.F., Letovsky S., Milos P., Causey M.. Quantification of the yeast transcriptome by single-molecule sequencing. Nat. Biotechnol. 2009; 27:652–658. [DOI] [PubMed] [Google Scholar]
- 16. Miura F., Kawaguchi N., Yoshida M., Uematsu C., Kito K., Sakaki Y., Ito T.. Absolute quantification of the budding yeast transcriptome by means of competitive PCR between genomic and complementary DNAs. BMC Genomics. 2008; 9:574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O'Shea E.K., Weissman J.S.. Global analysis of protein expression in yeast. Nature. 2003; 425:737–741. [DOI] [PubMed] [Google Scholar]
- 18. Newman J.R., Ghaemmaghami S., Ihmels J., Breslow D.K., Noble M., DeRisi J.L., Weissman J.S.. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006; 441:840–846. [DOI] [PubMed] [Google Scholar]
- 19. Lawless C., Holman S.W., Brownridge P., Lanthaler K., Harman V.M., Watkins R., Hammond D.E., Miller R.L., Sims P.F.G., Grant C.M. et al. . Direct and absolute quantification of over 1800 yeast proteins via selected reaction monitoring. Mol. Cell. Proteomics. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. von der Haar T. A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst. Biol. 2008; 2:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Picotti P., Bodenmiller B., Mueller L.N., Domon B., Aebersold R.. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009; 138:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Firczuk H., Kannambath S., Pahle J., Claydon A., Beynon R., Duncan J., Westerhoff H., Mendes P., McCarthy J.E.. An in vivo control map for the eukaryotic mRNA translation machinery. Mol. Syst. Biol. 2013; 9:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carroll K.M., Simpson D.M., Eyers C.E., Knight C.G., Brownridge P., Dunn W.B., Winder C.L., Lanthaler K., Pir P., Malys N.. Absolute quantification of the glycolytic pathway in yeast: deployment of a complete QconCAT approach. Mol. Cell. Proteomics. 2011; 10, M111 007633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brownridge P., Lawless C., Payapilly A.B., Lanthaler K., Holman S.W., Harman V.M., Grant C.M., Beynon R.J., Hubbard S.J.. Quantitative analysis of chaperone network throughput in budding yeast. Proteomics. 2013; 13:1276–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smallbone K., Messiha H.L., Carroll K.M., Winder C.L., Malys N., Dunn W.B., Murabito E., Swainston N., Dada J.O., Khan F. et al. . A model of yeast glycolysis based on a consistent kinetic characterisation of all its enzymes. FEBS Lett. 2013; 587:2832–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mackenzie R.J., Lawless C., Holman S.W., Lanthaler K., Beynon R.J., Grant C.M., Hubbard S.J., Eyers C.E.. Absolute protein quantification of the yeast chaperome under conditions of heat shock. Proteomics. 2016; 16:2128–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weinberg D., Shah P., Eichhorn S., Hussmann J., Plotkin J., Bartel D.. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016; 14:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Christiano R., Nagaraj N., Frohlich F., Walther T.C.. Global proteome turnover analyses of the Yeasts S. cerevisiae and S. pombe. Cell Rep. 2014; 9:1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subtelny A.O., Eichhorn S.W., Chen G.R., Sive H., Bartel D.P.. Poly(A)-tail lengths and a developmental switch in translational control. Nature. 2014; 508:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K.E., Graveley B.R. et al. . Codon optimality is a major determinant of mRNA stability. Cell. 2015; 160:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahrne E., Molzahn L., Glatter T., Schmidt A.. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics. 2013; 13:2567–2578. [DOI] [PubMed] [Google Scholar]
- 32. Holland M.J. Transcript abundance in yeast varies over six orders of magnitude. J. Biol. Chem. 2002; 277:14363–14366. [DOI] [PubMed] [Google Scholar]
- 33. Dudley A.M., Aach J., Steffen M.A., Church G.M.. Measuring absolute expression with microarrays with a calibrated reference sample and an extended signal intensity range. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7554–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith R.J. Use and misuse of the reduced major axis for line-fitting. Am. J. Phys. Anthropol. 2009; 140:476–486. [DOI] [PubMed] [Google Scholar]
- 35. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McManus C.J., May G.E., Spealman P., Shteyman A.. Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res. 2014; 24:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gerashchenko M.V., Lobanov A.V., Gladyshev V.N.. Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:17394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakagawa S., Niimura Y., Gojobori T., Tanaka H., Miura K.. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 2008; 36:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E.. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010; 467:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robbins-Pianka A., Rice M.D., Weir M.P.. The mRNA landscape at yeast translation initiation sites. Bioinformatics. 2010; 26:2651–2655. [DOI] [PubMed] [Google Scholar]
- 41. Gingold H., Pilpel Y.. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011; 7:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dvir S., Velten L., Sharon E., Zeevi D., Carey L.B., Weinberger A., Segal E.. Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E2792–E2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chew G.L., Pauli A., Schier A.F.. Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat. Commun. 2016; 7:11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hinnebusch A.G., Ivanov I.P., Sonenberg N.. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016; 352:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dana A., Tuller T.. Mean of the typical decoding rates: a new translation efficiency index based on the analysis of ribosome profiling data. G3 (Bethesda). 2014; 5:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sorensen M.A., Pedersen S.. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J. Mol. Biol. 1991; 222:265–280. [DOI] [PubMed] [Google Scholar]
- 47. Varenne S., Buc J., Lloubes R., Lazdunski C.. Translation is a non-uniform process. Effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J. Mol. Biol. 1984; 180:549–576. [DOI] [PubMed] [Google Scholar]
- 48. Brockmann R., Beyer A., Heinisch J.J., Wilhelm T.. Posttranscriptional expression regulation: what determines translation rates?. PLoS Comp. Biol. 2007; 3:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guimaraes J.C., Rocha M., Arkin A.P.. Transcript level and sequence determinants of protein abundance and noise in Escherichia coli. Nucleic Acids Res. 2014; 42:4791–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dacheux E., Malys N., Meng X., Ramachandran V., Mendes P., McCarthy J.E.. Translation initiation events on structured eukaryotic mRNAs generate gene expression noise. Nucleic Acids Res. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hinnebusch A.G., Lorsch J.R.. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb. Perspect. Biol. 2012; 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Radhakrishnan A., Green R.. Connections underlying translation and mRNA stability. J. Mol. Biol. 2016; 428:3558–3564. [DOI] [PubMed] [Google Scholar]
- 53. Arribere J.A., Gilbert W.V.. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res. 2013; 23:977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rojas-Duran M.F., Gilbert W.V.. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA. 2012; 18:2299–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thompson M.K., Gilbert W.V.. mRNA length-sensing in eukaryotic translation: reconsidering the “closed loop" and its implications for translational control. Curr. Genet. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang C.P., Chen S.J., Lin C.H., Wang T.L., Wang C.C.. A single sequence context cannot satisfy all non-AUG initiator codons in yeast. BMC Microbiol. 2010; 10:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boel G., Letso R., Neely H., Price W.N., Wong K.H., Su M., Luff J., Valecha M., Everett J.K., Acton T.B. et al. . Codon influence on protein expression in E. coli correlates with mRNA levels. Nature. 2016; 529:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bazzini A.A., Del Viso F., Moreno-Mateos M.A., Johnstone T.G., Vejnar C.E., Qin Y., Yao J., Khokha M.K., Giraldez A.J.. Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J. 2016; 35:2087–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mishima Y., Tomari Y.. Codon usage and 3′ UTR length determine maternal mRNA stability in zebrafish. Mol. Cell. 2016; 61:874–885. [DOI] [PubMed] [Google Scholar]
- 60. Payne T., Hanfrey C., Bishop A.L., Michael A.J., Avery S.V., Archer D.B.. Transcript-specific translational regulation in the unfolded protein response of Saccharomyces cerevisiae. FEBS Lett. 2008; 582:503–509. [DOI] [PubMed] [Google Scholar]
- 61. Drinnenberg I.A., Fink G.R., Bartel D.P.. Compatibility with killer explains the rise of RNAi-deficient fungi. Science. 2011; 333:1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Christensen A.K., Kahn L.E., Bourne C.M.. Circular polysomes predominate on the rough endoplasmic reticulum of somatotropes and mammotropes in the rat anterior pituitary. Am. J. Anat. 1987; 178:1–10. [DOI] [PubMed] [Google Scholar]
- 63. Arava Y., Wang Y., Storey J.D., Liu C.L., Brown P.O., Herschlag D.. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cavener D.R., Ray S.C.. Eukaryotic start and stop translation sites. Nucleic Acids Res. 1991; 19:3185–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gilbert W.V., Zhou K., Butler T.K., Doudna J.A.. Cap-independent translation is required for starvation-induced differentiation in yeast. Science. 2007; 317:1224–1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.