

Abstract
We recently described the isolation of a novel influenza virus from swine exhibiting respiratory disease in the United States that is distantly related to human influenza C virus. Based on genetic, biochemical and morphological analysis, the virus was provisionally classified as C/swine/Oklahoma/1334/2011 (C/OK). To further understand the genetics and evolution of this novel pathogen, we performed a comprehensive analysis of its sequence and phylogeny. The results demonstrated that C/OK and human influenza C viruses share a conserved array of predicted functional domains in the viral RNA genome replication and viral entry machinery but vary at key functional sites. Furthermore, our evolutionary analysis showed that homologous genes of C/OK and human influenza C viruses diverged from each other an estimated several hundred to several thousand years ago. Taken together, the findings described in this study support and extend our previous observations that C/OK is a genetically and evolutionarily distinct influenza virus in the family Orthomyxoviridae.
Introduction
Influenza viruses are divided into three genera in the family Orthomyxoviridae. There are numerous subtypes of influenza A virus (IAV), and these have been isolated from various species of birds and mammals [16]. Co-infection and genetic reassortment of influenza A viruses can generate new strains or subtypes that evade preexisting immunity and as a result cause seasonal epidemics or global pandemics [28]. Swine, which can be infected by both avian and human influenza A viruses due to the fact that they contain two types of viral receptors (α2,3- and α2,6- Gal receptors) [12], are proposed to be an intermediate host for genetic reassortment of different influenza A viruses [28]. New reassortants from swine can spread and be transmitted to and among humans, causing influenza epidemics or pandemics, was as seen with the H1N1 pandemic of 2009 [19]. In contrast, influenza B (IBV) and C (ICV) viruses mainly exist in humans, although influenza B infection of seals and influenza C infection of pigs have been reported previously [9, 15].
Evolutionarily, IBV and ICV are proposed to be in stasis, characterized by a low evolutionary rate, particularly at non-synonymous sites [17, 28]. According to this hypothesis, IBV and ICV are well adapted to the human host, and viral evolution has slowed. Several studies have suggested that most mutations that cause IBV and ICV to deviate from the phenotypically most fit state are frequently eliminated. Thus, these viruses generally experience strong purifying selection [7, 29].
Recently, members of our research group isolated a novel swine influenza virus that is phylogenetically related to human ICV [10]. This virus was isolated from a diseased pig with respiratory symptoms from a swine farm located in Oklahoma, USA, and has been provisionally designated C/swine/Oklahoma/1334/2011 (C/OK). Like human ICV, C/OK has seven genomic segments, unlike IAV and IBV, which have eight segments. Nonetheless, the sequence identity between C/OK and human influenza C viruses, ranging from 72 % for PB1 to 29 % for NS1, with an average of 50 % for the whole genome, is much lower if compared with >95 % sequence identity observed among the six lineages of human ICV. A serological survey indicated that C/OK circulates in pigs in the United States but is not widespread. In addition to swine, this virus can infect and be transmitted in ferrets through direct contact. Interestingly, the cellular tropism of C/OK is much wider than that of human ICV, which is supported by the observed difference in the receptor-binding pockets of the two viruses.
To further characterize C/OK virus, we analyzed the genome, phylogeny, and evolution of C/OK and compared them with those of members of the other genera of orthomyxoviruses. The results of these qualitative and quantitative studies provide novel insights into the evolution of this recently emerged influenza virus in swine and provide a framework for further investigation of this new pathogen with zoonotic potential.
Materials and methods
Sequence similarity analysis
A sliding-window method was used to calculate the similarity score for each site between homologous genes of C/OK and other viruses in a pairwise manner [27]. In each sliding window (25 amino acids in this study), the similarity score of the middle site (13th amino acid) is the summed similarity of the 25 pair sites scored by BLO-SUM62 matrix divided by the similarity score of one of the sequences in the sliding window to itself (the smaller score was chosen). Gaps were not scored.
Glycosylation site, transmembrane region, and signal peptide prediction
The N-glycosylation sites of C/OK and human influenza C HE proteins were predicted using Glycam (http://glycam.ccrc.uga.edu/ccrc/gp/index.jsp?tool=crystallography&option=ff99:glycam06). Glycam identifies potential N-glycosylation sites by calculating the solvent-accessible surface area of Asn in Asn-X-Thr/Ser motifs (X represents any amino acid except proline). For illustration purposes, a β-D-Manp-(1-4)-β-D-GlcpNAc was added to the potential glycosylation sites. The transmembrane and fusion peptide regions of HE protein were predicted using TMHMM [14]. SignaIP 4.1 and Phobius were used to predict the signal peptide sequence [13, 18].
Sequence alignment
Full-length cDNA sequences of influenza viruses were retrieved from the Influenza Virus Resource database (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Sequences from lab strains were excluded. Identical sequences were removed by retaining only the oldest one. For each genome segment, the cDNA sequences of human influenza C and C/OK were first translated to amino acid sequences and aligned using muscle within MEGA5 [3, 25], and then cDNA sequences were aligned according to the amino acid alignment. The cDNA alignments were curated manually. The GenBank accession numbers for C/OK segments 1–7 are JQ922305 to JQ922311, respectively [10].
Evolutionary analysis
Recombination sites were detected using a single-breakpoint recombination method [20]. A phylogenetic tree was estimated using the Bayesian method in Beast [2] (http://beast.bio.ed.ac.uk/Main_Page). For all of the segments, the tree prior was set to a constant size coalescent, the evolutionary rate was calculated separately for the three codon positions, and a uniform distribution was specified as a prior for relative mutation rate sampling. 2 × 109 Markov chain Monte Carlo iterations were run for PB1 segment, while 1 × 108 were used for the other segments, and trees were sampled every 2000 iterations. Convergence was achieved for all datasets, and the first 10 % iterations were used as burn-in for tree construction and parameter estimation. Parameter estimation and divergence time inference were performed using Tracer (http://beast.bio.ed.ac.uk/Tracer).
Results
Genomic, functional, and structural analysis
To explore the genomic and structural relationship between C/OK and other viruses of the family Orthomyxoviridae (influenza A, B, C viruses, infectious salmon anemia virus (ISAV) and Thogoto virus), we compared sequence positional similarity and structural feature of the polymerase and HE genes. During viral evolution, it is generally believed that the polymerase genes are highly conserved, while the genes encoding the surface proteins such as HE are considerably more diverse. Analysis of the HE gene was confined to C/OK and human ICV and did not include other members of the Orthomyxoviridae, because only these two viruses code for the HE protein.
Multiple sequence alignments of the polymerase genes (PB2, PB1, and PA or P3) showed that C/OK has more identical positions with human ICV, followed by IBV, IAV, ISAV and Thogoto virus (Fig. 1). Interestingly, in the region of the P3 or PA protein spanning amino acids 400–500, C/OK is more closely related to human ICV and ISAV than to IAV and IBV. Extensive investigation of the influenza A RNA replication machinery has established that PB2 is responsible for cap binding, and PB1 has RNA-dependent polymerase activity [1]. The PA protein is very versatile [1, 30], and its functions include protein stability, endonuclease activity, cap binding and promoter binding. Considering that the crystal structures of the enzymatic domain of PB1, the cap binding domain of PB2, and the endonuclease domain of PA in influenza A have been determined, we next performed structural modeling (Fig. 1) to investigate the conservation of known functional sites of polymerase proteins in C/OK [8, 11, 26, 30].
Fig. 1.

Schematic diagram of positional similarity and modeled structures of viral polymerase proteins. For the position similarity, the colored bar below in each plot shows domains and known functional sites. Domain regions and functional sites were predicted according to previous studies on human ICV and IAV [8, 10, 11, 26, 30]. Positions are numbered according to C/OK proteins. Virus strains: influenza A (H1N1/1918/Brevig Mission and H5N1/1996/GuangDong), influenza B (B/Brisbane/2008), influenza C (C/1966/Johannesburg), infectious salmon anemia virus (ISAV/1990/Glesvaer) and Thogoto virus (Dhori/1313/61). For the modeled structures, the side chains of active-site residues of C/OK and IAV P3 or PA are colored gray and magenta, respectively. Magnesium is shown as a green ball. Hydrogen bonds and salt bridges are shown with dashed yellow lines. Residues of C/OK and IAV PB2 are colored gray and magenta, respectively. The guanosine-binding residues are conserved, but the phosphate-binding resides (underlined) are different between C/OK virus and IAV. The structures were modeled using Modeller [5] with the structures of the endonuclease domain and cap-binding domain of influenza A virus as template (PDBID: 3HW5 and 2VQZ, respectively) [8]. The quality of the modeled structure was verified by Verify_3D [4]
The PB1 protein of C/OK possesses a set of key RNA polymerase active-site residues that are identical to those observed in other orthomyxoviruses representative of five genera (R241, K310, G411, D446, K482 and F493) (Figs. 1 and S1). In contrast to this highly conserved polymerase, some levels of sequence divergence were observed in functional domains of P3 or PA and PB2 proteins among different viruses. The endonuclease active site of P3 or PA was identical between C/OK and other influenza viruses (Figs. 1 and S2), but its counterparts in the non-influenza ISAV and Thogoto virus exhibited some sequence variations. For example, E has replaced K at P3 or PA position 120 of ISAV, while multiple substitutions have occurred in Thogoto virus (H41K, E66M, E105K). For PB2, the cap-binding domain (m7GTP binding site) of C/OK is more similar to that of human ICV (two substitutions: T351K, K366E), followed by IBV (four substitutions) and IAV (five substitutions), ISAV (seven substitutions) and Thogoto virus (completely different binding site) (Figs. 1 and S3). The guanine-binding residues of cap-binding domain are conserved between C/OK and IAV, but the phosphate-binding residues are different. The m7GTP phosphate-binding site of the cap-binding domain of IAV consists of four positively charged residues (K339, R355, H357 and H432) and one polar residue (N429) (Fig. 1). The cap-binding domain of PB2 of C/OK and human ICV differs in that it has fewer positively charged residues and includes more non-polar residues (T351, K366, W368, Y445 and P442, respectively, Fig. 1). Consequently, the phosphate-binding site of the C/OK cap-binding domain may use more hydrogen bonds to interact with the phosphate of m7GTP, while those of IAV and IBV use more salt bridges.
Our previous structural modeling work has indicated that the different sialic-acid-binding site may be the primary reason for the different cellular tropism observed between C/OK and human ICV [10]. By analyzing HE in more detail, we found that C/OK exhibits additional sequence variation from human ICV in the fusion domain 1 (F1) and fusion domain 3 (F3) (Fig. 2A). Notably, most of the variable residue positions are located within the N-and C-terminal regions of F1 and F3, which are believed to function as linkers to connect neighboring domains. In addition, we analyzed the fusion peptide and transmembrane domain (TM) of the C/OK HE protein using TMHMM (confidence posterior probability >0.95, Fig. S4). The fusion peptide of C/OK is highly similar to that of human ICV, while the TM harbors multiple amino acid changes (Fig. 2A). Intriguingly, C/OK, like human ICV, had a very short cytoplasmic tail containing only three amino acids (CKK) (Fig. S4). However, two of the three amino acids in the cytoplasmic tails of the two HE proteins were not conserved at their respective positions (CKK in C/OK vs. RTK in human ICV). Compared to C/OK and human ICV, influenza A and B viruses have a longer cytoplasmic tail, comprising 10 amino acids [23]. We also noted that C/OK possessed a 16-amino-acid signal peptide, while human ICV had a signal peptide that was 14 amino acids in length. Significant sequence variation was observed between these two signal peptides, but the sequences where they are cleaved by signal peptide peptidase were identical (A/E) (data not shown).
Fig. 2.

Schematic diagram of positional similarity and modeled structures of the viral HE protein. (A) Sequence position similarity between human ICV and C/OK. The F′ and TM regions of the C/OK HE protein were predicted using TMHMM [14]. The alignment of the TM region is colored according to sequence similarity (conserved residue: blue). S, signal peptide; F1, fusion domain 1; F2, fusion domain 2; F3, fusion domain 3; R, receptor-binding domain; E1, esterase domain 1; E2, esterase domain 2; C, cleavage site; F′, fusion peptide; TM, transmembrane region. (B) Modeled structure. Transparent cartoon and surface view of predicted glycosylation sites in human ICV and C/OK HE proteins. The modeled structure of C/OK HE is from our previous study [10]. Glycan (β-D-Manp-(1-4)- β-D-GlcpNAc, orange) and modified asparagine (magenta) are shown in stick mode. Sialic acid (cyan) is added to the receptor-binding pocket and esterase active site
Glycosylation of virion surface proteins such as HA or HE is one of the strategies frequently used by influenza viruses in escaping antibody-mediated neutralization [22]. For influenza viruses, glycans are added to asparagine in asparagine-X-serine/threonine motifs, where X represents any amino acid except proline. The HE proteins of C/OK and human ICV contain six and seven potential glycosylation sites, respectively (Fig. 2B). Two conserved glycosylation sites were observed: N26 and N144 in human ICV and N28 and N146 in C/OK. Although their sequences are not conserved, N61 of human ICV HE is located in a structural region similar to that of N390 in C/OK HE. In contrast to human ICV, which has two glycosylation sites in the receptor-binding domain of HE, C/OK virus HE possesses none of these N-linked glycan modifications. Numerous studies have previously shown that the glycosylation sites in the receptor-binding domain play a role in masking antigenic epitopes and facilitating viral evasion of antibody recognition, and they can influence receptor-binding avidity [6, 21, 22]. This observation warrants future study to address the potential impact of lacking an N-linked glycan modification in the receptor-binding domain on the susceptibility of C/OK to antibody neutralization as well as on other properties of C/OK such as virulence and virus-host interaction.
Divergence time inference
To explore the evolutionary relationship between C/OK and human ICV, we estimated the divergence time for five genes (HE, PA, PB1, PB2 and NP) using their respective nucleotide sequences. The recombination sites were analyzed by a single-breakpoint recombination method [20]. No recombination sites were found for any of the genes analyzed.
For each of the five genes, the estimated mean time to the most recent common ancestor (t-MRCA) of human ICV are consistent with those determined in a previous study [7], suggesting that our phylogenetic analyses are unbiased (Fig. 3). The t-MRCA of C/OK and human ICV ranges from 334 years for PB1 to 1299 years for HE (Fig. 3; Table 1). The t-MRCA of HE has the widest 95 % highest posterior densities (HPD) (727–1912 years), followed by PA. The mean age of HE falls outside of the 95 % HPD of the other genes, suggesting that HE is significantly older than the other genes at the 95 % significance level (Fig. 3). The more ancestral nature of HE was also observed for human influenza C viruses [7]. In addition to PB1 and HE, the high overlap of the 95 % HPD of divergence times suggests that the other three genes (PB2, P3 and NP) of C/OK and human ICV diverged on a similar time scale.
Fig. 3.
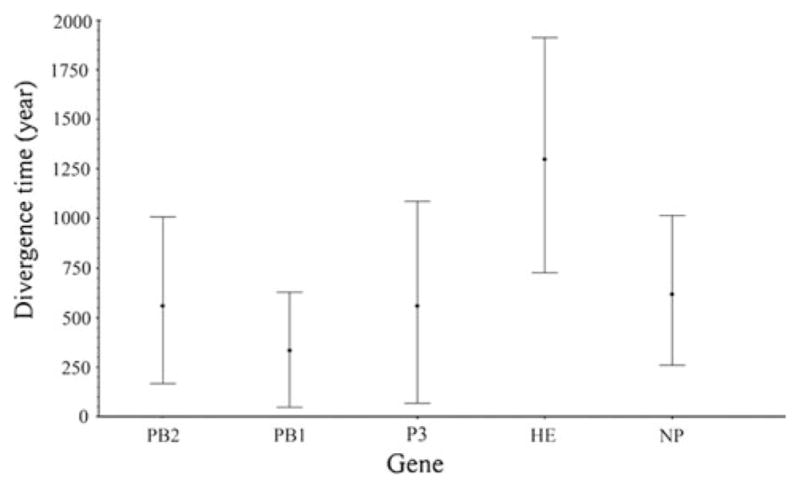
Estimated divergence time to the most recent common ancestor of C/OK and human ICV using the Bayesian probability method. The mean time is marked by a dot. The upper and lower lines are the 95 % highest posterior density (HPD) limits
Table 1.
Length and number of sequences for phylogenetic analysis and mean divergence timea
| Gene | Length (nucleotides) | Sequences | t-MRCA |
|---|---|---|---|
| PB2 | 1404 | 55 | 541 |
| PB1 | 1107 | 55 | 334 |
| P3 | 1125 | 40 | 545 |
| HE | 1950 | 134 | 1299 |
| NP | 1800 | 73 | 610 |
tMRCA, time to the most recent common ancestor (years)
Discussion
C/OK is a newly identified influenza virus in swine. Our previous study indicated that this novel virus is distantly related to human influenza C virus, and we proposed that C/OK should be provisionally classified as a distinct subtype of influenza C virus in the family Orthomyxoviridae. The comprehensive analysis of its genomic sequence and phylogeny reported here supports and further extends this previous classification. Despite the agreement, several features analyzed in this study demonstrate significant divergences between C/OK and human ICV.
Human ICV, like IBV, largely infects and utilizes humans as its primary host. The lack of an animal reservoir or the inability to replicate in multiple hosts is believed to contribute to their stable nature in genetic and antigenic evolution and their slow evolutionary rate. In contrast, IAV infects multiple mammal species and has diverse hosts as its reservoir. It is this unique property that accounts for the faster evolutionary rate observed for IAV, accompanied by frequent antigenic and genetic changes. C/OK can infect and be transmitted by direct contact in ferrets, a surrogate for human influenza infection studies, as well as in swine. The observed genetic differences between C/OK and ICV viruses coupled with their differences in cellular tropism as well as in response to growth temperature highlight that C/OK is extremely divergent from ICV and may argue that C/OK virus could be considered the prototype of a new genus with seven segments. Because we only sequenced C/OK virus, and no other similar viruses have been obtained so far, this limitation has prevented us from performing an evolutionary rate analysis like those that have been conducted previously for IAV, IBV, and ICV. When more C/OK-like viruses are isolated and become available, such a study is needed in order to determine the precise evolutionary rate, which is critical to understanding the evolution and host range of C/OK virus.
Our analysis of the viral entry machinery generated a number of interesting findings about this novel pathogen. First, C/OK varies significantly from human ICV in two domains of the HE protein responsible for virus-cell fusion. A recent study on human ICV showed that the HE protein is a restricting factor for its inefficient replication at high temperature (37 °C) and HE-associated poor fusion activity at 37 °C contributes to this intrinsic restriction. Intriguingly, C/OK is not restricted at 37 °C in its growth and it replicates at this temperature as efficiently as at 33 °C, which is the optimal temperature to propagate human influenza C viruses. Therefore, it is possible that the sequence variation in fusion domains may influence the replication of C/OK and human influenza C viruses at different temperatures as suggested previously [24]. Second, the HE protein of C/OK virus possesses fewer N-linked glycosylation sites than other members of the genus Influenzavirus C. A particularly interesting finding is that C/OK lacks a predicted glycosylation site in the receptor-binding domain, in contrast to other influenza C viruses. This may suggest that glycosylation of the C/OK receptor-binding domain is not important for preventing neutralization, or there may be a difference in receptor-binding kinetics. Additionally, the conservation of N26, N61 and N144 glycosylation sites in the fusion domains imply their importance in viral fitness, but more experimental studies are required to determine the function of these sites. Finally, both C/OK and human ICV have a short, three-amino-acid cytoplasmic tail, in contrast to human IAV and IBV. Significant sequence divergence was demonstrated in the cytoplasmic tail between C/OK and human ICV HE proteins. It will be interesting to address in future experiments the impact of sequence variation in the cytoplasmic portion of the HE protein on the biology of C/OK and human ICV.
Supplementary Material
Acknowledgments
This research was supported in part by the SDSU AES Fund 3AH203, a Public Health Service grant (AI076125 to F.L.), and SD 2010 Research Center (Biological Control and Analysis of Applied Photonics, BCAAP) Fund (3SJ163 to F.L.). Research in the S.C. laboratory is supported by the SDSU AES to S.C. and the South Dakota 2010 Research Center, BCAAP (Biological Control and Analysis of Applied Photonics) Fund (3SG163 to S.C.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00705-013-1815-3) contains supplementary material, which is available to authorized users.
Contributor Information
Zizhang Sheng, Department of Chemistry and Biochemistry, South Dakota State University, Brookings, SD 57007, USA.
Zhiguang Ran, Department of Veterinary and Biomedical Sciences, South Dakota State University, Brookings, SD 57007, USA. Department of Biology and Microbiology, South Dakota State University, Brookings, SD 57007, USA.
Dan Wang, Department of Health and Nutrition Sciences, South Dakota State University, Brookings, SD 57007, USA.
Adam D. Hoppe, Department of Chemistry and Biochemistry, South Dakota State University, Brookings, SD 57007, USA
Randy Simonson, Newport Laboratories, Worthington, MN 56187, USA.
Suvobrata Chakravarty, Department of Chemistry and Biochemistry, South Dakota State University, Brookings, SD 57007, USA.
Ben M. Hause, Department of Veterinary and Biomedical Sciences, South Dakota State University, Brookings, SD 57007, USA. Newport Laboratories, Worthington, MN 56187, USA
Feng Li, Department of Veterinary and Biomedical Sciences, South Dakota State University, Brookings, SD 57007, USA. Department of Biology and Microbiology, South Dakota State University, Brookings, SD 57007, USA. Shandong Poultry Institute, Shandong Academy of Agricultural Sciences, Jinan 250023, China.
References
- 1.Boivin S, Cusack S, Ruigrok RWH, Hart DJ. Influenza A virus polymerase: structural insights into replication and host adaptation mechanisms. J Biol Chem. 2010;285:28411–28417. doi: 10.1074/jbc.R110.117531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 5.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2(Unit 2.9) doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 6.Gambaryan AS, Marinina VP, Tuzikov AB, Bovin NV, Rudneva IA, Sinitsyn BV, Shilov AA, Matrosovich MN. Effects of host-dependent glycosylation of hemagglutinin on receptor-binding properties on H1N1 human influenza A virus grown in MDCK cells and in embryonated eggs. Virology. 1998;247:170–177. doi: 10.1006/viro.1998.9224. [DOI] [PubMed] [Google Scholar]
- 7.Gatherer D. Tempo and mode in the molecular evolution of influenza C. PLoS Curr. 2010;2:RRN1199. doi: 10.1371/currents.RRN1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ, Cusack S. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol. 2008;15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 9.Guo YJ, Jin FG, Wang P, Wang M, Zhu JM. Isolation of influenza C virus from pigs and experimental infection of pigs with influenza C virus. J Gen Virol. 1983;64(Pt 1):177–182. doi: 10.1099/0022-1317-64-1-177. [DOI] [PubMed] [Google Scholar]
- 10.Hause BM, Ducatez M, Collin EA, Ran Z, Liu R, Sheng Z, Armien A, Kaplan B, Chakravarty S, Hoppe AD, Webby RJ, Simonson RR, Li F. Isolation of a Novel Swine Influenza Virus from Oklahoma in 2011 Which Is Distantly Related to Human Influenza C Viruses. PLoS pathog. 2013;9:e1003176. doi: 10.1371/journal.ppat.1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He XJ, Zhou J, Bartlam M, Zhang RG, Ma JY, Lou ZY, Li XM, Li JJ, Joachimiak A, Zeng ZH, Ge RW, Rao ZH, Liu YF. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 12.Ito T, Kawaoka Y, Vines A, Ishikawa H, Asai T, Kida H. Continued circulation of reassortant H1N2 influenza viruses in pigs in Japan. Arch Virol. 1998;143:1773–1782. doi: 10.1007/s007050050415. [DOI] [PubMed] [Google Scholar]
- 13.Kall L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 14.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 15.Osterhaus ADME, Rimmelzwaan GF, Martina BEE, Bestebroer TM, Fouchier RAM. Influenza B virus in seals. Science. 2000;288:1051–1053. doi: 10.1126/science.288.5468.1051. [DOI] [PubMed] [Google Scholar]
- 16.Palese PSM. Orthomyxoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1647–1690. [Google Scholar]
- 17.Pease CM. An evolutionary epidemiological mechanism, with applications to type A influenza. Theor Popul Biol. 1987;31:422–452. doi: 10.1016/0040-5809(87)90014-1. [DOI] [PubMed] [Google Scholar]
- 18.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 19.Poeppl W, Hell M, Herkner H, Stoiser B, Fritsche G, Schurz-Bamieh N, Poeppl G, Gattringer R, Jones N, Maass M, Egle A, Burgmann H. Clinical aspects of 2009 pandemic influenza A (H1N1) virus infection in Austria. Infection. 2011;39:341–352. doi: 10.1007/s15010-011-0121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006;23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- 21.Schulze IT. Effects of glycosylation on the properties and functions of influenza virus hemagglutinin. J Infect Dis. 1997;176(Suppl 1):S24–S28. doi: 10.1086/514170. [DOI] [PubMed] [Google Scholar]
- 22.Sun SS, Wang QZ, Zhao F, Chen WT, Li Z. Glycosylation Site Alteration in the Evolution of Influenza A (H1N1) Viruses. PLoS One. 2011;6(7):e22844. doi: 10.1371/journal.pone.0022844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szepanski S, Veit M, Pleschka S, Klenk HD, Schmidt MF, Herrler G. Post-translational folding of the influenza C virus glycoprotein HEF: defective processing in cells expressing the cloned gene. J Gen Virol. 1994;75(Pt 5):1023–1030. doi: 10.1099/0022-1317-75-5-1023. [DOI] [PubMed] [Google Scholar]
- 24.Takashita E, Muraki Y, Sugawara K, Asao H, Nishimura H, Suzuki K, Tsuji T, Hongo S, Ohara Y, Kawaoka Y, Ozawa M, Matsuzaki Y. Intrinsic temperature sensitivity of influenza C virus hemagglutinin-esterase-fusion protein. J Virol. 2012;86:13108–13111. doi: 10.1128/JVI.01925-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarendeau F, Boudet J, Guilligay D, Mas PJ, Bougault CM, Boulo S, Baudin F, Ruigrok RWH, Daigle N, Ellenberg J, Cusack S, Simorre JP, Hart DJ. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat Struct Mol Biol. 2007;14:229–233. doi: 10.1038/nsmb1212. [DOI] [PubMed] [Google Scholar]
- 27.Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu X, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci USA. 2012;109(11):4269–4274. doi: 10.1073/pnas.1116200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita M, Krystal M, Fitch WM, Palese P. Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology. 1988;163:112–122. doi: 10.1016/0042-6822(88)90238-3. [DOI] [PubMed] [Google Scholar]
- 30.Yuan PW, Bartlam M, Lou ZY, Chen SD, Zhou J, He XJ, Lv ZY, Ge RW, Li XM, Deng T, Fodor E, Rao ZH, Liu YF. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.