

Commentary
A Deleterious Nav1.1 Mutation Selectively Impairs Telencephalic Inhibitory Neurons Derived From Dravet Syndrome Patients.
Sun Y, Pasca, SP, Portmann, T, Goold C, Worringer KA, Guan W, Chan KC, Gai H, Vogt D, Madison DV, Hallmayer J, Froehlich-Santino WM, Bernstein JA, Dolmetsch RE. eLife 2016;5:e13073.27458797
Dravet Syndrome is an intractable form of childhood epilepsy associated with deleterious mutations in SCN1A, the gene encoding neuronal sodium channel Nav1.1. Earlier studies using human induced pluripotent stem cells (iPSCs) have produced mixed results regarding the importance of Nav1.1 in human inhibitory versus excitatory neurons. We studied a Nav1.1 mutation (p.S1328P) identified in a pair of twins with Dravet Syndrome and generated iPSC-derived neurons from these patients. Characterization of the mutant channel revealed a decrease in current amplitude and hypersensitivity to steady-state inactivation. We then differentiated Dravet-Syndrome and control iPSCs into telencephalic excitatory neurons or medial ganglionic eminence (MGE)-like inhibitory neurons. Dravet inhibitory neurons showed deficits in sodium currents and action potential firing, which were rescued by a Nav1.1 transgene, whereas Dravet excitatory neurons were normal. Our study identifies biophysical impairments underlying a deleterious Nav1.1 mutation and supports the hypothesis that Dravet Syndrome arises from defective inhibitory neurons.
Dravet syndrome (DS) is a catastrophic pediatric epileptic encephalopathy with cognitive, behavioral, and motor impairments, as well as a high risk of sudden unexpected death in epilepsy (SUDEP). The majority of DS patients have de novo mutations in SCN1A, the gene encoding the a subunit of the voltage-gated sodium channel NaV1.1, that result in haploinsufficiency (1). While the field has made key advances toward the goal of understanding how reduced expression of a major brain sodium channel gene results in hyperexcitability and network synchrony, critical gaps remain. Importantly, what is the best experimental model of DS and how do we interpret seemingly conflicting results between and within model systems?
The first model of DS was developed by the Catterall group (2). Scn1a deletion in mice resulted in a severe seizure phenotype in heterozygous animals and was the first in vivo confirmation that Scn1a haploinsufficiency resulted in seizures. In this study, postnatal day (P) 14–16 Scn1a+/− bipolar neurons—but not pyramidal neurons—had significantly reduced sodium current density, resulting in reduced inter-neuron firing frequency. Further work showed that Dlx1/2 Cre-mediated inactivation of Scn1a in parvalbumin-positive (PV+) interneurons phenocopied the severe seizures and SUDEP (3). Based on these results, it was hypothesized that pathophysiology in DS could be explained by disinhibition, or dysfunctional inhibitory circuits, that involve PV+ fast-spiking interneurons. Kearney and George extended this work to demonstrate, first, that genetic background is key to the severity of pathophysiology in DS mouse models, and, second, that changes in sodium current density are dependent on the stage of brain development in mice. They reported that sodium current density was elevated in Scn1a+/− pyramidal neurons, but not GABAergic neurons, at P21–24. Moreover, pyramidal neurons isolated from Scn1a+/− mice on the more phenotypically severe background strain displayed a larger increase in sodium current density than pyramidal neurons from the less severe strain. Scn1a+/− P21–24 pyramidal neurons from either strain exhibited spontaneous firing and hyperexcitability. To add further complexity, more recent results demonstrated that, while interneuron hypoexcitability is observed in Scn1a+/− mouse brain slices, it is not observed in Scn1a−/+ DS mice during spontaneous activities in vivo, suggesting that alternative mechanisms must be considered (4).
Clearly, transgenic mouse models are critical to understanding the mechanisms of neurological disease. However, mice are not small humans, and there are important differences between mouse and human brain development (5, 6). To understand the mechanism of DS in human brain, a number of groups set out to model DS using patient-derived induced pluripotent stem cell (iPSC) neurons. This remarkable technique allows the study of DS mutations in native cells in the context of the patient's unique genetic background. In addition, iPSCs have the potential to generate all human neuronal subpopulations. In spite of this, results obtained by different groups using this powerful technique are as inconsistent as those gleaned from the mouse models. Table 1 summarizes DS patient iPSC neuron results to date. Similar to the mouse results, changes in neuronal excitability and sodium current density appear to be dependent on a number of critical factors that likely include the patient's genetic background, the specific mutation tested, the method of differentiation, and the developmental maturity of the neurons.
TABLE 1.
Summary of DS Patient Derived iPSC Neuron Phenotypes
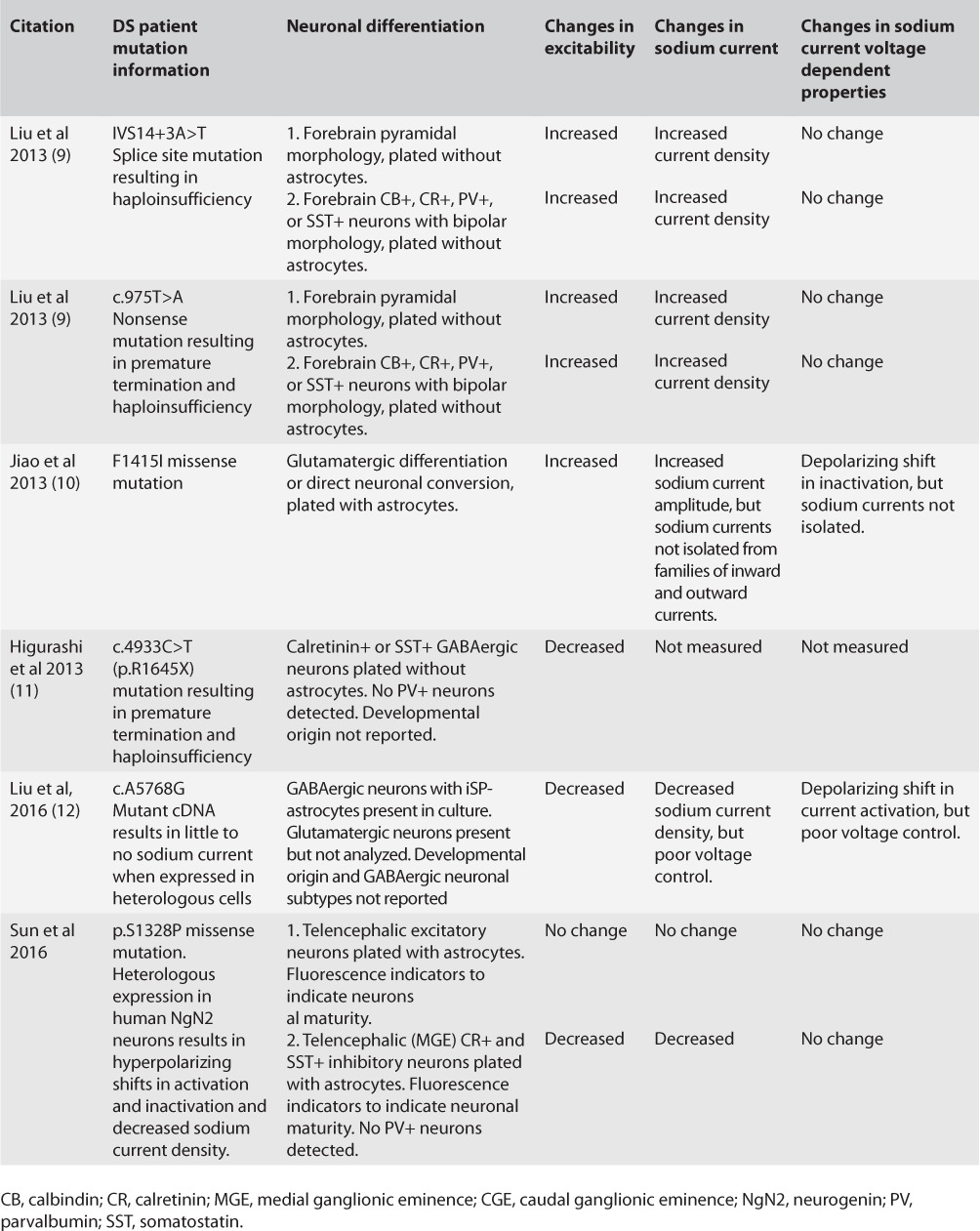
Importantly, interpreting the significance of electrophysiological recordings from single iPSC interneurons in terms of network excitability requires a detailed understanding of interneuron identity. Our knowledge of interneuron diversity and origins has significantly advanced since the development of the first DS mouse model. GABAergic interneurons have been classified into a variety of subpopulations with distinct embryonic origins, targets, and functions (7). For example, of the telencephalic GABAergic interneurons studied by Sun et al., 50% were calretinin+ (CR+), with a small percentage expressing somatostatin (SST). Importantly, none of the neurons were PV+. Action potential firing properties shown for these neurons are consistent with a CR+ regular-spiking nonpyramidal cell designation. These data suggest that the differentiation protocol may have enriched for GABAergic CR+ interneurons with a caudal (CGE), rather than medial (MGE), ganglionic eminence origin. MGE-derived interneurons are primarily PV+ and SST+, with a minor population of CR+ (7). CR+ interneurons, which are primarily derived from the CGE, are considered to be interneuron-specific interneurons (8). Thus, reduced sodium current density and reduced firing in these interneurons would be predicted to functionally result in the loss of inhibitory tone onto other inhibitory neurons, allowing for an overall increase in inhibition of excitatory neurons. This would lead to decreased network excitability and a decreased propensity toward seizures—the opposite of DS—and would be inconsistent with mouse data demonstrating that Cre-mediated inactivation of Scn1a in PV+ interneurons, which primarily target excitatory neurons, results in epilepsy (3).
Neuronal maturity plays a factor in iPSC neuron electro-physiology results. iPSC neurons described in earlier work (9) were less mature and had smaller sodium current densities than reported in more recent papers using newer differentiation protocols. Also, in contrast to the Sun et al. paper, the 2013 Liu et al. study did not include astrocytes, which enhance neuronal maturity. While sodium current densities reported in the Sun et al. study are more in line with results expected from acutely dissociated mouse brain neurons, they may not have been fully mature. The firing rates reported for interneurons and excitatory neurons in this study appear to be similar, while fully mature interneurons would be expected to have higher firing frequencies.
In spite of the caveats and controversies associated with the use of iPSC neurons, all of the studies described in Table 1 provide important information regarding DS mechanisms. It is reasonable to propose that a pediatric epileptic encepha-lopathy like DS may involve differential excitability changes in diverse neuronal subpopulations within a given network at multiple developmental time points. Each study may capture specific neuronal subpopulations at key stages of brain development. The immaturity of iPSC neurons may be useful for understanding the effects of sodium channel mutations on the prenatal brain. For example, during critical early developmental periods when GABAergic signaling results in neuronal depolarization rather than hyperpolarization, the interpretation of changes in interneuron and pyramidal sodium current density in terms of neuronal excitability and network synchrony takes on a distinct perspective. It is interesting to consider that the functional effects of DS mutations on sodium channels may change over the lifetime of a given neuron, depending on the expression time course of other channels, receptors, and signaling molecules. Mutant sodium channels may also differentially affect excitability in subcellular compartments, for instance, axon initial segment, cell body, or dendrites, of the same neuron.
In conclusion, the iPSC field is moving at lightning speed. Emerging methods to generate MGE-derived PV+ fast-spiking interneurons, brain organoids, and cortical spheroids will allow sophisticated analyses of specific neuronal subpopulations and network properties. Continued advancement of this fascinating technology will be essential to discovering epilepsy cures through precision medicine.
References
- 1. Marini C, Scheffer IE, Nabbout R, Suls A, De Jonghe P, Zara F, Guerrini R.. The genetics of Dravet syndrome. Epilepsia 2011; 52 suppl 2: 24– 29. [DOI] [PubMed] [Google Scholar]
- 2. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA.. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006; 9: 1142– 1149. [DOI] [PubMed] [Google Scholar]
- 3. Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA.. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U.S.A. 2012; 109: 14646– 14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Stasi AM, Farisello P, Marcon I, Cavallari S, Forli A, Vecchia D, Losi G, Mantegazza M, Panzeri S, Carmignoto G, Bacci A, Fellin T.. Unaltered network activity and interneuronal firing during spontaneous cortical dynamics in vivo in a mouse model of severe myoclonic epilepsy of infancy. Cereb Cortex 2016; 26: 1778– 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parent JM, Anderson SA.. Reprogramming patient-derived cells to study the epilepsies. Nat Neurosci 2015; 18: 360– 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Du X, Parent JM.. Using patient-derived induced pluripotent stem cells to model and treat epilepsies. Curr Neurol Neurosci Rep 2015; 15: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wamsley B, Fishell G.. Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat Rev Neurosci 2017; 18: 299– 309. [DOI] [PubMed] [Google Scholar]
- 8. Cauli B, Zhou X, Tricoire L, Toussay X, Staiger JF.. Revisiting enigmatic cortical calretinin-expressing interneurons. Front Neuroanat 2014; 8: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O'Malley HA, Patino GA, O'Brien JE, Rusconi R, Gupta A, Thompson RC, Natowicz MR, Meisler MH, Isom LL, Parent JM.. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol 2013; 74: 128– 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan Y, Yao H, Liao W, Sun XF, Gao S.. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet 2013; 22: 4241– 4252. [DOI] [PubMed] [Google Scholar]
- 11. Higurashi N, Uchida T, Lossin C, Misumi Y, Okada Y, Akamatsu W, Imaizumi Y, Zhang B, Nabeshima K, Mori MX, Katsurabayashi S, Shirasaka Y, Okano H, Hirose S.. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol Brain 2013; 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu J, Gao C, Chen W, Ma W, Li X, Shi Y, Zhang H, Zhang L, Long Y, Xu H, Guo X, Deng S, Yan X, Yu D, Pan G, Chen Y, Lai L, Liao W, Li Z.. CRISPR/Cas9 facilitates investigation of neural circuit disease using human iPSCs: Mechanism of epilepsy caused by an SCN1A loss-of-function mutation. Transl Psychiatry 2016; 6: e703. [DOI] [PMC free article] [PubMed] [Google Scholar]