

Abstract
Cardiovascular disease, predominantly ischemic heart disease (IHD), is the leading cause of death in diabetes mellitus (DM). In addition to eliciting cardiomyopathy, DM induces a ‘wicked triumvirate’: (i) increasing the risk and incidence of IHD and myocardial ischemia; (ii) decreasing myocardial tolerance to ischemia–reperfusion (I–R) injury; and (iii) inhibiting or eliminating responses to cardioprotective stimuli. Changes in ischemic tolerance and cardioprotective signaling may contribute to substantially higher mortality and morbidity following ischemic insult in DM patients. Among the diverse mechanisms implicated in diabetic impairment of ischemic tolerance and cardioprotection, changes in sarcolemmal makeup may play an overarching role and are considered in detail in the current review. Observations predominantly in animal models reveal DM-dependent changes in membrane lipid composition (cholesterol and triglyceride accumulation, fatty acid saturation vs. reduced desaturation, phospholipid remodeling) that contribute to modulation of caveolar domains, gap junctions and T-tubules. These modifications influence sarcolemmal biophysical properties, receptor and phospholipid signaling, ion channel and transporter functions, contributing to contractile and electrophysiological dysfunction, cardiomyopathy, ischemic intolerance and suppression of protective signaling. A better understanding of these sarcolemmal abnormalities in types I and II DM (T1DM, T2DM) can inform approaches to limiting cardiomyopathy, associated IHD and their consequences. Key knowledge gaps include details of sarcolemmal changes in models of T2DM, temporal patterns of lipid, microdomain and T-tubule changes during disease development, and the precise impacts of these diverse sarcolemmal modifications. Importantly, exercise, dietary, pharmacological and gene approaches have potential for improving sarcolemmal makeup, and thus myocyte function and stress-resistance in this ubiquitous metabolic disorder.
Keywords: Caveolae, Cardioprotection, Cholesterol, Diabetes, Fatty acids, Glucose transport, Infarction, Phospholipids
Diabetes impacts myocardial ischemic tolerance and cardioprotection
Clinical evidence indicates DM sensitizes human hearts to I–R injury [1, 2], which is generally consistent with experimental findings in animal models, though conflicting observations arise. Compounding the problem of infarct intolerance, DM may also render hearts broadly refractory to established cardioprotective stimuli that include ischemic pre- and post-conditioning (direct or remote) and protective G protein-coupled receptor (GPCR) agonism, together with the anti-infarct effects of ATP-gated K+ channel (KATP) openers, anesthetics, phosphodiesterase-5 (PDE-5) inhibition and heat shock activation [3–6]. Thus, while elusive cardioprotective therapies [6–8] are of particular value in the high-risk DM population, implementation appears an even greater challenge in this cohort. Prevalence of DM and insulin-resistance in those suffering IHD may in turn contribute to poor translation of experimental cardioprotection in these patients. Relatively few studies specifically address the conundrum of I–R sensitivity and cardioprotective insensitivity in DM [6]. Investigations to date implicate a diversity of mechanisms extending beyond fundamental alterations in glucose and lipid metabolism, including associated glycation/glycosylation [9], oxidative stress [10, 11], abnormal survival kinase signaling [12–14] and exosome dysfunction [15], excessive ubiquitin–proteasome system activity [16], suppression of sirtuin-1 expression [17], and changes in miRNA expression [18], among others. Considerable attention has focused on mitochondrial dysfunction, including shifts in quality control mechanisms (mitophagy, fission/fusion), as a point of convergence in the complex pathogenesis of diabetic cardiomyopathy [10, 19, 20]. However, the sarcolemma is also a critical though under-appreciated nexus, influencing DM progression and its impacts [21]. Indeed, transcriptomic profiling indicates that the largest group of diabetes-modified cardiac genes encode membrane/plasma membrane components [22], consistent with more recent studies identifying DM-dependent changes in transcripts for membrane and structural proteins, sarcolemmal receptors and ion channels [23]. Transporters for glucose and fatty acids, ion channels and exchangers, and receptor systems governing insulin responses, inflammation, mitochondrial quality control, and cell stress, growth and death are all located within the sarcolemma, while mitochondrial function is also sensitive to sarcolemmal domains and proteins. Perturbations in membrane composition and architecture may thus be critical to the dysfunctional stress responses characteristic of diabetic myocardium, together with other cardiac outcomes including hypertrophy and contractile dysfunction. We herein review the clinical and experimental evidence of DM-dependent changes in myocardial ischemic tolerance and cardioprotection, before focusing specifically on sarcolemmal changes and their contribution to the cardiac sequelae of DM.
Effects of DM in human myocardium
Diabetes induces a spectrum of abnormalities within the myocardium and coronary vasculature. Diastolic dysfunction, fibrosis and hypertrophy functionally and structurally underpin diabetic cardiomyopathy [10]. These changes are linked to reactive oxygen species (ROS) generation, inflammation, mitochondrial dysfunction, and abnormalities in molecular quality control, including autophagy, fission/fusion, endoplasmic reticulum (ER) stress and unfolded protein responses. Coronary endothelial dysfunction and vascular remodeling exaggerate atherosclerosis and impair vascular control and coronary perfusion, potentially contributing to cardiac dysfunction. These changes in myocardial and coronary phenotypes (and underlying molecular mechanisms) may participate in impairment of myocardial stress tolerance, hormesis and protective signaling, which may in turn further exacerbate these phenotypic changes.
Myocardial ischemic tolerance
The impacts of DM on myocardial ischemic tolerance and infarction remain somewhat contentious. Certainly DM worsens long-term outcomes from ischemic insult, including increased incidence of heart failure and all-cause mortality [24–26]. There is some evidence these poor outcomes may involve diabetic impairment of myocardial reperfusion [27, 28], consistent with vascular dysfunction and reduced coronary reserve [29–31]. The contribution of worsened infarction to poor post-ischemic prognosis awaits further clarification, with some contrasting data acquired. Diabetes can significantly increase infarct size as assessed via scintigraphy [1, 32] and magnetic resonance imaging (MRI) [33]. Insulin-treated DM patients also exhibit worsened myocardial infarction, mortality, major adverse cardiac events and thrombosis compared with untreated or non-DM subjects, potentially reflecting negative impacts of more complex and prolonged disease [34–36]. On the other hand, some myocardial scintigraphic [37] and MRI analyses [38, 39] report no significant differences in infarction in DM vs. non-DM STEMI patients post angioplasty.
Other evidence strongly supports exaggerated myocardial damage and cell death in DM patients: DM markedly increases morbidity and mortality (up to 90%) following cardioplegic arrest [40–42]; DM promotes pro-apoptotic signaling, apoptosis and contractile dysfunction in reperfused human myocardium [43–45]; and DM exaggerates oxidative damage and anti-oxidant depletion [45, 46], transcriptional changes and pro-inflammatory signaling [45, 47]. Analysis of I–R injury in ex vivo tissue reveals significantly impaired resistance of myocardium from T1 and T2DM patients, including increased apoptosis (partially caspase- and PARP-dependent) and oncosis [48]. Anderson et al. [49] more recently provided evidence that myocardium from DM patients has a greater propensity for mitochondria-dependent cell death. There is also evidence of exaggeration of post-ischemic contractile dysfunction in DM: the studies of Hoogslag et al. [50] and Dimitriu-Leen et al. [51] reveal worsened myocardial longitudinal strain independently of infarct size, supporting greater mechanical disruption in DM. Hyperglycemia itself has been shown to increase infarct size and mortality in infarct patients [1, 28, 52–54]. This may also involve impaired reperfusion, though there is evidence hyperglycemia exaggerates infarction by increasing the area at risk [55]. Use of insulin and sulfonylureas to manage hyperglycemia may additionally worsen ischemic injury, morbidity and mortality [34–36, 56, 57].
Conversely, there is some limited evidence myocyte ischemic tolerance might be increased in T1DM patients, though for skeletal and not cardiac tissue [58]. This is consistent with some rodent studies in acute T1DM models (see below). Nonetheless, the weight of experimental evidence supports reduced myocardial I–R tolerance in DM, encompassing exaggerated apoptosis, oncosis and infarction, contractile dysfunction and markers of oxidative damage. It remains unclear to what extent poor post-ischemic prognosis reflects exaggerated ischemic insult, impaired reperfusion, and increased propensity to cell death. The roles of individual metabolic disturbances (hyperglycemia, hyperinsulinemia, insulin-resistance, dyslipidemia), coronary dysfunction and compromised reflow, together with intrinsic myocardial stress-resistance, thus require further detailed analysis.
Cardioprotection
Studies broadly support the desensitization or elimination of diverse cardioprotective responses in DM myocardium, though again this is not universal. There are relatively few studies of diabetic impacts on cardioprotective responses in human myocardium. Ishihara et al. [59] reported that DM inhibits ischemic preconditioning in anterior wall infarct patients, while Lee et al. [4] present evidence of impaired preconditioning responses in DM patients undergoing angioplasty. Galiñanes and colleagues found that ex vivo myocardium from DM patients was insensitive to ischemic preconditioning [3], and subsequently identified loss of responsiveness not only to preconditioning but to phenylephrine, adenosine and diazoxide (implicating signal dysfunction proximal to protein kinase C (PKC) and p38 mitogen-activated protein kinase (MAPK) [5]. More recent studies support desensitization of DM myocardium to hypoxic preconditioning in association with impaired phosphatidylinositol 3 kinase (PI3K) and Akt signaling [60], and failure of ischemic preconditioning in myocardium from DM patients [61]. On the other hand, some studies confirm protective efficacies of anesthetic post-conditioning in ex vivo myocardium [62, 63] and of ischemic preconditioning in vivo [64] in DM patients. Additionally, a meta-analysis assessing influences of risk factor across ten trials of post-conditioning in STEMI [65] verified significant interactions with age and sex (reduced efficacy in older and/or female patients) yet not with DM. The authors concede analytical limitations may lead to an under-estimation of the influences of co-morbidities such as DM. A subsequent focused albeit smaller analysis also failed to identify interaction between DM and post-conditioning in STEMI patients [66], though also failed to detect the sex and age effects revealed by Zhou et al., highlighting limited power to detect effects in small sample sizes via posteriori statistical analysis.
Complicating effects of anti-hyperglycemia therapies
In addition to the underlying DM cardio-pathology, there is evidence clinical approaches to managing hyperglycemia may impair cardioprotective signaling and worsen ischemic outcomes. Sulfonylurea use is associated with greater ischemic injury and infarction in DM [56, 57], and inhibition of ischemic preconditioning in both non-DM and DM patients [67, 68] and ex vivo myocardium from DM patients [69]. Glinide also impairs preconditioning in DM patients [70, 71]. These negative impacts are consistent with their ability to inhibit KATP channels implicated in transducing or mediating cardiac protection [72].
In addition, insulin treatment has been linked to a paradoxic worsening of complications, all-cause mortality and cardiac outcomes in DM [34, 35]. Concern regarding potentially untoward effects of glycemic control arose from epidemiological evidence of increased mortality in insulin-treated vs. untreated T2DM patients [73, 74], together with observations of insulin effects on cardiac events [75, 76] and mortality in heart failure complicated by T2DM [77]. Evidence of worsened outcomes with insulin and sulfonylureas over metformin [34] suggests direct insulin- and KATP channel dependent actions rather than simple glucose-lowering. However, whether involving direct effects of insulin, influences of acutely reduced glucose (or overt hypoglycemia) following chronic hyperglycemia, or the fact insulin-treated patients often exhibit greater comorbidities and suffer more protracted disease, awaits further clarification. There are potential mechanisms by which insulin might worsen cardiovascular outcomes despite normalization of glucose. For example, insulin can induce weight gain which can exaggerate cardiovascular (and also cancer) risks, while atherogenic and mitogenic effects may accelerate atherosclerosis/IHD. Moreover, there is evidence insulin treatment up-regulates pro-inflammatory tumor necrosis factor α and interleukin-1 to a greater extent in T1DM vs. healthy animals [78], and insulin-dependent NO generation may promote oxidative stress [79], together with vascular damage through increased circulatory pulsatility [80]. Hypoglycemia as a result of poor glycemic control may also increase arrhythmogenesis, cardiac events and mortality [81], though whether this reflects a causal relationship is unclear, with other studies reporting no association between hypoglycemia and cardiac or all-cause mortality in T2DM [82]. Conversely, there is evidence hyperglycemia can promote compensatory mechanisms that protect against I–R injury, including improvements in anti- vs. pro-oxidant balance and protein integrity [83], which might be countered by reductions in glucose levels. The hearts of diabetic patients do appear desensitized to the injurious effects of elevated glucose [38]. However, further work is needed in disentangling these complexities.
Effects of DM in animal and in vitro models
There are some conflicting reports regarding impacts of DM on myocardial infarction and cardioprotection in animal models. Reviewed previously [2, 84, 85], studies in different species and models report increases, no change, or reductions in infarct size with DM. Similarly, despite a substantial body of evidence supporting impaired protection via pre- or post-conditioning and GPCR agonists, some report preserved responses to similar stimuli [86, 87]. Reasons for these discrepancies are debated, though disease progression and the presence of dyslipidemia appear to be important. While infarct enlargement is observed across species and models of T1DM and T2DM [2], infarct reduction is predominantly identified in rodent models of acute streptozotocin (STZ) dependent hyperglycemia [2, 86, 88–90]. This may reflect distinct impacts of acute (0–6 week) vs. established or chronic disease. While some also report apparent cardioprotection in models of T2DM [91], this may similarly reflect distinct changes on early transition to T2DM vs. established disease [12, 92]. Presence or absence of dyslipidemia may also be important, with some evidence hypercholesterolemia has opposing effects on infarct tolerance compared with hyperglycemia alone [93]. Mechanisms implicated in differing ischemic tolerance in acute vs. chronic DM include shifts in PI3K/Akt [12, 94, 95] and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling [90], mitochondrial glucose oxidation and malate-aspartate shuttle function [92], and capillary density, vascular endothelial growth factor (VEGF) expression and endothelial nitric oxide synthase (eNOS) signaling [94]. Clinically, the negative impacts of chronic disease are most relevant regarding infarction and cardioprotection, with acute effects relevant only during transition to disease and potentially on cessation of therapy. Almost universally, observations support worsened myocardial ischemic tolerance in models of chronic T1DM or T2DM, with the weight of evidence supporting associated failure in diverse cardioprotective responses.
T1DM and infarction
A range of studies report worsened infarction in experimental models of T1DM [96–100] while some report no effect on infarct tolerance [6, 13, 101–112], or protection against both infarction [86, 88, 89, 113] and contractile dysfunction [114]. However, as alluded to above, a biphasic pattern may emerge in STZ-dependent rodent models with evidence of early protection followed by restoration or worsening of infarct tolerance beyond 6–8 weeks. Protection against infarction evident 1–4 weeks after STZ challenge is lost from 8 weeks [115], while reduced ischemic tolerance may emerge by 20 weeks [90] (in association with impaired ERK1/2 phospho-activation). Ma et al. [94] report that protection against infarction and caspase-3 activation in T1DM rats is transient, apparent at 2 weeks and lost by 6 weeks, in association with transient changes in capillary density, VEGF expression, Akt phosphorylation, and eNOS expression. Similarly, early protection against arrhythmogenesis at 2 weeks (with improved maintenance of Na+, Ca2+, K+ and Mg2+) transitions to worsened outcomes after 8 weeks in T1DM rats [116]. Acute hyperglycemia itself has been shown to worsen myocardial infarction [96, 102, 117–124], exert no effect [125–128], or less commonly to reduce infarction [129]. Reasons for these disparate observations are unclear, and together with the basis of apparently opposing effects of early vs. late hyperglycemia in rat models of T1DM, warrant further investigation.
T1DM and cardioprotection
Beyond a transient intrinsic protection in the early stages of STZ-induced hyperglycemia [2, 86, 88–90], studies report inhibition or complete loss of cardioprotective responses in rodent models of T1DM [91, 130–132]. Protective ‘conditioning’ responses negated or inhibited include ischemic pre- [110, 116, 133] and post-conditioning [102, 104, 106, 108, 134, 135], delayed protection with ischemic preconditioning [102], hyperoxic preconditioning [113], and remote post-conditioning [109]. Protective responses to pharmacological stimuli including anesthetic post-conditioning [101, 104, 111], ACE inhibition [108], opioid [103, 107, 112] and adenosine GPCR agonism [100], and adiponectin [135] and cytokine [13] receptor activation are also lost in T1DM. Przyklenk and colleagues [134] present evidence post-conditioning may actually exaggerate injury in the context of T1DM. Acute hyperglycemia also inhibits cardioprotective responses, blocking ischemic pre- [102, 120] and post-conditioning [121], remote ischemic perconditioning [127], anesthetic pre- [119] and post-conditioning [125, 128], together with glucose-insulin-potassium (GIK) protection [118]. Nonetheless, there are some reports of preserved protection in models of T1DM, including exercise [136] and ischemic preconditioning [86], while Potier et al. [108] identify a specific shift to protective efficacy of B2 bradykinin receptors in T2DM hearts (vs. B1 receptors in non-DM tissue). Atorvastatin is also reportedly cardioprotective in T1DM rats [137], involving a glycogen synthase kinase 3β (GSK3β) dependent protection linked to heat shock factor 1 and heat shock protein 70 (HSP70).
T2DM and infarction
Elements of T2DM individually modify infarct tolerance and cardioprotective signaling, including dyslipidemia [138–140], insulin-resistance and hyperglycemia [102, 118, 120–122]. Studies identify exaggerated infarction and contractile dysfunction in different models of T2DM [132, 141–146]. Nonetheless, there are also reports of unchanged infarct tolerance [87, 126, 134, 147–150] or reduced infarction in models of T2DM. The latter reductions are observed 5 days post STZ injection in high-fat fed rats (a protection negated by hypercholesterolemia) [93], and at 16 weeks in Zucker diabetic fatty (ZDF) and lean Goto-Kakizaki (GK) T2DM rats [91, 94]. As for T1DM, disease progression appears to be key, with evidence of a transient protection during disease onset that is lost with T2DM progression in GK rats [90], while infarct-intolerance also emerges with chronic T2DM in ZDF rats [92]. These latter studies link the evolution of infarct tolerance with established T2DM to shifts in Akt signaling, suppression of malate-aspartate shuttle proteins and impaired post-ischemic recovery of glucose oxidation.
T2DM and cardioprotection
Diverse cardioprotective responses are impaired or negated in models of T2DM, including loss of ischemic pre- [141] and post-conditioning [134, 142, 145], He-induced pre- and post-conditioning [147], and protection via anesthetic [49], erythropoietin [13, 132], adiponectin [151], and β3-adrenergic receptor activation [146]. Interestingly, and consistent with membrane dysfunction, T2DM abolishes the cardioprotective potential of human and rat exosomes [15]. Exosomes are ~ 100 nm lipid bilayer vesicle derivatives of endosomes that play a role in transmitting protective signals between cells and tissues [152]. Cardioprotection may involve exosomal HSP70-dependent activation of myocyte toll like receptor 4 and reperfusion injury salvage kinase (RISK) signaling [153]. Failure of exosomes to induce protection in DM appears to involve abnormal vesicle structure/function rather than impaired protective signaling since exosomes from healthy donors are protective [15]. While exosome size was unaltered, contents of CD81 and HSP70 were increased in DM. Given evidence of exosome involvement in endogenous protection, this dysfunction may contribute to impairment of both conditioning responses and intrinsic ischemic tolerance.
Conversely, there are reports of preserved cardioprotective responses in T2DM, including efficacy of far red/near infrared light [126], sphingosine-1-phosphate [87], peroxisome proliferator-activated receptor γ (PPARγ) activation [150], post-ischemic glutamate [154] and H2S preconditioning [155]. Exercise may also retain efficacy, improving ischemic tolerance in the hearts of T2DM (GK) rats [146, 156], and glycemic state and ischemic tolerance in obese mice subjected to 20 weeks of high-fat feeding [157]. Those cardioprotective modalities consistently preserved in different models of DM demand further focused study as potentially efficacious therapeutic candidates.
Summary
While somewhat contentious, studies of human and animal myocardium generally support detrimental effects of both T1 and T2DM on myocardial ischemic tolerance and cardioprotection (Table 1). Mechanistic interrogation supports a complex pathogenesis, including signaling dysfunction (e.g. impaired PI3K/Akt signaling) [5, 12–14, 60, 90, 94], and abnormalities in mitochondrial function and quality control [10, 19, 20], ubiquitin–proteasome system activity [16], oxidant/anti-oxidant systems [10, 11], and gene and miRNA expression [18]. Influencing many of these potential effector mechanisms, the sarcolemma plays an overarching role in governing ischemic tolerance and cardioprotection. Cardiac sarcolemmal changes arise in DM (see Table 2), reflecting altered lipid metabolism and incorporation, modification of resident lipids and proteins, and significant structural and functional remodeling of caveolae [158, 159], T-tubules [160, 161] and gap junctions [162]. Detailed further below, such changes modify the fundamental biophysical properties of the membrane, glucose and fatty acid utilization, ion channel function, propensity to membrane disruption, and signaling via the insulin receptor (InsR) and receptors governing cardiac stress, growth and death responses.
Table 1.
Changes in myocardial ischemic tolerance and cardioprotection in animal models of DM
| Species—model | Duration or age | Ischemic tolerance | Effect on cardioprotection | Ref. |
|---|---|---|---|---|
| Type 1 DM | ||||
| Mouse—STZ | 1 week | ⇓ | ⇓ RPostC | [109] |
| 2 week | ⇔ | ⇓ IPostC | [134] | |
| 4–5 week | ⇔ | ⇓ IPostC, ⇓ ACE inhibition | [108] | |
| Rat—STZ | 1 week | ⇑ | ⇓ HOPreC | [113] |
| 2 week | ⇑ | ⇔ IPreC | [116] | |
| 2 week | ⇔ | ⇓ Opioid | [107] | |
| 2 week | ⇔ | ⇓ Opioid | [112] | |
| 2 week | ⇔ | ⇓ Opioid | [298] | |
| 2 week | ⇓ | ⇓ Sevoflurane | [106] | |
| 4 week | ⇔ | ⇓ Erythropoietin | [13] | |
| 4 week | ⇓ | ⇓ APN, ⇓ IPostC | [135] | |
| 4–5 week | ⇓ | ⇓ IPostC, ⇓ Sevoflurane | [104] | |
| 6 week | ⇓ | ⇓ IPreC | [133] | |
| 6 week | ⇔ | ⇓ IPreC | [110] | |
| 8 week | ⇓ | ⇓ IPostC | [99] | |
| 8 week | ⇓ | ⇓ APN, ⇓ IPostC | [135] | |
| 8 week | ⇓ | ⇓ IPreC | [116] | |
| 8 week | ⇓ | ⇓ Adenosine | [90] | |
| 9 week | ⇔ | ⇓ Sevoflurane | [111] | |
| 12 week | ⇓ | ⇓ IPostC | [121] | |
| Unreported | ⇔ | ⇓ Opioid | [103] | |
| Dog—alloxan/STZ | 3 week | ⇔ | ⇓ Isoflurane | [101] |
| 3 week | ⇔ | ⇓ IPreC | [130] | |
| Rabbit—alloxan | 5–6 week | ⇔ | ⇓ LPreC | [102] |
| TYPE 2 DM | ||||
| Mouse—HFD | 8 week | ⇓ | ⇓APN | [151] |
| 12 week | ⇓ | ⇓β3-AR | [146] | |
| Mouse—ob/ob | 8–10 week old | ⇓ | ⇓IPreC | [142] |
| Mouse—db/db | 10–12 week old | ⇓ | ⇓IPostC | [145] |
| 12–14 week old | ⇔ | ⇓IPostC | [134] | |
| Unreported | ⇔ | ⇔Infra-red light | [126] | |
| 12 week old | Not tested | ⇔H2S PreC | [155] | |
| Rat—STZ/HFD | 6 week | ⇔ | ⇔S1P | [87] |
| Rat—HFD | 4 week | ⇔ | ⇔Erythropoietin | [13] |
| 8 week | ⇑ | ⇓Sevoflurane | [496] | |
| Rat—ZDF | 12 week old | ⇔ | ⇔Glutamate | [154] |
| 16 week old | ⇑ | ⇓IPreC | [91] | |
| Rat—ZO | 10–12 week old | ⇓ | ⇓IPreC, ⇓Diazoxide, ⇓HePreC | [141] |
| Rat—GK | 12 week old | ⇔ | ⇓PPAR | [150] |
| Rat—OLETF | 25–30 week old | ⇓ | ⇓Erythropoietin | [132] |
| Rat—mtFHH | 12–14 week old | ⇔ | ⇓Isoflurane | [149] |
HFD high fat diet, ZDF Zucker diabetic fatty, ZO Zucker obese, GK Goto-Kakizaki, OLETF Otsuka Long-Evans-Tokushima fatty, mtFHH T2D crossbreed with mtDNA from fawn hooded hypertensive rats, IPreC ischaemic preconditioning, IPostC ischaemic postconditioning, HOPreC hyperoxic preconditioning, HePreC helium preconditioning, LPreC ischaemic late preconditioning, RPreC remote preconditioning, S1P sphingosine-1-phosphate, APN adiponectin, β 3 -AR β3-adrenergic receptor, w weeks
Table 2.
Cardiac sarcolemmal composition changes in models of T1DM
| Sample | Chol | FFA | TRI | Phospholipid | Saturated FAs | Unsaturated FAs | Ref. |
|---|---|---|---|---|---|---|---|
| Heart | ⇑ | ⇔ ∑PL | [191] | ||||
| Ventricle | ⇑ | ⇑ | [192] | ||||
| Ventricle | ⇑ LPC ⇓ PE, CL ⇔ PI, PS |
[196] | |||||
| Heart | ⇓ 20:4, 22:4, 22:5 ⇑ 18:2, 20:3, 20:5 |
[197] | |||||
| Heart | ⇑ | ⇔ PC ⇔ PE |
(PC) ⇓16:0 ⇑18:0 | (PC) ⇓ 20:4, ⇑ 18:2 (PE) ⇓ 20:4 |
[193] | ||
| Heart | (PE) ⇓ 18:0 (PC) ⇓ 16:0 |
⇓ 22:4 (PE) | [201] | ||||
| Heart | ⇔ CGP ⇔ EGP |
(CGP) ⇓ 16:0 | (CGP) ⇓ 20:4, ⇑ 18:2 (EGP) ⇑ 18:2 |
[200] | |||
| Ventricle | ⇑ EGP | (CGP, EGP) ⇓ 16:0 (EGP) ⇓ 18:0 |
(CGP, EGP) ⇓ 22:6, 20:4 ⇑ 18:2 |
[360] | |||
| Heart | ⇑ EGP, PME, PI | (EGP) ⇑ 18:0, 16:0 (TRI, NEFA) ⇑ 16:0 |
(EGP) ⇑ 18:2 (TRI) ⇑ 18:1, ⇓ 18:2 |
[202] | |||
| Sarcol-emma | ⇑ | ⇑ ∑PL, CGP, EGP, SGP ⇓ SM |
(PC) ⇑ 16:0 (PMC) ⇑ 18:0 (PE) ⇓ 16:0 (PME) ⇓ 18:0, ⇑ 16:0 (PS) ⇑ 16:0, ⇓ 18:0 |
(PC) ⇓ 20:4, ⇑ 18:2, 18:3 (PMC) ⇑ 18:2, ⇓ 20:4 (PE) ⇓ 20:4 (PME) ⇑ 18:2, ⇓ 20:4 (PS) ⇓ 22:6, 20:4, ⇑ C18:1 |
[199] | ||
| Heart | ⇑ | ⇑ | ⇑ | ⇑ ∑PL | [194] | ||
| Heart | ⇑ | ⇑ | ⇑ | ⇑ PE, SM, LPL ⇓ PC, PI + PS |
⇑ ∑Sat FA | ⇓ ∑Unsat FA, ⇓ ∑n − 3, ⇓ ∑n − 6 | [195] |
Changes (up or down) in levels of myocardial or sarcolemmal lipids in models of T1DM are summarized. Sarcolemmal lipid changes are not well defined in models of T2DM. Changes in specific saturated and unsaturated fatty acids species are indicated, with shortened numerical descriptions reflecting numbers of carbons and double bonds (e.g. palmitic acid, 16:0; stearic acid, 18:0; linoleic acid, 18:2; docosahexaenoic acid, 22:6)
CGP choline glycerophospholipids, EGP ethanolamine glycerophospholipids, IGP inositol glycerophospholipids, Chol cholesterol, CL cardiolipin, FAs fatty acids, FFA free fatty acid, LPC lysophosphatidylcholine, LPL lysophospholipid, NEFA non-esterified fatty acid, PC phosphatidylcholine, PE phosphatidylethanolamine, PI phosphatidylinositol, PL phospholipid, PMC plasmenylcholine, PME plasmenylethanolamine, PS phosphatidylserine, SGP serine glycerophospholipids, SM sphingomyelin, TRI triglyceride
Sarcolemmal changes in DM
Though research has largely focused on intracellular and metabolic determinants of cardiac stress responses in DM, the sarcolemma plays a key role in governing these and other changes and warrants further research attention [21]. The sarcolemma represents the myocytes structural bounds, and is the primary environmental and inter-cellular interface; a scaffold for ion channel, receptor, transport and mechano-transduction complexes, and medium for detection of intra- and extra-cellular stressors. It is thus intimately involved in receptor signaling, ion homeostasis, substrate delivery, inflammatory and immune function, and detection and transduction of physico-chemical changes. As the site of glucose and fatty acid uptake and InsR signaling, the sarcolemma and its microdomains are a fundamental substrate for the metabolic dysregulation in DM. Molecular modification and disruption of the sarcolemma can thus contribute to multiple aspects of myocardial dysfunction and pathology in DM.
Structurally the sarcolemma is a dynamic fluid bilayer of phospholipids, comprising complex assemblies of proteins, cholesterol and other lipids (Fig. 1a). Within this lipid sea float organized clusters of sphingolipids and cholesterol that form distinct microdomains known as lipid rafts. An important sub-set of these rafts, the caveolae are small invaginations (50–100 nm in diameter) that appear particularly relevant in DM and its cardiac sequelae [158, 159, 163]. Among other functions these ‘little caves’ serve as structural and regulatory platforms for receptor, ion channel and transporter proteins [164–166]; participate in mechanotransduction, protection against disruption and regulation of membrane repair [167]; and govern cardioprotective signaling [168–170]. Lipid rafts can also serve as redox signaling platforms that recruit and assemble nicotinamide adenine dinucleotide (NADPH) oxidase subunits and related proteins [171, 172]. The functional properties of the sarcolemma and its microdomains are governed by molecular composition, which is sensitive to diet, physical activity, genetic makeup and disease, and is significantly disturbed in DM (Fig. 1b, Table 2).
Fig. 1.
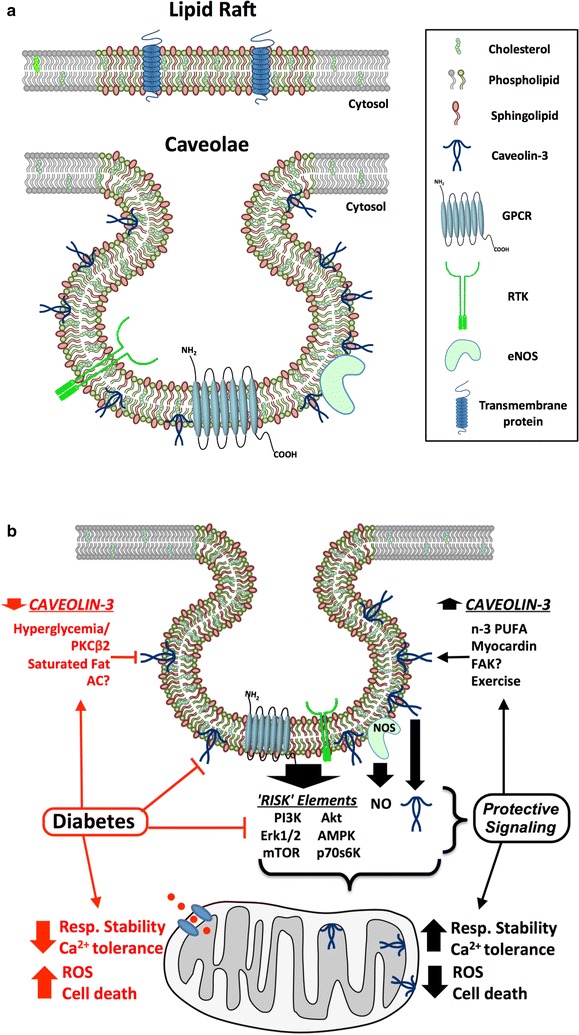
a Sarcolemmal makeup and caveolar domains. Planar lipid rafts are more ordered elements of the sarcolemma, containing greater sphingolipid and cholesterol levels and forming signaling microdomain platforms. A subset of rafts, caveolae, localize signaling integral to ischemic tolerance and cardioprotection, including NOS, GPCRs, RTKs and coupled effector molecules. Caveolins are critical to caveolae formation and function and protective signaling. b Modulation of caveolae/caveolins and related cardioprotective signaling in DM. Diabetes may exaggerate mitochondrial dysfunction and associated death, while individual elements of DM may disrupt caveolar control and caveolin expression: (i) hyperglycemia-dependent PKCβ2 activation may suppress caveolin-3 expression/localization; (ii) saturated fats (e.g. palmitate) may displace or depress caveolin-3. Disruption of caveolar control and caveolins will limit protective signaling to mitochondria, including caveolin-3 translocation/modulation. Potential determinants of caveolin-3 expression and caveolar function include PKCβ2, saturated fats vs. n-3 PUFAs, AC (adenylate cyclase) and FAK (focal adhesion kinase) signaling, myocardin activity and physical activity
Changes in sarcolemmal lipid profiles and function in DM
The biophysical properties of the membrane determine resident protein conformation, mobility and function. Fluidity or viscosity governs molecular motion and interactions within this lipid bilayer, thereby influencing the functionality of receptors, transporters and ion channels [173]. Fluidity is determined by lipid makeup, including: the tightness of acyl-chain assembly on phospholipid molecules; degree of phospholipid saturation; and local ratios of cholesterol, lipids and proteins. Membranes rich in cholesterol and tightly packed acyl chains possess greater rigidity, impacting movement and interaction of receptors and other molecules. Changes in sarcolemmal biophysics as a result of altered lipid metabolism do appear causally important in DM [174]: InsR signaling is inhibited by reductions in membrane fluidity, and both glucose transporter type 4 (GLUT-4) transport to the membrane [21, 174] and glucose uptake [175–177] parallel membrane fluidity changes. Analysis of hepatic cells supports causal involvement of sphingomyelin-dependent lipid microdomain changes in insulin-resistance and T2DM [178]. Inflammation, important in DM and cardiovascular dysfunction, is also promoted by abnormalities in membrane phospholipid and polyunsaturated fatty acid (PUFA) composition [179–181]. Positive feedbacks arise, whereby effects of DM and dyslipidemia on sarcolemmal signaling evoke further lipid accumulation and membrane dysfunction. For example, up-regulation of G protein-coupled receptor kinase 2 (GRK-2) with DM or high-fat feeding inhibits GPCR and InsR signaling, promoting further lipid accumulation, insulin-resistance and hypertrophy [182], with recent evidence these changes in GRK-2 also inhibits cardioprotective opioid receptor signaling [183]. The fundamental biophysical properties of the membrane can thus strongly influence the development and pathological impacts of DM across cell types.
Initial studies in DM identified reduced erythrocytes membrane fluidity [184, 185], a change evident even in newly formed cells [186] and subsequently confirmed via different approaches in multiple cell types [21, 187, 188], including cardiomyocytes [189]. Reduced fluidity is broadly consistent with increased membrane content of lipids promoting rigidity, including cholesterol, sphingomyelin and saturated fatty acids [187, 190] (Table 2). It is nonetheless interesting to note that erythrocytes normally possess relatively little cholesterol and lack caveolins. Specific myocardial analyses support increased levels of cholesterol and fatty acid saturation vs. desaturation and differential changes in phospholipids and PUFAs, though studies are limited to models of T1DM/hyperglycemia.
In early work, Denton and Randle [191] found a twofold increase in myocardial glycerides (predominantly triglycerides) in alloxan-induced T1DM in rats without significant changes in phospholipid content, though sarcolemmal fractions were not specifically examined. Increased cardiac triglyceride were confirmed in T1DM rat hearts, together with elevations in free fatty acid levels [192–195]. Early study of phospholipid content revealed reductions in sarcolemmal cardiolipin and phosphatidylethanolamine (PE) vs. elevated lysophosphatidylcholine levels in T1DM hearts, while phosphatidylinositol (PI) and phosphatidylserine (PS) were unchanged [196]. Later analysis of phospholipid makeup in hearts from T1DM rats revealed phospholipid depletion of n-6 arachidonic acid (AA; C20:4), docosatetraenoic acid (C22:4) and docosapentaenoic acid (C22:5) species, whereas contents of n-6 linoleic (C18:2) and dihomo-γ-linolenic acids (C20:3) and n-3 eicosapentaenoic acid (C20:5) were increased [197]. Subsequent studies confirm phospholipid depletion of AA and also palmitic acid (16:0) vs. enrichment with linoleic and dihomo-γ-linolenic acids [193, 198–200] (Table 2), though Black et al. [201] found no change in phospholipid AA content in T1DM rat hearts (while phospholipid stearic acid and palmitate levels fell). Han et al. [202] reported three major sarcolemmal changes in T1DM rats: a reduced ratio of saturated:unsaturated PE species; increased PI and plasmenylethanolamine; and remodeling of triacylglycerol species. More recent analysis in alloxan-induced T1DM in rats confirms increased cardiac cholesterol, free fatty acids, triglycerides and lipid saturation, reduced de-saturation and n-3 and n-6 PUFA levels, and differential changes in phospholipids including increased PE, sphingomyelin and lysophospholipid vs. reduced phosphatidylcholine (PC) and PI + PS [194, 195]. Collectively, these studies support cholesterol, triglyceride and free fatty acid accumulation, increased saturation vs. desaturation, and remodeling of the major choline and ethanolamine phospholipids, with loss of AA and accumulation of linoleic acid and dihomo-γ-linolenic species (Table 2).
Shifts in cholesterol and fatty acid saturation are important to changes in membrane biophysics. Membrane fluidity is particularly dependent upon cholesterol content, which is consistently increased in models of T1DM [194, 195], in association with reduced fluidity and Ca2+ influx [173, 203]. Indeed, membrane cholesterol changes are likely to contribute in multiple cardiovascular disorders [204]. Cholesterol molecules provide structural support, function as molecular ‘glue’ for lipid raft assembly, promote curvature of the membrane [205], and are important to caveolae formation [206]. Control of cholesterol is therefore essential to maintenance of membrane architecture, fluidity and microdomain formation. Shifts in the degree of fatty acid saturation also influence fluidity, with DM consistently increasing cardiac fatty acid levels and saturation vs. desaturation [193, 198]. Changes in phospholipid profiles additionally alter fundamental biophysical properties [207], together with protein activity [208, 209], recruitment of signal proteins [210], propensity for fusion [211] and production of lipid second messengers [209, 212]. Sarcolemmal phospholipids may also undergo relevant post-translation modification in DM, for example altered N-methylation [213] may alter cardiac sarcolemmal Ca2+ fluxes [214, 215].
Polyunsaturated fatty acids exert complex effects on membrane structure and function [216]. For example, n-3 PUFAs such as eicosapentaenoic (EPA) and docosahexaenoic acid (DHA) remodel cholesterol-enriched lipid microdomains, with evidence their incorporation increases molecular order (despite their high disorder). Differences in lipid microdomain interactions of EPA and DHA may lead to differential changes in ‘bioactivity’ [216]. Both in vitro [217, 218] and in vivo studies [219, 220] indicate n-3 PUFAs incorporate into membrane fractions corresponding to rafts, generally within the most abundant and DM-sensitive phospholipids (PE and PC), and magnetic resonance studies show differential changes in membrane structure when n-3 PUFAs are incorporated into PE vs. PC, the phospholipid fractions predominantly modified in DM [193, 195, 198, 199]. Lipid raft incorporation of n-3 PUFAs is accompanied by reduced levels of the highly disordered n-6 PUFA AA [179], consistent with declining sarcolemmal AA species in DM [193, 198, 199]. Several studies show n-3 PUFAs can lower raft cholesterol levels, which may underlie effects on protein lateral organization and signaling [221]. Impacts on cholesterol may include shunting from raft to detergent-soluble membrane fractions in some cell types [221, 222], potentially reflecting the poor affinity of n-3 PUFAs for cholesterol [223]. However, in some cell types reductions in raft cholesterol are not matched by changes in the detergent-soluble fraction [224], which may be related to the ability of n-3 PUFA to promote internalization of lipid microdomains (including raft cholesterol) [225]. An area of interest has been the influence of PUFAs on ion channel function, electrical stability and arrhythmogenesis, though mechanisms underlying such effects are yet to be fully detailed [226, 227].
Membrane dynamics are also influenced by glycation and associated free radical production [173, 203]. Treatment with the anti-glycation and anti-oxidant compound resorcylidene aminoguanidine (RAG) reverses DM-dependent reductions in cell membrane fluidity [173]. Profoundly reduced sensitivity to Ca2+ overload in myocardium from DM rats is also inhibited [173], additionally highlighting the importance of fluidity to ion homeostasis in DM hearts. Roles of such post-translational changes are discussed in more detail further below.
Unfortunately, studies of sarcolemmal changes in DM have focused to date on models of T1DM, with little to no information regarding changes in T2DM (Table 2). Moreover, investigations have yet to detail temporal patterns of sarcolemmal change during DM development and progression. It would be of great value to undertake such time-course analyses across what appears the critical range for variable shifts in ischemic tolerance (i.e. no effects or improved tolerance from weeks 1–6; reduced tolerance at later times), permitting correlation of membrane makeup and ischemic tolerance changes. Membrane lipid analyses have been undertaken within yet not across these differing time periods (Table 2), with broadly similar lipid changes reported at 2–6 weeks [195, 201, 202] and 8–9 weeks [197, 199, 200]. No distinguishing feature is evident in later membrane profiles, though there is only a single study at ≥ 12 weeks [193]. There is also no information on the time-course of changes in caveolae and caveolin proteins, another knowledge gap deserving attention.
Remodeling of sarcolemmal microdomains
Membrane proteins and signaling are compartmentalized between specialized microdomains rich in cholesterol and sphingolipids vs. other membrane regions [228]. Though a simplified model given the true complexity of the plasma membrane [229], sarcolemmal assemblies of lipids and proteins may be divided into either lipid rafts or planar platforms (loosely corresponding to detergent insoluble membrane fractions) and non-raft domains (corresponding to detergent soluble membrane). These domains differ markedly in their ion channel, transporter, receptor and signaling protein profiles [230–232]. Altered membrane composition can thus disrupt signaling, ion movement and substrate transport through differential changes in lipid raft/caveolar vs. non-raft domains. However, the compartmentation of proteins between raft and non-raft regions remains a controversial topic, reflecting in part varying outcomes with different membrane fractionation methodologies.
Lipid raft vs. non-raft proteins
These distinct regions may be differentially modified in DM, although studies of the cardiac sarcolemma are limited. For example T2DM db/db mice exhibit ~ tenfold elevations in the area of raft clusters in aortic endothelium, confirming that increased cellular lipid content can drive raft cluster formation [233]. Both horizontal and vertical clustering of rafts is observed, increasing the height of these aggregates [233]. Such changes will modify resident protein integration and function. Sequestration of specific ion channel, receptor and transporter proteins within raft microdomains is an important means of compartmentalizing and specifying downstream signal transduction by the sarcolemma. For example, caveolar localization ensures signaling specificity of cardiomyocyte β-adrenoceptors, limiting non-selective effects on sarcoplasmic reticulum (SR) and myofilament function [234]. A sub-population of L-type Ca2+ channels (LTCCs) has also been identified in caveolae domains that appears critical in regulating β-adrenoceptor [235] and hypertrophic calcineurin/nuclear factor of activated T-cells (NFAT) signaling [236]. Evidence also supports functional localization of LTCCs to caveolae in human and rodent atrial myocytes, with a caveolae-targeted LTCC antagonist inhibiting Ca2+ fluxes [237]. In contrast, a recent report concludes that neither a caveolae targeted LTCC activator or inhibitor modifies function or hypertrophic responses in murine hearts [238].
Regulation of other important channels and pumps may be dependent upon caveolae domains. For example, despite evidence cardiac Na+-K+-ATPase localizes to non-raft regions [239], there is also evidence for caveolae/caveolin association and control [240]. Almost half of the Na+-K+-ATPase α1-subunit and nearly all of the glycosylated β1-subunit reportedly localizes to cardiac caveolae [241], and caveolin-1 deletion inhibits interactions between Na+-K+-ATPase, caveolin-3 and PI3K in cardiomyocytes [242]. Thus, inhibition of sarcolemmal Na+-K+-ATPase sub-unit expression and activity in STZ-dependent T1DM models [243, 244] may reflect a caveolae specific response. Such an effect is consistent with predicted outcomes of cholesterol accumulation [245]. Indeed, high dietary cholesterol also reduces Na+-K+-ATPase [246], and cholesterol may render sarcolemmal penetration of the ATPase complex energetically unfavorable, while reducing surface charge density is chemically unfavorable [245]. Similarly, the cardiac Na+/Ca2+ exchanger is suppressed in DM [247], and despite some evidence it does not localize to rafts or caveolae [239], there is also evidence the exchanger interacts with caveolin-3 in sarcolemmal vesicles [248], and its activity is depressed with cholesterol depletion [249]. Further work may clarify the impacts of DM on cardiac raft and non-raft ion channels and pumps.
Caveolar membrane microdomains
Evidence accumulated over the past decade highlights a particular importance of caveolae in protection of myocardium against metabolic (ischemia, hypoxia) and mechanical stressors [167–170], together with perturbations and potential involvement in cardiac disease [159, 163]. Few studies have examined effects of DM on caveolar structure and density, although constituent caveolin proteins are significantly modified [135, 250–253] and caveolar localization of signaling molecules altered [250, 251, 254]. Evidence implicates abnormal caveolar control in the development and cardiac-specific effects of DM [158, 159].
Caveolae have at least four major functions: (i) as signaling platforms in the membrane, for example for receptor tyrosine kinases (RTKs) including the InsR [131], GPCRs [165], eNOS [255], other signaling proteins [256] and ion channels [166]; (ii) regulating fatty acid transport [257, 258] and glucose handling [158]; (iii) participating in mechanotransduction and acting as membrane ‘reservoirs’ to limit damage with mechanical stress [167, 259]; and (iv) functioning as membrane transport vesicles, budding from the membrane in response to specific cues and participating in membrane repair [260]. Abnormalities within these regulatory domains will thus influence ion and substrate movement, protective signaling and myocyte responses to mechanical perturbation, impairing cardiac responses to both pathologic insult and potential therapies.
As for lipid rafts in general, caveolae formation and function are dependent upon lipid composition, particularly cholesterol and sphingolipid content [228, 261, 262]. A key distinguishing feature is the presence of cholesterol-associated caveolin proteins, involved in stabilizing the physical architecture of these flasks and regulating signaling and transport processes [228]. Depletion of membrane cholesterol [263] or caveolins [264] inhibits caveolae formation and negates myocardial responses to diverse protective stimuli [265, 266]. Cholesterol depletion also disrupts the Z-band localization of caveolin-3 in cardiomyocytes, and alters cytoskeletal architecture [267]. Highly abundant PS and phosphatidylinositol (4,5)-bisphosphate [PIP2] may also be important, concentrating within caveolae and functionally compartmentalizing lipid pools [268–270]. Caveolae depletion with caveolin-1 knockout or depletion leads to re-organization of plasma membrane PS domains [271], and consistently down-regulates pathways of lipid metabolism across cells [272]. There is evidence the caveolin scaffolding domain—a 20-amino acid sequence initially implicated in controlling signal molecules—has an intrinsic capacity to concentrate local cholesterol, PS and PIP2 [273]. Effectively enriching caveolar oligomers in PS and PIP2, this process is proposed as a means of attracting membrane-sensing cavin proteins to initiate a cascade of further caveolin, PIP2 and PS recruitment to membrane rafts [274]. Cholesterol concentration around caveolin oligomers may modify biophysical properties to favor membrane bending by the cavin-caveolin coat complex. These inter-dependencies provide a basis for the sensitivity of caveolae formation and function to cholesterol and phospholipids, the levels of which are perturbed in DM [195, 196, 199–202]. Given evidence of cholesterol accumulation in the sarcolemma of DM hearts, a scenario of both caveolar disruption via caveolin suppression and reduced membrane fluidity via cholesterol accumulation could arise. How accumulated cholesterol is distributed between microdomains in DM is not clear, however it is possible fluidity within depleted caveolae populations is compromised.
Caveolar “coat proteins”—the caveolins and cavin families
While there are few analyses of cardiac caveolar architecture and density in DM, significant changes in constituent caveolins and cavins are observed and likely disrupt caveolae function and formation. Hyperglycemia may suppress myocardial caveolin-3 in a PKCβ2 dependent manner [250] and H9c2 cardiomyoblast caveolin-3 in an oxidant-dependent manner [275], while hyperinsulinemia also depresses caveolar caveolin-3 in H9c2 cells [254]. Moreover, saturated fats reduce cardiac caveolin-3 [276], as does aging [277], whereas PUFA supplementation can up-regulate caveolin-3 expression [278]. Caveolin-1, in contrast, may be significantly up-regulated in DM [252, 279].
The caveolin proteins are primary structural and regulatory elements of caveolae [168], though also play important non-caveolar roles [259]. For example, sequestration of active caspase-3 by extra-caveolar caveolins may underlie protective effects of β-receptor antagonism in DM hearts [253]. Three caveolin isoforms have been identified with differing functions and tissue distributions [158, 164, 206, 264]. All are expressed in the central nervous system, with ubiquitous caveolin-1 most highly expressed in endothelium, fibroblasts and pneumocytes, where it appears structurally supported by caveolin-2 hetero-oligomerization. In contrast, caveolin-3 is highly specific to striated muscle and plays crucial roles in cardiac stress sensing/responses and cardioprotection [167–170, 256, 259, 264]. Caveolins preferentially arrange in homo-oligomers of 2 to ~ 16 monomers, forming caveolar assembly units, and may require cholesterol for effective insertion into the membrane [264]. A common feature of all isoforms are scaffold domains where signal molecules including G proteins, PKC and eNOS are proposed to physically interact. However, the basis of regulatory molecular interactions with caveolins remains to be defined [280]. Noted above, these domains also appear important in locally concentrating cholesterol and phospholipids [273]. Caveolin-3 is not only essential to myocardial caveolae formation but is particularly important to stress tolerance and cardioprotection. Myocardial [266] and mitochondrial [281] stress responses are strongly caveolin-3 dependent, as is cardiac protection via ischemic and anesthetic preconditioning [169, 265] and opioid GPCRs [266]. Caveolin-3 also influences cholesterol transport [282], ion handling [235, 283, 284], GLUT4 and glucose metabolism [252, 285, 286], and hypertrophic remodeling [287, 288].
Despite these key roles, the control of myocardial caveolin expression remains to be detailed, though studies in other cells support transcriptional regulation by myocardin. A member of a family of transcriptional co-activators responsive to stress, myocardin up-regulates caveolins and caveolae in smooth muscle cells [289]. Specific cardiac studies are lacking, however human expression data support a close association between Myocd and Cav1 gene levels across tissues, including heart [289]. Myocardin control of caveolin-1 and -2 and cavin-2 appears independent of serum response factor whereas control of cavin-1 is dependent on this transcription factor, providing for differential control of cavin-1 vs. caveolins [289]. Myocyte myocardin may be up-regulated by hyperinsulinemia [290], and in other muscle cell types myocardin is up-regulated by oxidative stress sensitive miR-145 [291]. Importantly, emerging evidence reveals new roles for myocardins in glucose and lipid homeostasis (including via caveolins) [292].
The few studies analyzing myocardial caveolins in T1DM have employed relatively acute models (0–6 weeks), and report a hyperglycemic depression of caveolin-3 [135, 250–253] that may contribute to diastolic dysfunction [250], impaired GLUT4 translocation [252] and I–R intolerance [251]. Nonetheless, the acuteness of STZ-induced hyperglycemia and variable ischemic tolerance in these T1DM models raise questions regarding relevance: paradoxical cardioprotection in the initial weeks in rat T1DM models [2, 84, 293] is not relevant to the ischemic intolerance observed in chronic disease and T2DM. Hyperglycemia also acutely depresses caveolin-3 expression in cardiac myoblasts [275], and hyperinsulinemia suppresses caveolar levels of caveolin-3 in H9c2 myoblasts, which may dysregulate Akt-dependent InsR signaling [254]. No study has comprehensively assessed mechanistic involvement of caveolin-3 in the cardiac sequelae of T2DM, with only a single report of an insignificant fall in cardiac Cav3 mRNA in the non-obese GK rat model [294].
Inhibitory effects of saturated fatty acids [276, 295] and glucose [250] on cardiac caveolin-3 expression and caveolin-dependent eNOS signaling present plausible mechanisms for reduced cardioprotection in DM. Impaired PI3K/Akt/NOS signaling is characteristic in DM myocardium [12, 90, 94, 95, 146, 250], and these signal elements cluster in caveolae [164–166] where eNOS is regulated by caveolin-1 and -3 [250, 275, 296–298], and Akt signaling is promoted by caveolin-3 [250–252, 299]. Studies in rodent models indicate that DM dysregulation of RISK signaling, including PI3K/Akt and glycogen synthase kinase-3β, underlies impaired protection via cytokine receptors [13], GPCRs [14] and progestin and adiponectin receptors [35]; and RISK-dependent pre- and post-conditioning responses are also inhibited in DM [2, 116, 117, 130]. Inhibition of Akt signaling and ischemic tolerance in T1DM has been linked to caveolin-3 depletion [251], as has disruption of adiponectin receptor cardioprotection [135]. Recent work also implicates oxidant-mediated dysregulation of caveolin-3/eNOS signaling in the ischemic intolerance in T1DM hearts [275]. An increase in caveolin-1, reported by Penumathsa et al. [252] in hearts of T1DM rats and Bucci et al. [279] in aortic tissue, may also inhibit protective signaling, suppressing eNOS activity [296–298, 300] and promoting dephosphorylation of sarcolemma-associated Akt [301]. In support of this, Ajmani et al. [300] report that a ‘caveolin inhibitor’ and sodium nitrite both restore preconditioning in T1DM rat hearts, however significant limitations include multiple non-specific biological actions of the inhibitor employed, and failure to measure caveolin-1 expression or establish diabetic inhibition of preconditioning.
Less is known regarding potential roles of more recently identified cavin proteins [206, 302]. These coat proteins homo- and heteroligomerize (independently of membrane and caveolins) to form specific caveolar sub-complexes, and are involved in orchestrating the cell-specific formation, caveolin/cavin incorporation and structural modeling of caveolae [206, 302]. They may also be released intracellularly with different stressors/stimuli to regulate gene expression and non-caveolar processes. Depletion of cavin-1 (with attendant loss of caveolae) results in elevations in circulating triglycerides, glucose intolerance and hyperinsulinemia [303], and inhibits cardiac ischemic tolerance and stretch responses while exaggerating cellular permeability (potentially via NOS overactivity) [304]. Perturbation of the caveolar system via caveolin-1 depletion or knockout also dysregulates cardiac stress responses [305, 306]. Whether these gene deletion effects reflect distinct roles and influences of cavins and caveolins, or highlight the broader importance of caveolae is presently unclear. However, differences do emerge in the cardiac effects of cavin-1 vs. caveolin knockout [304]. Intriguingly, effects of cavin-1 and caveolin-1 knockout suggest the diastolic dysfunction in DM could involve disruption of sarcolemmal caveolae: caveolar depletion in both cavin-1 [304] and caveolin-1 [307] knockout hearts is associated with significant diastolic dysfunction or stiffening. Caveolae provide an effective membrane reserve to accommodate physical deformation or stretch [167], potentially influencing the compliance of cardiac cells. Although diabetic diastolic dysfunction is attributed to fibrosis/hypertrophy [20], sarcolemmal makeup and specifically caveolae and associated signaling may contribute to this dysfunction [304, 307].
Changes in sarcolemmal caveolae influence substrate handling
Glucose transport
Caveolar domains are important in glucose and lipid transport, and InsR receptor signaling [158, 274]. Myocardial glucose transport via GLUT4 is spatially confined to caveolar domains [158, 308], where InsRs are also localized [309]. Cardiac insulin-resistance and impaired GLUT4 expression and transport in T2DM [310] may involve disruption of caveolae and caveolin-3 with DM [135, 250–253] and high-fat feeding [276, 295]. Indeed, Penumathsa et al. [252] report reduced expression and association of GLUT4 and caveolin-3 in lipid-rafts of T1DM rat hearts.
Activation of the InsR normally leads to a cascade of Akt phospho-activation and phosphorylation of the Rab-GTPase activating TBC1D4/AS160 protein, a distal effector maintaining GLUT4 vesicles within an inactive intracellular pool [311]. This initiates pathways mediating docking and diffusion of GLUT4 vesicles at the plasma membrane [311]. This path not only increases GLUT4 exocytosis but can limit endocytosis to re-distribute plasma membrane GLUT4. However, in cardiac [312] and skeletal myocytes [313] insulin does not influence endocytosis. Nonetheless, GLUT4 endocytosis in skeletal myoblasts is sensitive to energy state (inhibited by mitochondrial uncoupling), and both clathrin-dependent and clathrin/caveolae-independent (yet cholesterol-dependent) endocytosis paths are involved [313]. This energy-sensitive endocytosis reveals non-caveolae effects of cholesterol, for example promoting negative membrane curvature [205]. This not only further highlights the importance of membrane cholesterol, but shows that distinct membrane changes may independently modify GLUT4 exocytosis and GLUT4 endocytosis.
Not only is GLUT4 movement influenced by caveolae and caveolins, but signaling via the InsR is strongly dependent upon these raft elements. Yamamoto et al. [314] first demonstrated positive control of InsR signaling via caveolin-1 and -3, including evidence of direct caveolin interaction with the InsR kinase domain to promote insulin receptor substrate 1 phosphorylation. Caveolin-3, caveolin-1 and the InsR all interact in cardiac myoblasts, and caveolin-3 depletion renders myocytes insulin-resistant while caveolin-3 haplo-insufficiency increases susceptibility to fatty acid induced insulin-resistance [286]. Disruption of caveolae or caveolin-3 expression in DM [135, 250–253] is thus predicted to limit cardiac InsR signaling, although a parallel elevation in caveolin-1 as reported in a rat T1DM model [252] may modulate such effects. Supporting the value of targeting caveolins, insulin-resistance in obese and DM mice is reversed by hepatic overexpression of caveolin-3, which substantially enhances InsR signaling [315]. Nonetheless, basal glucose metabolism appears largely unaltered in hearts lacking either caveolin-3 [288] or caveolin-1 [316], and thus also devoid of caveolae, although skeletal muscle insulin-resistance arises in both models [308, 317]. While suggesting distinct caveolin/caveolar control of substrate metabolism in cardiac vs. skeletal muscle, cardiac InsR signaling and insulin-resistance have yet to be detailed in these knockout models. Lifelong absence of both caveolae and caveolins in these models may also limit their relevance to more moderate and progressive changes in DM. Other analyses confirm that reductions in caveolin-3 inhibit insulin-stimulated glucose uptake in cardiac myoblasts and myocytes [286], and that hyperinsulinemia in cardiac myoblasts reduces caveolar levels of caveolin-3 and insulin-dependent phospho-Akt [254]. Insulin-dependent myocardial glucose uptake is thus predicted to be impaired with reductions in caveolin-3 expression in DM hearts, though this has yet to be directly assessed.
Ubiquitously expressed caveolin-1 may additionally modulate InsR signaling in DM, and cardiac expression is reportedly increased in T1DM rat hearts [252]. Caveolin-1 is also induced by micro-RNAs up-regulated in obesity (miR103, miR107), and their overexpression induces insulin-resistance in an entirely caveolin-1 dependent manner [318]. However, changes in caveolin-1 are not universal in obesity, some dietary interventions may also augment caveolin-1 [319], and distinct from caveolin-3, cardiac expression of caveolin-1 appears repressed with medium-chain triglyceride but not palmitate supplementation [276]. Further work is needed to clarify effects of caveolin-3 and -1 on insulin-dependent glucose uptake and metabolism in myocardium and cardiac myocytes, identifying specific roles of the caveolins themselves vs. caveolae as regulatory platforms, and the effects of moderate and acute vs. prolonged changes in expression (modeling changes in DM, and avoiding limitations of lifelong gene deletion).
Fatty acid uptake
Fatty acid transport is also compartmentalized within lipid rafts and caveolae [320], with the regulatory InsR [309]. Accumulation of long-chain fatty acid metabolites is important in development of myocardial insulin-resistance [321, 322], with more prolonged changes involved in later development of heart failure. The major cardiac fatty acid transporters CD36 and fatty acid binding protein (FABP) normally relocate to the sarcolemma from intracellular stores in response to insulin or contraction [322, 323]. Active CD36 specifically localizes to lipid rafts and caveolae where fatty acid uptake activity is promoted, while inactive intracellular CD36 is associated with non-raft fractions [320]. Overexpression of CD36 enhances skeletal muscle fatty acid oxidation and decreases plasma lipids [324], while deletion impairs cardiac fatty acid uptake, though this may be metabolically compensated by increased glucose oxidation [325]. Sarcolemmal CD36 not only governs uptake but targets fatty acids to specific metabolic sites including mitochondria [326], and plays roles in promoting 5′-AMP activated protein kinase (AMPK) signaling, regulating Ca2+ signaling and levels, and acting as co-receptor for toll-like receptors [327]. Permanent sarcolemmal relocation of transporters in obesity and DM thus greatly promotes cardiac lipid and lipid metabolite accumulation to impair insulin signaling and glucose utilization [322, 323]. As critical sites of control, sarcolemmal CD36 and FABP are important therapeutic targets for countering myocardial insulin-resistance and cardiomyopathy.
Changes to caveolae and caveolin-1 and -3 in DM are predicted to impact CD36-dependent uptake given functionally relevant caveolar localization and caveolin control. Hearts from caveolin-3 haplo-insufficient mice do express less CD36 in line with differing caveolin-3 levels, though a twofold rise in caveolin-1 suggests potentially complicating adaptation [286]. Lipid raft targeting of CD36 may involve interaction with caveolin-1 based on effects in non-muscle cells [328], and cardiac lipids and fatty acid uptake are also reduced with caveolin-1 knockout [316]. Diabetic up-regulation of caveolin-1 [252] could thus promote lipid uptake, though myocardial CD36 and caveolin-1 are not always linked: for example, cardioprotective isoflurane increases caveolin-1 and caveolae [305] while reducing caveolar CD36 levels [329]. Although lifelong absence of caveolin-3 does not reduce cardiac fatty acid uptake [288], a halving of cholesterol levels and a 40–50% increase in triglycerides confirm major perturbations of fatty acid handling. Importantly, and as noted above, this model reflects a complex phenotype encompassing lifelong absence of caveolin-3 and caveolae (thus caveolae-localized transporters), which likely disrupts potential caveolin-1 control.
Membrane cholesterol—beyond fluidity and caveolar domains
Changes in membrane cholesterol do not only influence membrane fluidity [189], curvature [205], and caveolar endowment [223, 262, 267], but also govern T-tubule system integrity and excitation–contraction (E–C) coupling [330, 331], contractile function [267, 332], glucose and fatty acid transport [320, 331, 333–336], and functionality of membrane ion channels, receptors and transporters [337–339]. These diverse effects of cholesterol on sarcolemmal architecture and the functionality of associated proteins may contribute to impairment of cardioprotection and ischemic tolerance with hypercholesterolemia [138, 140, 340] and are relevant to the DM myocardium.
For example, there is evidence that increased membrane cholesterol is key to impaired GLUT4 traffic in insulin-resistance and T2DM, though studies have focused on skeletal muscle given its contribution to systemic insulin sensitivity and glucose homeostasis: glucose-intolerant animal models and humans accumulate cholesterol in skeletal muscle membranes [331, 335]; high-fat diets also increase skeletal muscle cholesterol [332]; DM also increases cardiac cholesterol levels [195]; cholesterol depletion with methyl-β-cyclodextrin reversibly and dose-dependently increases plasma membrane GLUT4 incorporation in myotubes [334]; and cholesterol depletion improves glucose homeostasis in high-fat fed animals, together with insulin-dependent GLUT4 translocation and glucose uptake in muscle fibers [335]. The cholesterol depleting agent chromium also improves glycemic control in T2DM patients [341], and activates GLUT4 trafficking and insulin-stimulated glucose transport in a cholesterol- and AMPK-dependent manner [342]. This is consistent with evidence AMPK improves insulin-stimulated GLUT4 control by lowering membrane cholesterol [335]. These observations support regulation of insulin-stimulated GLUT4 translocation via tissue cholesterol content, and suggest cholesterol removal may be useful in countering myocyte insulin-resistance, although cardiac studies are lacking.
Additional to indirect influences on protein confirmation and function, cholesterol recognition/interaction amino acid consensus (CRAC) and more recently CARC (similar to CRAC, with an opposite orientation—hence “CARC”) domains have been identified in transmembrane proteins, including receptors regulating cellular stress responses [337, 343]. Sometimes located within the same transmembrane segment, these CRAC and CARC domains can directly interact with cholesterol in the cytoplasmic leaflet of the plasma membrane. Modulating multiple ion channels [284, 338] and receptors [339, 343–345], the cardiac significance of sarcolemmal cholesterol:protein interactions awaits further study, particularly in the context of DM and metabolic syndrome.
Potential influences of DM on cardiac phospholipid signaling
Membrane lipids not only serve structural roles but are substrates in cell signaling (Fig. 2). Sarcolemmal phospholipids are targeted by three primary phospholipase groups to generate lipid signaling molecules: phospholipases A2, C and D (PLA2, PLC and PLD, respectively). Phospholipid signaling is implicated in cardiac hypertrophy/cardiomyopathy and is perturbed in cardiovascular disease states including DM [346]. Changes observed in sarcolemmal glycerol-phospholipid species in DM rat hearts likely contribute to membrane and contractile dysfunction [195, 199, 202]. In terms of ischemic tolerance, phospholipases are implicated both in mediating and protecting against ischemia–reperfusion injury [347–349]. This may reflect isoform specific effects, including protection via PLCγ1 and injury via PLCδ1. Shifts in membrane phospholipase signaling may thus contribute to alterations in both infarct tolerance and cardioprotection in DM.
Fig. 2.
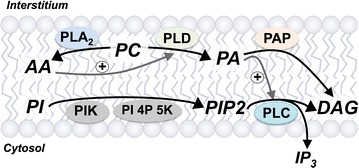
Sarcolemmal phospholipid signaling via phospholipases. AA arachidonic acid, DAG 1,2-diacylglycerol, IP3 inositol 1,4,5-triphosphate, PA phosphatidic acid, PC phosphatidylcholine, PIP 2 phosphatidylinositol-4,5-bisphosphate, PLA 2 phospholipase A2, PLC phospholipase C, PLD phospholipase D
Cardiac PLC activities are reduced in STZ-induced T1DM rats, and basal and phosphatidic acid induced IP3 generation are reduced in cardiomyocytes from DM rats [350]. In contrast, increased PLC activity is implicated in exaggerated α1-adrenergic receptor mediated inotropy with acute (3 day) hyperglycemia [351]. Distinct outcomes with acute vs. chronic DM may be relevant to patterns of early protection and later ischemic intolerance in models of STZ dependent T1DM [2]. A reduced rather than increased PLC activity may contribute to abnormalities in contractility and α1-adrenergic receptor responses with more protracted disease [352]. Reduced PLC generation of 1,2-diacylglycerol (DAG) may impact other cellular processes [353, 354], although myocardial DAG levels are increased with more acute STZ-dependent T1DM and in autoimmune DM (biobreeding) rats [355, 356], which could destabilize the membrane [357, 358].
Phospholipase D activity reportedly declines in DM cardiomyopathy [359], which may limit PA generation and thereby PLC activation. Alterations in AA content of sarcolemmal phospholipids could also reflect dysfunctional PLC signaling [346]. Relatively little is known regarding cardiac PLA2 signaling in DM, however cardiac membrane associated PLA2 activity is increased in rat models of DM [360]. The local environment of caveolae is also important to phospholipid signaling. Up to half of cellular PIP2 is located in caveolin-enriched membrane fractions [268, 361, 362], and this pool is specifically sensitive to GPCR and RTK activation [268] in a cholesterol-dependent manner [263]. For example, the α1 adrenergic receptor (AR) and its Gq effector protein are caveolae localized in adult cardiomyocytes [363, 364], and analysis in neonatal cells possessing both caveolar and non-caveolar PIP2 fractions supports select caveolar depletion upon α1-AR stimulation [365]. Coupled with localized PLC-dependent hydrolysis to DAG, this compartmentation provides for select regulation of caveolar populations of PIP2- and DAG-sensitive ion channels and exchangers [366, 367]. Shifts in caveolar makeup and localized phospholipids thus have capacity to selectively disturb cardiomyocyte receptor signaling and ion channel function.
Critical to cellular growth, substrate metabolism, stress responses and cardioprotection, PI3K isoforms catalyze production of the 3-phosphorylated phosphoinositides phosphatidylinositol 3-phosphate, phosphatidylinositol (3,4)-bisphosphate, and phosphatidylinositol (3,4,5)-triphosphate. While membrane phospholipid pools are modified in DM, it is unclear whether changes are sufficient to influence PI3K signaling. Certainly, dysfunctional PI3K/Akt signaling is implicated in altered InsR control and impaired cardioprotection, among other cardiac changes.
Membrane glycation, glycosylation, palmitoylation and oxidation in DM
Glycation and enzymatic glycosylation are major factors in the cardiac abnormalities arising in DM [9], as is oxidative stress [10, 11]. Palmitoylation is also an important determinant of sarcolemmal protein function [368–370] and is highly relevant in metabolic disorders such as DM [371, 372], however, diabetic perturbations have been largely studied in non-cardiac tissues. Other modifications may also be relevant in DM, for example reductions in phospholipid N-methylation [213] depress Na+-dependent Ca2+ uptake [214] and enhance ATP-dependent Ca2+ efflux [215] in the cardiac sarcolemma of STZ-dependent T1DM models. Such effects may be mechanistically relevant to paradoxical resistance to external Ca2+ overload in DM hearts [189].
Advanced glycation end-products (AGEs) and the receptor for AGE (RAGE)
Chronic hyperglycemia promotes glycation, the non-enzymatic covalent bonding of carbohydrates to proteins and lipids. Glycation products in turn can form cross-linked structures known as AGEs. These modified proteins/lipids activate cell surface RAGE to trigger ROS generation, activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and pro-inflammatory cytokine production. Positive feedback between NFκB and RAGE expression exaggerates ROS and cytokine generation. These processes are implicated in vascular dysfunction in DM, and have been shown to contribute to myocardial changes and dysfunction [373, 374]. Targeting AGE accumulation has also been shown to improve myocardial ischemic tolerance in different models of DM. For example, cardioprotection in rat T1DM models with natural xanthonoid and flavonoids [375, 376] and anti-hyperglycemic glitazones [377] appear to involve inhibition of the AGE-RAGE axis and AGE accumulation. However, cytoplasmic AGE accumulation is typically documented in animal and human tissues, and specific sarcolemmal targets of glycation have not been investigated in detail.
Glycosylation
Diabetes increases fluxes through accessory paths of glucose metabolism, including the hexosamine biosynthetic pathway (HBP) that produces the sugar donor for enzyme-mediated β-O-linked-N-acetylglucosamine (O-GlcNAc) modification of proteins or lipids. Studies confirm that increased protein O-GlcNAc levels contribute to the cardiac abnormalities of DM. This modulation is complex, however, with O-GlcNAc mediating both beneficial and detrimental effects [378]. Transient elevations in O-GlcNAc may provide cytoprotection [379], with acutely increased O-GlcNAc prior to ischemia or in reperfusion reducing infarction and dysfunction [380]. Inhibition of O-linked β-N-acetylglucosamine transferase (OGT) can also inhibit cardioprotection [381] while inhibition of protein O-GlcNAcase (OGA) may improve cardiac ischemic tolerance [380]. Such effects might be relevant to observations of acute protection early in STZ-dependent hyperglycemia. Indeed, Jensen et al. [382] present evidence O-GlcNAc signaling participates in remote ischemic preconditioning and activates cardioprotection in DM myocardium from T2DM patients (based on functional I–R tolerance of atrial trabeculae).
Other evidence indicates chronic elevations in O-GlcNAc are detrimental to the heart. Hyperglycemia mediated HBP activation increases cardiomyoblast death [383], and inhibitors of OGA have been shown to improve I–R tolerance in DM hearts, potentially via preserved integrity of O-GlcNAc associated Z-line protein structures [380]. Activation of the HBP and protein O-GlcNAcylation modulates hypertrophic and cell signaling pathways in T2DM [378]. Increased protein O-GlcNAcylation in non-DM cardiomyocytes also decreased hypertrophic signaling responses, while HBP inhibition partly restored hypertrophic signaling in DM cardiomyocytes. Cardiac beclin-1 and Bcl-2 have also been recently identified as targets for O-GlcNAcylation [384], with blunted autophagy in cardiomyocytes from T2DM db/db mice partly reversed by inhibiting the HBP. Ramirez-Correa et al. [385] present evidence that Z-line localization of O-GlcNAc and OGT and A-band localization of OGA is disrupted, consistent with changes in human DM hearts. Their data indicate subcellular redistribution of OGT and OGA rather than changes in overall activities are responsible for altered O-GlcNAcylation in DM. On the other hand, Dassanayaka et al. [386] show O-GlcNAcylation is not involved in inhibition of mitochondrial metabolism in hyperglycemic cardiomyocytes.
There is only limited evidence for glycosylation modifications of plasma membrane proteins. In coronary endothelium OGA expression is decreased and OGT expression and O-GlcNAcylation increased with DM [387], with CX40 identified as a potential target of O-GlcNAcylation regulating cell function. Effects of glucosamine and OGT blockade on post-ischemic Ca2+ levels also implicate modulation of sarcolemmal channels [388]. Further studies of cardiac sarcolemmal targets of O-GlcNAcylation are required to clarify the role of this process in membrane changes and dysfunction in T1 and T2DM.
Palmitoylation
Reversible S-palmitoylation (thioester attachment of palmitic acid to cysteine) is an important protein ‘sorting’ signal, governing trafficking and membrane localization [389]. Palmitoylation enhances membrane affinity of many proteins to facilitate membrane accumulation [390]. N-myristoylation (amide bond attachment of myristoyl group to N-terminal glycine residues) may also facilitate protein localization to membrane palmitoylases [390, 391]. Within the membrane, palmitoylated proteins have high affinities for cholesterol and sphingolipid-rich domains [392], which is important in targeting proteins to membrane raft regions [393]. Some GPCRs are palmitoylated down-stream of the 7th transmembrane domain [394], which may be required for efficient plasma membrane delivery [395, 396]. Palmitoylation may regulate internalization of some GPCRs and promote trafficking of internalized proteins to the plasma membrane. Ion channel and exchanger functions are also modified with palmitoylation. For example, the cardiac Na+/K+-ATPase is targeted by palmitoylation, though functional outcomes await detailed study [369]. The inactivation of the Na+/Ca2+ exchanger is also strongly dependent on palmitoylation [370].
Recent data support induction of endocytosis via membrane protein palmitoylation. Massive endocytosis (MEND) is an adapter-independent form of endocytosis that can rapidly internalize up to 70% of the plasma membrane in response to stressors such as Ca2+ overload [397]. Increased plasma membrane palmitoylation promotes MEND in response to mitochondrial stress [398], likely due to clustering of palmitoylated proteins in lipid-ordered domains as a result of palmitoyl chain affinity for the ordered lipid environment [399]. Reilly et al. [370] show that elevations in palmitoylated Na+/Ca2+ exchanger 1 protein in the plasma membrane accelerates MEND, mirroring effects of palmitoylated phospholemman [397, 398] and suggesting palmitoylated proteins promote formation of lipid-protein domains to trigger endocytosis. Since acyl groups of palmitoylated proteins insert more readily between the phospholipid head groups of curved rather than planar membrane regions [400], palmitoylated proteins will cluster in invaginated lipid-ordered domains that may include caveolae [401], the curved domains formed in endocytosis, and potentially curved junctions between T-tubule and surface sarcolemma. Clustering of palmitoylated membrane proteins with large cytoplasmic domains (e.g. Na+/K+-ATPase, Na+/Ca2+ exchanger) may itself promote membrane curvature [402]. Unfortunately, despite such effects and the importance of palmitoylation to sarcolemmal protein trafficking and function, few studies have examined potential roles of altered palmitoylation in the cardiac abnormalities of DM.
Membrane oxidation
It is well established that oxidative stress is involved in development and progression of DM and its organ-specific complications [10, 403, 404], and shifts in cardiac stress responses may involve oxidative modification of sarcolemmal elements. Oxidative stress may underlie changes in caveolae and caveolins: Su et al. [275] show the anti-oxidant N-acetylcysteine (NAC) limits changes in caveolin-3 together with phosphorylated eNOS known to localize to caveolae. Diabetes reduces association of caveolin-3 and eNOS in cardiomyocytes, an effect countered by antioxidant treatment. Protective effects of NAC on hyperglycemic and hypoxic cell injury were also abolished by knockdown of either caveolin-3 or eNOS, supporting the notion hyperglycemic inhibition of eNOS results from impaired caveolin-3 expression. Membrane lipid metabolism also contributes to oxidative stress: lipoxygenases oxidatively metabolize AA released from the plasma membrane following PC hydrolysis, generating ROS in the process. Hyperglycemia-induced activation of 12/15-lipoxygenase is associated with cardiac oxidative stress and DM cardiomyopathy [404]. However, beyond largely indirect evidence (e.g. preventing caveolar changes with anti-oxidant intervention), there is relatively little information available regarding the specific sarcolemmal targets of oxidative modification in DM hearts, and their roles in associated ischemic intolerance. As for glycation/glycosylation and palmitoylation, further studies are needed to clarify modifications to sarcolemmal proteins in T1 and T2DM, and their roles in altered stress responses.
Remodeling of the T-tubule system in DM
Despite limited studies, and none in human cardiomyocytes, evidence supports significant remodeling of T-tubules in DM. Studies in skeletal muscle confirm the T-tubule system is a functionally important target governing glucose handling [336, 405, 406]. In heart, McGrath et al. [160] report a pronounced fall in functionally intact SR/T-tubular junctions together with an increased T-tubule area (longitudinal rather than transverse orientation) in rat models of T2DM. A subsequent study in db/db mice identified a fall in T-tubule density in this model of T2DM [161]. Despite differing morphological outcomes, both studies highlight diabetic disruption of T-tubule organization and functionality, likely perturbing E–C coupling and contractile function. For example, the synchrony of cardiomyocyte Ca2+ release (influencing contractile function and arrhythmogenesis) depends on T-tubule integrity, and disorganization underlies cardiac dyssynchrony in different settings [407–409]. Disruption of T-tubule structure and function may thus mediate the reduction in synchrony observed in DM [161, 410].
Changes specifically within the T-tubule system may also be important in altered substrate handling. Magnetic resonance spectroscopic [411] and biochemical analyses [412] confirm defective GLUT4 translocation in muscle of T2DM patients, while studies in animal models confirm impaired translocation in skeletal [405] and cardiac tissue [413, 414]. Dissociation of T-tubules has been shown to reduce basal and abolish insulin-dependent glucose transport in skeletal muscle [415], confirming a critical role in glucose metabolism and homeostasis. Since the majority of GLUT4 translocation occurs specifically within T-tubules [415, 416] and cholesterol-rich microdomains [253], T-tubule disruption and changes in cholesterol will modify insulin-stimulated GLUT4 exocytosis in DM.
Changes in both membrane cholesterol and caveolae/caveolins may contribute to the T-tubule dysfunction in DM. In skeletal myocytes, cholesterol is more concentrated within T-tubules compared to surface membrane regions [417–419], which may contribute to lower fluidity in the lipid phase of T-tubules compared with most cell membranes [420]. While data are lacking for cardiomyocytes, similar compartmentation in T-tubules is likely. Cardiac Ca2+ levels and contractility are sensitive to membrane cholesterol [267, 332], and Zhu et al. [330] recently confirmed cholesterols importance to cardiomyocyte T-tubule stability and E–C coupling, an effect apparently independent of caveolin-3/caveolae. The integrity of intercalated disks and intercellular communication were also sensitive to cholesterol. Caveolae and caveolins are also important in T-tubule development and maintenance of functional integrity [421], including the co-localization and interaction between junctophilin-2 and caveolin-3 in dyadic structures to establish efficient, synchronous EC coupling. Depletion of caveolin-3 could contribute to loss of dyadic integrity, and junctophilin/caveolin-3 interactions are known to be suppressed in cardiomyopathy [422].
Changes in gap junctions and sarcolemmal ion channels in DM
Gap junctions
Abnormal conduction and arrhythmogenesis is evident in both DM patients [423, 424] and animal models of T1 and T2DM [425–427]. Together with changes to the T-tubule system, shifts in gap junctions [162] and sarcolemmal ion (Ca2+, Na+, K+) channels will disrupt electrophysiology in DM, and influence cardiac responses to I–R. Specialized gap junction pores provide effective electrical coupling of adjacent cardiomyocytes, and are critical not only to conduction and electrical stability but responses to ischemia and cardioprotective stimuli [428]. Principle connexin (CX) protein components are altered in DM, including evidence of modified expression and phosphorylation. The latter post-translational changes are functionally important: PKC inhibits cardiac gap junction conductance [429, 430] via CX-43 phosphorylation [431, 432]; dephosphorylation of gap junction elements results in their uncoupling [433] and lateralization [432, 434]; and excess phosphorylation of CX-43 by PKCε may promote proteolysis to deplete junction channels in DM myocardium [435].
In cultured myocytes CX-43 expression is suppressed by hyperglycemia [436], potentially involving PKC-dependent miR-1/206 expression [437]; and by the AGE-RAGE system, potentially involving PKC and ERK signaling [438]. In STZ-dependent T1DM in rats an increased SA nodal expression of CX-43 (and -40 and -45) is associated with nodal conduction delay [439], while ventricular expression is reportedly unaltered [440], reduced [435] or increased [441, 442]. Olsen et al. [426] observe reduced lateralization of CX-43 in hearts from ZDF rats exhibiting reduced conduction velocity. Phosphorylation of atrial and ventricular CX-43 declines in models of DM [439, 442, 443], potentially as a result of impaired PKCε expression [444], though these investigators also report increased PKCε mediated CX-43 phosphorylation in DM myocardium, which may promote proteolytic degradation [435]. The extent of cardiac CX-43 phosphorylation reportedly declines with progression of DM while protein nitration increases [443]. Zhu et al. [330] also recently found that cholesterol depletion not only destabilized cardiomyocyte T-tubules, but disrupted the integrity of intercalated disks and intercellular communication. Gap junction function and inter-cellular communication may therefore be influenced by sarcolemmal cholesterol changes in DM. Supporting mechanistic involvement of gap junction changes in the myocardial abnormalities of DM, benefit with exercise in T2DM db/db mice is attributed to restoration of CX-43 networks [445], and beneficial effects of n-3 PUFA feeding on DM cardiomyopathy are linked to increased CX-43 expression and phosphorylation (associated with up-regulated PKCε) [414]. In contrast, it has also been reported that moderate exercise reduces ventricular CX-43 phosphorylation [446].
Ion channels
Sarcolemmal ion channels fundamental to E–C coupling and relevant to I–R injury are modified in DM myocardium, including changes in Ca2+ channels, levels and contractile responses [447–451], K+ currents and channels [452–454], and Na+ pumps [247, 455]. These may participate in enhanced arrhythmogenesis and risk of sudden cardiac death [423, 425, 427, 452]. Altered membrane lipids and biophysical properties in DM will influence ion channel function, and changes in channel transcription and expression patterns also arise. There is evidence of increased transcription of Ca2+ channels (Cacna1c, Cacna1g, Cacnb1) and Gja4 (CX-37), and differential changes in K+ channels (Kcnj11 up, Kcnb1 down) in GK T2DM rat hearts [450]. Sucrose feeding induces K+ channels (Kcnj2, Kcnj8) and Gja1 (CX-43) and Gja4 in non-DM rats [450]. This group also reports up-regulation of ventricular Cacna1h, Scn1b and Hcn2 vs. down-regulation of Hcn4, Kcna2, Kcna4 and Kcnj2 in this model [449], and up-regulation of genes encoding cardiac LTCC proteins (Cacna1c, Cacna1g, Cacna1h and Cacna2d1) in association with prolongation of Ca2+ transients in the ZDF rat model of T2DM [448].
These transcriptional changes do translate to altered channel expression, with shifts in Ca2+, K+ and Na+ channels all potentially contributing to electrophysiological perturbations in DM hearts. Abnormal Ca2+ currents in cardiomyocytes from T1DM Akita mice involve reduced sarcolemmal levels of the LTCC, potentially related to impaired PI3K control [456]. The decline in sarcolemmal Ca2+ permeability in T2DM db/db mice is associated with reduced expression of the pore-forming α1C subunit of the LTCC [447]. Though less well studied, cardiac T-type Ca2+ channel expression/function may also be modified in DM given caveolar localization and sensitivity to caveolin-3 [457], and evidence of changes in other cell types with chronic DM [458]. Reductions in cardiomyocyte K+ current density in models of T1DM also involve defective channel expression (potentially involving AMPK signaling) [459], and action potential prolongation in Otsuka-Long-Evans-Tokushima Fatty rats is linked to down-regulation of endocardial Kv4.2 (voltage-gated K+ channel subfamily D) and transmural KChIP2 (K+ channel interacting protein) expression [454]. Impaired insulin signaling has been shown to reduce the amplitude of the transient outward K+ current fast component in cardiomyocytes in association with reduced Kv4.2 and KChIP2 expression [453]. Broadened ventricular repolarization and reduced ‘repolarization reserve’ in alloxan-induced T1DM in dogs may also involve impaired K+ currents as a result of reduced Kv4.3 (voltage-gated K+ channel subfamily D) and MinK (voltage-gated K+ channel sub-family E subunit) expression, while Kv1.4 (voltage-gated K+ channel subfamily A), KChIP2 and KvLQT1 (voltage-gated K+ channel subfamily D) were increased [452]. Such changes may significantly predispose to sudden cardiac death.
Depressed I Na may additionally play a role in altered electrical activity in DM cardiomyocytes, with less Na+ influx during contraction linked to reduced expression of both the Na+/K+-ATPase and Na+/Ca2+ exchanger [247]. This is consistent with sarcolemmal Na+-K+ pump inhibition in other models of T1DM [455]. Changes in Ca2+, K+, and Na+ channels are not only likely to increase susceptibility to arrhythmias in I–R, but may well modulate cell death processes. Moreover, expression of subunits for the sarcolemmal KATP channel implicated in multiple cardioprotective responses [72] is also disrupted in DM, with SUR2A and Kir6.2 decreased both in myocytes from T1DM rats and isolated myocytes subjected to hyperglycemia [460]. Such a change will not only desensitize cardiac myocytes to KATP openers, but impair transduction of cardioprotective signaling. The impacts of these varied channel expression changes in DM will be exacerbated by membrane lipid changes and structural modifications to the sarcolemma, including shifts in microdomains in which select channels cluster, and T-tubule and gap-junction remodeling.
Potential ‘membrane-targeted’ therapies?
Based on the array of detrimental sarcolemmal changes evident in DM, a number of therapeutic approaches present themselves, including modifications to diet and physical activity, cholesterol manipulation, and modulation of caveolins and caveolar biology.
Targeting caveolae and caveolins
Given evidence of abnormal caveolin-3 expression in models of DM, this caveolar protein has appeal as a therapeutic target [158, 159, 169]. Beneficial anti-diabetic effects of hepatic caveolin-3 gene transfer supports the therapeutic potential of caveolin-3 in DM [315]. Although the regulation of cardiac caveolin-3 expression is not well understood, there is evidence from non-cardiac cells for transcriptional control by myogenin, ID2, miR-22 and myocardin [289]. Myocardins are important to formation of caveolae, and in glucose and lipid homeostasis [292]. Whether transcriptional control of caveolins might be targetable is not clear. However, hyperinsulinemia does up-regulate myocardin in cardiac myoblasts [290], which additional to modulating hypertrophy could up-regulate caveolins and caveolae [289]. Conversely, insulin-resistance may reduce myocardin expression and thereby caveolins and caveolae. Aortic myocardin is substantially induced in GK (T2DM) rats, which appears to involve a miR-145 dependent response to oxidative stress [291].
Interestingly, cardiac caveolin-3 may be differentially modifiable via dietary saturated fat [276] and PUFA [278, 461], and hyperglycemic depression of caveolin-3 may also be PKCβ2-dependent, providing a potential pharmacological target. Lei et al. [240] show that inhibition or knockdown of PKCβ2 counters hyperglycemic depression of caveolin-3 in hearts and myocytes, and improves cardiac Akt phosphorylation and diastolic function. This group subsequently showed that PKCβ2 inhibition also improved cardiac I–R tolerance together with caveolin-3 levels and control of Akt signaling in STZ-dependent T1DM rats [241]. Supplementation with the anti-oxidant NAC also attenuates PKCβ2 expression and hypertrophy [462] while enhancing ischemic tolerance [275] in STZ-dependent T1DM rats. Caveolin-3 levels were not measured, though a reduction in PKCβ2 activity is predicted to improve caveolin-3 based on other work [250].
Other potential targets include adenylyl cyclase (AC) and focal adhesion kinase (FAK) signaling paths: in vitro studies suggest AC can repress caveolin-3 in cardiac myoblasts [463] while FAK up-regulates caveolin-3 in skeletal myoblasts [464]. No data are available regarding cardiac FAK signaling in DM, however, FAK may be activated in hyperglycemic conditions [465], and FAK induction in hypertrophied skeletal muscle is exaggerated in T1DM rats [466]. In skeletal myotubes FAK also appears important in insulin-dependent GLUT4 translocation and glucose uptake [467]. Adenylate cyclase itself appears functionally unaltered in DM hearts, while adrenergic receptor mediated control is impaired [468]. However, vascular AC expression/function may be altered in DM [469]. Inhibition of cardiac AC5 activity does protect against cardiac abnormalities in T2DM and obesity [470].
Targeting caveolin-3 expression via acute gene therapy with adeno-associated virus (AAV) for Cav3 improves I–R and Ca2+ tolerance, preserves mitochondrial stability and reduces reactive oxygen species [281]. In addition, cardiac specific caveolin-3 overexpression enhances functional outcomes post-I–R and reduces infarct size (similar to effects of ischemic preconditioning), which may be due to improved mitochondrial Ca2+ tolerance and respiratory rates with reduced ROS generation [281]. Fridolfsson et al. [281] identified that increased O2 consumption in caveolin-3 overexpressing hearts improved energy production without a parallel increase in ROS generation. Further experiments targeting caveolin-3 to mitochondria confirmed improved mitochondrial stability during Ca2+ challenge, and delayed mitochondrial depolarization and improved respiratory complex activity associated with enhanced ischemic tolerance. Conversely, deletion results in mitochondrial dysfunction [281] and hypertrophy [287, 288]. How caveolin-3 migrates to/communicates with mitochondria and subsequently promotes mitochondrial and ischemic tolerance remains to be further detailed.
Cholesterol lowering therapies
Reducing membrane cholesterol has capacity to improve fluidity and counter some sarcolemmal abnormalities evident in DM hearts. On the other hand, whether reductions in cholesterol might adversely impact caveolae, caveolins and T-tubules is unclear. Certainly, statins are of value in DM, with low-dose treatment significantly reducing cardiovascular events in T2DM patients [471]. However, while pleiotropic effects of statins include ‘anti-diabetic’ actions such as reduced inflammation in T2DM patients [472], they may also include induction of insulin-resistance and promotion of DM [473, 474]. That said, such effects appear modest relative to the benefits of statins, and may only be a factor in those at particular risk of new onset DM [475]. Experimental studies show statins do protect against myocardial ischemic injury in hearts from DM and healthy animals, though again this reflects pleiotropic effects of the drugs independent of cholesterol lowering [476, 477].
Dietary intervention
Modifiable diet and physical activity have long been appreciated as major determinants of DM severity and complications. Dietary modification can alter sarcolemmal makeup and function, and inflammatory, glycation/glycosylation and oxidative processes in the heart and vessels. For example, homeostatic control of inflammation is mediated by eicosanoids (prostaglandins, leukotrienes, thromboxanes) whose generation is dependent on the n-6 PUFA AA [179]. Shifts in saturated vs. unsaturated fat intake can modify fundamental membrane properties together with caveolar components, while limitations in caloric intake may profoundly influence the DM phenotype and promote protective outcomes.
Unsaturated vs. saturated fats
Mammalian species are unable to produce n-3 PUFAs, thus must acquire these essential fatty acids via the diet. Edible seeds such as flaxseed and chia seeds are rich sources of the 18C n-3 PUFA α-linolenic acid, while longer chain n-3 PUFAs (EPA, DHA) can be synthesized from α-linolenic acid or consumption of fish oils. Once acquired, n-3 PUFAs can integrate into the sarcolemma to displace membrane AA: dietary n-3 PUFA incorporation in myocardium and myocytes occurs at the expense of n-6 PUFAs [478]. Consumption of n-3 PUFAs thus reduces inflammation via disrupting production of AA-derived eicosanoids [479, 480]. However, it is worth noting that AA-derived eicosanoids (including prostaglandin E2) exhibit both pro- and anti-inflammatory capabilities.
Dietary α-linolenic acid is cardioprotective in a rat model of T2D [143], with 4 weeks of α-linolenic acid supplementation improving ischemic tolerance including enhanced functional outcomes and reductions in infarction and markers of cell death (whereas no protection was evident in non-DM rats). Cardioprotection was linked to anti-inflammatory (reduced tumor necrosis factor-α and interleukin-6) and anti-oxidative (reduced superoxide and enhanced anti-oxidant capacity) actions, possibly involving PI3K/Akt signaling [143]. Insulin-resistance, glucose intolerance, dyslipidemia and cardiac lipid accumulation after 3–6 months of a high-sugar diet are also reversed by transition to a chia seed-rich diet [481]. Consumption of n-3 PUFAs improves sarcolemmal functions, critical to the management of DM cardiomyopathy. For example, consumption of fish oils: enhances EPA and DHA in cardiac membranes while reducing AA [479, 480]; prevents translocation of CD36, limiting fatty acid uptake and lipid storage [482]; and counters abnormal membrane fluidity in T1DM mice [195]. A vegetarian diet improvement in linoleic acid content is also associated with improved insulin sensitivity in subjects with T2DM [483].
Diets containing high ratios of PUFA/mono-unsaturated fatty acid (MUFA) improve insulin-binding and glucose uptake in adipose cells from healthy and T1DM rats [484]. Membranous phospholipid content is also altered, with enhanced PUFA and reduced MUFA (though no effect on total saturated phospholipids) [484]. Interestingly, even at very high insulin levels (1000 ng/mL), cells from T1DM rats fed low PUFA/MUFA diets bind less insulin than those fed high PUFA/MUFA diets and exposed to lower insulin levels. This suggests that insulin has greater affinity for cells with more unsaturated membranes, which may be particularly useful in management of insulin-resistant T2DM.
Enriched n-3 PUFA diets also modify ion exchange and action potential duration, which may limit cardiac propensity to I–R injury and arrhythmias. Isolated myocytes from rabbits fed fish oil for 3 weeks exhibit increased sarcolemmal EPA and DHA (vs. decreased MUFAs) and 20% shorter action potentials compared with myocytes from animals on a n-9 MUFA-rich diet [485]. Exposure of myocytes to EPA and DHA shortened action potentials in cells from n-9 MUFA and not n-3 PUFA fed rabbits. These findings indicate action potential shortening likely stems from altered membrane lipid composition and not direct ligand-like interaction with ion channels [485]. Other studies report inhibitory effects of PUFAs on sarcolemmal K+ [486] and Ca2+ channels [487], and the Na+/H+ exchanger [488], potentially limiting pathological Ca2+ overload in myocardial cells.
Dietary fats also influence caveolin expression and thus caveolae. A palmitate enriched diet significantly depresses cardiac caveolin-3 [276], whereas a flaxseed-enriched diet reverses reductions in cardiac caveolin-3 in cardiomyopathic hamsters [278], and prevents reductions in skeletal muscle caveolin-3 in a model of muscular dystrophy (also repairing sarcolemmal damage, reducing inflammation and cell death) [459]. Effectiveness of such diet intervention in a DM animal model awaits testing. In addition to n-3 PUFA supplementation, improved cardiac function in DM may be achievable through calorie restriction (CR).
Caloric limitation and time-restricted feeding
Calorie restriction or intermittent fasting may provide significant benefit in DM, and such interventions modify membrane composition in murine myocardium [489]. Though prolonged caloric limitation is a well-established protective stimulus, effects of brief or moderate fasting await detailed study in DM animals. Severe CR for 11 days generates unique I–R tolerance [490], and 24–72 h of fasting enhances cardiac I–R tolerance and mitochondrial viability in non-DM hearts [491, 492]. There are surprisingly few studies of caloric limitation in DM. A 30% limitation in calories for ≥ 2 months improves glucose homeostasis and markers of systemic or cardiac oxidative-stress in rodent models of T2DM [493, 494]. A similar CR regime fails to influence I–R tolerance in models of T2DM and metabolic syndrome, though benefit via ischemic preconditioning was restored [144]. Contrasting reported protection with fasting, one recent study suggests 18 h of fasting actually worsens ischemic tolerance in T2DM and also non-DM rat hearts [495], potentially linked to enhanced glucose vs. fatty acid metabolism. Another recent study [496] also found that loss of sevoflurane preconditioning with a high calorie western diet was unaltered with 4 week of control diet (though an apparently detrimental impact of sevoflurane with the western diet was countered).
Whether ischemic tolerance with caloric limitation involves membrane changes in either DM or non-DM hearts remains to be established. However, modest (12 h) fasting does induce membrane remodeling via a reduction in acyl chains, predominately lost from C22:6 (DHA) species [489]. While effects of CR on myocardial caveolar domains are unknown, it does prevent age-related reductions in caveolin-1 in liver tissue [497], and repression of caveolin-1 in breast tissue is mediated by a micro-RNA (miR-203) that is induced with CR [498].
Circadian biology is extremely important in the influences of fat and calorie intake on obesity and associated metabolic disturbances, and restricted feeding times rather than calorie intakes can be highly beneficial in cardiometabolic disorders [499]. The timing of food intake appears a key determinant of circadian rhythm, particularly in metabolic organs, and the impacts of high-fat feeding on body weight, insulin levels, glucose tolerance, inflammation and hepatic steatosis can be effectively countered by time-restricted feeding without caloric limitation [500]. Conversely, short-term feeding at the wrong time of day can desynchronize peripheral clocks and induce obesity and metabolic disorder [501]. Time-restricted feeding also counters cardiac aging changes in the Drosophila model [502], however effects on myocardial ischemic tolerance, or the cardiomyopathy and sarcolemmal changes in DM, have yet to be tested.
Exercise in DM—membrane involvement?
Physical activity and VO2 have been identified as perhaps the most important factors governing chronic disease risk, particularly CVD and DM [503]. Up to 50% of coronary artery disease can be prevented by 30 min of moderate exercise daily (assessed in middle-aged women) [504–506], and as little as 3 weeks of exercise can reduce the clinical impact of metabolic syndrome (a combination of coronary heart disease, hypertension and T2DM) by 50% [507]. Not only substantially reducing risk/incidence, exercise can be applied ‘therapeutically’ in existing disease states to alleviate symptoms and counter progression. Broadly beneficial systemic effects render physical activity an effective therapy in disorders including cancer [508], depression [509] and cardiovascular disease [510]. Exercise induces obvious metabolic advantages, improving tissue and whole body VO2/oxidative capacity and vascularity, cardiac functional reserve and efficiency, insulin signaling and sensitivity, glucose and fat handling, anti-oxidant status, inflammation and immune function [510–512]. Analyses confirm benefits of physical activity in patients with T2DM, though questions regarding effective exercise prescription remain [513, 514]. Not only reducing the incidence of infarction, exercise also boosts myocardial tolerance to infarction [515] and may improve or restore conventional protective responses in models of stress, disease and aging [516]. Effects on the DM heart revolve around improved glucose and fatty acid metabolism, mitochondrial function and oxidative stress, however sarcolemmal abnormalities are also influenced. Exercise does modify fatty acid composition of phospholipids and triglycerides in cardiac and skeletal muscle [517, 518], and beneficial remodeling of plasma membrane lipids is reported in other cell types [519]. Sarcolemmal effects in DM are less well defined.
Studies confirm exercise-dependent improvements in cardiac function, survival signaling and ischemic tolerance in models of T1DM [136] and T2DM [146, 520, 521]. While improvements in substrate metabolism are broadly implicated, Pons et al. [520] report cardioprotection in ob/ob mice independent of hyperglycemia, hypercholesterolemia, hyperinsulinemia, fat mass or body weight. Schrauwen-Hinderling et al. [522] found that 12 week endurance/strength training improves systemic insulin sensitivity and cardiac function in T2DM patients without modifying cardiac lipid content. Altered myocardial O-GlcNAcylation may participate, with evidence swimming in T1DM rats increases OGA activity and reduces cardiac protein O-GlcNAcylation [523]. However, this also reduces O-GlcNAcylation in non-DM hearts [524]. Indeed, Medford et al. [525] show as little as 15 min of exercise can alter myocardial O-GlcNAcylation. Exercise protection in models of DM has been linked to normalization of nitro-oxidative stress and eNOS control [146], and improvements in PPARγ coactivator-1α and Akt signaling [521], both effects that may arise via restoration of sarcolemmal caveolae and caveolin control of eNOS [250, 275, 296–298] and Akt signaling [250–252, 299]. Studies in non-DM [526] and DM hearts [294] do support up-regulation of caveolin-3, though the contribution of this change to exercise cardioprotection awaits analysis. Indirectly supporting targeting of sarcolemmal elements, da Silva et al. [449] show that altered Ca2+ transients (and mitochondrial uptake) in T1DM rat hearts are countered by swimming, which also enhanced benefit via insulin.
More directly supporting improved sarcolemmal makeup, Hesari et al. [444] report that exercise reduces CX-43 phosphorylation in hearts from T1DM rats, and Veeranki et al. [443] demonstrate beneficial effects of exercise on CX-43 levels and gap-junction function in db/db mice, associated with preservation of mitochondrial function. This is consistent with evidence exercise modulates sarcolemmal determinants of signaling and E–C coupling in T2DM rats, including transcriptional up-regulation of caveolin-3 and CX-43, and differential changes in K+ channels (Hcn2, Kcnk3) [294]. Our unpublished findings support up-regulation of cardiac caveolin-3 and protection against I–R with swim training in mice, coupled with powerful anti-inflammatory effects of exercise (data not shown).
Conclusions and perspectives
A diversity of mechanisms are involved in the cardiac and coronary abnormalities arising in DM, and evolution of DM cardiomyopathy. However, the sarcolemma is a nexus for many fundamental mechanistic elements and sequelae of DM. The ability of the sarcolemma to withstand rupture is fundamentally important to cell survival and stress tolerance and is governed by molecular makeup and caveolar membrane ‘reserve’. The sarcolemma is also the seat of glucose and fatty acid transport and InsR control, and therefore fundamentally participates in the pathogenesis of DM complications. Furthermore, the functionality of ion channels and cell surface receptors is determined by membrane makeup. Diabetes impacts sarcolemmal architecture, remodeling T-tubules, caveolar domains and gap junctions, disrupting E–C coupling and promoting injury and arrhythmogenesis in I–R. Specific molecular changes include increased cholesterol and fatty acid saturation vs. reduced desaturation, and differential shifts in phospholipids and PUFAs. Caveolar proteins are a particularly important target in DM, with evidence for caveolin-3 depletion and caveolae dysfunction in dysregulation of GLUT4 and CD36 function, survival kinase and eNOS signaling. Importantly, the sarcolemma is malleable, responsive to dietary modification, physical activity and other interventions. A further unraveling of the roles of sarcolemmal changes in DM and its cardiac complications thus has potential to inform approaches to managing these disorders, improving ischemic tolerance and developing cardioprotective therapies for the DM population. This requires further focused investigation of sarcolemmal changes in animal models and particularly in sufferers of T1 and T2DM, though the latter presents a significant experimental challenge.
Authors’ contributions
Drafted manuscript: JR, JPH. Critical analysis: EFD, JNP, HHP. Approval of final manuscript: JR, JPH, EFD, JNP, HHP. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
JR was supported by a doctoral scholarship award from the Australian government.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jake Russell, Email: j.russell@griffith.edu.au.
Eugene F. Du Toit, Email: j.dutoit@griffith.edu.au
Jason N. Peart, Email: j.peart@griffith.edu.au
Hemal H. Patel, Email: Hepatel@ucsd.edu
John P. Headrick, Phone: +61 7 55528292, Email: j.headrick@griffith.edu.au
References
- 1.Marso SP, Miller T, Rutherford BD, Gibbons RJ, Qureshi M, Kalynych A, Turco M, Schultheiss HP, Mehran R, Krucoff MW, Lansky AJ, Stone GW. Comparison of myocardial reperfusion in patients undergoing percutaneous coronary intervention in ST-segment elevation acute myocardial infarction with versus without diabetes mellitus (from the EMERALD Trial) Am J Cardiol. 2007;100:206–210. doi: 10.1016/j.amjcard.2007.02.080. [DOI] [PubMed] [Google Scholar]
- 2.Miki T, Itoh T, Sunaga D, Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol. 2012;11:67. doi: 10.1186/1475-2840-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh S, Standen NB, Galiñanes M. Failure to precondition pathological human myocardium. J Am Coll Cardiol. 2001;37:711–718. doi: 10.1016/S0735-1097(00)01161-X. [DOI] [PubMed] [Google Scholar]
- 4.Lee TM, Chou TF. Impairment of myocardial protection in type 2 diabetic patients. J Clin Endocrinol Metab. 2003;88:531–537. doi: 10.1210/jc.2002-020904. [DOI] [PubMed] [Google Scholar]
- 5.Hassouna A, Loubani M, Matata BM, Fowler A, Standen NB, Galiñanes M. Mitochondrial dysfunction as the cause of the failure to precondition the diabetic human myocardium. Cardiovasc Res. 2006;69:450–458. doi: 10.1016/j.cardiores.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Przyklenk K. Efficacy of cardioprotective ‘conditioning’ strategies in aging and diabetic cohorts: the co-morbidity conundrum. Drugs Aging. 2011;28:331. doi: 10.2165/11587190-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Peart JN, Headrick JP. Clinical cardioprotection and the value of conditioning responses. Am J Physiol Heart Circ Physiol. 2009;296:H1705–H1720. doi: 10.1152/ajpheart.00162.2009. [DOI] [PubMed] [Google Scholar]
- 8.Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66:1142–1174. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- 9.Yamagishi S, Nakamura N, Matsui T. Glycation and cardiovascular disease in diabetes: a perspective on the concept of metabolic memory. J Diabetes. 2017;9:141–148. doi: 10.1111/1753-0407.12475. [DOI] [PubMed] [Google Scholar]
- 10.Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142:375–415. doi: 10.1016/j.pharmthera.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Zhao D, Yang J, Yang L. Insights for oxidative stress and mTOR signaling in myocardial ischemia/reperfusion injury under diabetes. Oxid Med Cell Longev. 2017;2017:6437467. doi: 10.1155/2017/6437467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whittington HJ, Harding I, Stephenson CI, Bell R, Hausenloy DJ, Mocanu MM, Yellon DM. Cardioprotection in the aging, diabetic heart: the loss of protective Akt signalling. Cardiovasc Res. 2013;99:694–704. doi: 10.1093/cvr/cvt140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghaboura N, Tamareille S, Ducluzeau PH, Grimaud L, Loufrani L, Croué A, Tourmen Y, Henrion D, Furber A, Prunier F. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3β signaling. Basic Res Cardiol. 2011;106:147–162. doi: 10.1007/s00395-010-0130-3. [DOI] [PubMed] [Google Scholar]
- 14.Gross ER, Hsu AK, Gross GJ. Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3beta. Diabetes. 2007;56:127. doi: 10.2337/db06-0907. [DOI] [PubMed] [Google Scholar]
- 15.Davidson SM, Riquelme JA, Takov K, Vicencio JM, Boi-Doku C, Khoo V, Doreth C, Radenkovic D, Lavandero S, Yellon DM. Cardioprotection mediated by exosomes is impaired in the setting of type II diabetes but can be rescued by the use of non-diabetic exosomes in vitro. J Cell Mol Med. 2017 doi: 10.1111/jcmm.13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams B, Mapanga RF, Essop MF. Partial inhibition of the ubiquitin-proteasome system ameliorates cardiac dysfunction following ischemia-reperfusion in the presence of high glucose. Cardiovasc Diabetol. 2015;14:94. doi: 10.1186/s12933-015-0258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding M, Lei J, Han H, Li W, Qu Y, Fu E, Fu F, Wang X. SIRT1 protects against myocardial ischemia-reperfusion injury via activating eNOS in diabetic rats. Cardiovasc Diabetol. 2015;14:143. doi: 10.1186/s12933-015-0299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Ren Y, Shi E, Tan Z, Xiong J, Yan L, Jiang X. Inhibition of the Let-7 family microRNAs induces cardioprotection against ischemia-reperfusion injury in diabetic rats. Ann Thorac Surg. 2016;102:829–835. doi: 10.1016/j.athoracsur.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Liang Q, Kobayashi S. Mitochondrial quality control in the diabetic heart. J Mol Cell Cardiol. 2016;95:57–69. doi: 10.1016/j.yjmcc.2015.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie J, Cui K, Hao H, Zhang Y, Lin H, Chen Z, Huang X, Cao S, Liao W, Bin J, Kitakaze M, Liao Y. Acute hyperglycemia suppresses left ventricular diastolic function and inhibits autophagic flux in mice under prohypertrophic stimulation. Cardiovasc Diabetol. 2016;15(1):136. doi: 10.1186/s12933-016-0452-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pilon M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016;15:167. doi: 10.1186/s12944-016-0342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glyn-Jones S, Song S, Black MA, Phillips AR, Choong SY, Cooper GJ. Transcriptomic analysis of the cardiac left ventricle in a rodent model of diabetic cardiomyopathy: molecular snapshot of a severe myocardial disease. Physiol Genom. 2007;28:284–293. doi: 10.1152/physiolgenomics.00204.2006. [DOI] [PubMed] [Google Scholar]
- 23.Sárközy M, Szűcs G, Fekete V, Pipicz M, Éder K, Gáspár R, Sója A, Pipis J, Ferdinandy P, Csonka C, Csont T. Transcriptomic alterations in the heart of non-obese type 2 diabetic Goto-Kakizaki rats. Cardiovasc Diabetol. 2016;15:110. doi: 10.1186/s12933-016-0424-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone PH, Muller JE, Hartwell T, York BJ, Rutherford JD, Parker CB, Turi ZG, Strauss HW, Willerson JT, Robertson T. The effect of diabetes mellitus on prognosis and serial left ventricular function after acute myocardial infarction: contribution of both coronary disease and diastolic left ventricular dysfunction to the adverse prognosis. The MILIS Study Group. J Am Coll Cardiol. 1989;14:49–57. doi: 10.1016/0735-1097(89)90053-3. [DOI] [PubMed] [Google Scholar]
- 25.Melchior T, Kober L, Madsen CR, Seibaek M, Jensen GV, Hildebrandt P, Torp-Pedersen C. Accelerating impact of diabetes mellitus on mortality in the years following an acute myocardial infarction. TRACE Study Group. Trandolapril Cardiac Evaluation. Eur Heart J. 1999;20:973–978. doi: 10.1053/euhj.1999.1530. [DOI] [PubMed] [Google Scholar]
- 26.Carrabba N, Valenti R, Parodi G, Santoro GM, Antoniucci D. Left ventricular remodeling and heart failure in diabetic patients treated with primary angioplasty for acute myocardial infarction. Circulation. 2004;110:1974–1979. doi: 10.1161/01.CIR.0000143376.64970.4A. [DOI] [PubMed] [Google Scholar]
- 27.Prasad A, Stone GW, Stuckey TD, Costantini CO, Zimetbaum PJ, McLaughlin M, Mehran R, Garcia E, Tcheng JE, Cox DA, Grines CL, Lansky AJ, Gersh BJ. Impact of diabetes mellitus on myocardial perfusion after primary angioplasty in patients with acute myocardial infarction. J Am Coll Cardiol. 2005;45:508–514. doi: 10.1016/j.jacc.2004.10.054. [DOI] [PubMed] [Google Scholar]
- 28.Timmer JR, van der Horst IC, de Luca G, Ottervanger JP, de Hoorntje JC, Boer MJ, Suryapranata H, Dambrink JH, Gosselink M, Zijlstra F, van’t Hof AW, Zwolle Myocardial Infarction Study Group Comparison of myocardial perfusion after successful primary percutaneous coronary intervention in patients with ST-elevation myocardial infarction with versus without diabetes mellitus. Am J Cardiol. 2005;95:1375–1377. doi: 10.1016/j.amjcard.2005.01.088. [DOI] [PubMed] [Google Scholar]
- 29.Nitenberg A, Valensi P, Sachs R, Dali M, Aptecar E, Attali J-R. Impairment of coronary vascular reserve and ACh-induced coronary vasodilation in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes. 1993;42:1017–1025. doi: 10.2337/diab.42.7.1017. [DOI] [PubMed] [Google Scholar]
- 30.Nahser PJ, Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation. 1995;91:635–640. doi: 10.1161/01.CIR.91.3.635. [DOI] [PubMed] [Google Scholar]
- 31.Feng J, Chu LM, Dobrilovic N, Liu Y, Singh AK, Sellke FW. Decreased coronary microvascular reactivity after cardioplegic arrest in patients with uncontrolled diabetes mellitus. Surgery. 2012;152:262–269. doi: 10.1016/j.surg.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alegria JR, Miller TD, Gibbons RJ, Yi QL, Yusuf S. Infarct size, ejection fraction, and mortality in diabetic patients with acute myocardial infarction treated with thrombolytic therapy. Am Heart J. 2007;154:743–750. doi: 10.1016/j.ahj.2007.06.020. [DOI] [PubMed] [Google Scholar]
- 33.Mather AN, Crean A, Abidin N, Worthy G, Ball SG, Plein S, Greenwood JP. Relationship of dysglycemia to acute myocardial infarct size and cardiovascular outcome as determined by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2010;12:61–66. doi: 10.1186/1532-429X-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Currie CJ, Poole CD, Evans M, Peters JR, Morgan CL. Mortality and other important diabetes-related outcomes with insulin vs other antihyperglycemic therapies in type 2 diabetes. J Clin Endocrinol Metab. 2013;98:668–677. doi: 10.1210/jc.2012-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Amsterdam EA, Young JN, Hoegh H, Armstrong EJ. Contemporary outcomes of coronary artery bypass grafting among patients with insulin-treated and non-insulin-treated diabetes. Ann Thorac Surg. 2015;100:2262–2269. doi: 10.1016/j.athoracsur.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 36.Munnee K, Bundhun PK, Quan H, Tang Z. Comparing the clinical outcomes between insulin-treated and non-insulin-treated patients with type 2 diabetes mellitus after coronary artery bypass surgery: a systematic review and meta-analysis. Medicine (Baltimore) 2016;95:e3006. doi: 10.1097/MD.0000000000003006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Luca G, Parodi G, Sciagrà R, Bellandi B, Vergara R, Migliorini A, Valenti R, Antoniucci D. Effect of diabetes on scintigraphic infarct size in STEMI patients undergoing primary angioplasty. Diabetes Metab Res Rev. 2015;31:322–328. doi: 10.1002/dmrr.2620. [DOI] [PubMed] [Google Scholar]
- 38.Eitel I, Hintze S, de Waha S, Fuernau G, Lurz P, Desch S, Schuler G, Thiele H. Prognostic impact of hyperglycemia in nondiabetic and diabetic patients with ST-elevation myocardial infarction: insights from contrast-enhanced magnetic resonance imaging. Circ Cardiovasc Imaging. 2012;5:708–718. doi: 10.1161/CIRCIMAGING.112.974998. [DOI] [PubMed] [Google Scholar]
- 39.Sanidas EA, Brener SJ, Maehara A, Généreux P, Witzenbichler B, El-Omar M, Fahy M, Mehran R, Gibson CM, Stone GW. Outcomes in diabetic patients undergoing primary percutaneous coronary intervention for acute anterior myocardial infarction: results from the INFUSE-AMI study. Catheter Cardiovasc Interv. 2014;83:704–710. doi: 10.1002/ccd.25203. [DOI] [PubMed] [Google Scholar]
- 40.Risum O, Abdelnoor M, Svennevig JL, Levorstad K, Gullestad L, Bjørnerheim R, Simonsen S, Nitter-Hauge S. Diabetes mellitus and morbidity and mortality risks after coronary artery bypass surgery. Scand J Thorac Cardiovasc Surg. 1996;30:71–75. doi: 10.3109/14017439609107245. [DOI] [PubMed] [Google Scholar]
- 41.Barsness GW, Peterson ED, Ohman EM, Nelson CL, DeLong ER, Reves JG, Smith PK, Anderson RD, Jones RH, Mark DB, Califf RM. Relationship between diabetes mellitus and long-term survival after coronary bypass and angioplasty. Circulation. 1997;96:2551–2556. doi: 10.1161/01.CIR.96.8.2551. [DOI] [PubMed] [Google Scholar]
- 42.Carson JL, Scholz PM, Chen AY, Peterson ED, Gold J, Schneider SH. Diabetes mellitus increases short-term mortality and morbidity in patients undergoing coronary artery bypass graft surgery. J Am Coll Cardiol. 2002;40:418–423. doi: 10.1016/S0735-1097(02)01969-1. [DOI] [PubMed] [Google Scholar]
- 43.Feng J, Liu Y, Dobrilovic N, Chu LM, Bianchi C, Singh AK, Sellke FW. Altered apoptosis-related signaling after cardioplegic arrest in patients with uncontrolled type 2 diabetes mellitus. Circulation. 2013;128(Suppl 1):S144–S151. doi: 10.1161/CIRCULATIONAHA.112.000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen-Scarabelli C, Knight R, Stephanou A, Scarabelli G, Onorati F, Tessari M, Rungatscher A, Narula J, Saravolatz L, Mazzucco A, Faggian G, Scarabelli TM. Diabetic hearts have lower basal urocortin levels that fail to increase after cardioplegic arrest: association with increased apoptosis and postsurgical cardiac dysfunction. J Thorac Cardiovasc Surg. 2014;148:2296–2308. doi: 10.1016/j.jtcvs.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 45.Owais K, Huang T, Mahmood F, Hubbard J, Saraf R, Bardia A, Khabbaz KR, Li Y, Bhasin M, Sabe AA, Sellke F, Matyal R. Cardiopulmonary bypass decreases activation of the signal transducer and activator of transcription 3 (STAT3) pathway in diabetic human myocardium. Ann Thorac Surg. 2015;100:1636–1645. doi: 10.1016/j.athoracsur.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 46.Matyal R, Sakamuri S, Huang T, Owais K, Parikh S, Khabbaz K, Wang A, Sellke F, Mahmood F. Oxidative stress and nerve function after cardiopulmonary bypass in patients with diabetes. Ann Thorac Surg. 2014;98:1635–1643. doi: 10.1016/j.athoracsur.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 47.Voisine P, Ruel M, Khan TA, Bianchi C, Xu SH, Kohane I, Libermann TA, Otu H, Saltiel AR, Sellke FW. Differences in gene expression profiles of diabetic and nondiabetic patients undergoing cardiopulmonary bypass and cardioplegic arrest. Circulation. 2004;110(Suppl 1):II280–II286. doi: 10.1161/01.CIR.0000138974.18839.02. [DOI] [PubMed] [Google Scholar]
- 48.Chowdhry MF, Vohra HA, Galiñanes M. Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: role of caspases and poly-adenosine diphosphate-ribose polymerase. J Thorac Cardiovasc Surg. 2007;134:124–131. doi: 10.1016/j.jtcvs.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 49.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol. 2011;300:H118–H124. doi: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoogslag GE, Abou R, Joyce E, Boden H, Kamperidis V, Regeer MV, van Rosendael PJ, Schalij MJ, Bax JJ, Marsan NA, Delgado V. Comparison of changes in global longitudinal peak systolic strain after ST-segment elevation myocardial infarction in patients with versus without diabetes mellitus. Am J Cardiol. 2015;116:1334–1339. doi: 10.1016/j.amjcard.2015.07.061. [DOI] [PubMed] [Google Scholar]
- 51.Dimitriu-Leen AC, Scholte AJ, Katsanos S, Hoogslag GE, van Rosendael AR, van Zwet EW, Bax JJ, Delgado V. Influence of myocardial ischemia extent on left ventricular global longitudinal strain in patients after ST-segment elevation myocardial infarction. Am J Cardiol. 2016;119:1–6. doi: 10.1016/j.amjcard.2016.08.091. [DOI] [PubMed] [Google Scholar]
- 52.Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355:773–778. doi: 10.1016/S0140-6736(99)08415-9. [DOI] [PubMed] [Google Scholar]
- 53.Bolk J, van der Ploeg T, Cornel JH, Arnold AE, Sepers J, Umans VA. Impaired glucose metabolism predicts mortality after a myocardial infarction. Int J Cardiol. 2001;79:207–214. doi: 10.1016/S0167-5273(01)00422-3. [DOI] [PubMed] [Google Scholar]
- 54.Brener SJ, Mehran R, Dressler O, Cristea E, Stone GW. Diabetes mellitus, myocardial reperfusion, and outcome in patients with acute ST-elevation myocardial infarction treated with primary angioplasty (from HORI-ZONS AMI) Am J Cardiol. 2012;109:11116. doi: 10.1016/j.amjcard.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 55.Lønborg J, Vejlstrup N, Kelbæk H, Nepper-Christensen L, Jørgensen E, Helqvist S, Holmvang L, Saunamäki K, Bøtker HE, Kim WY, Clemmensen P, Treiman M, Engstrøm T. Impact of acute hyperglycemia on myocardial infarct size, area at risk, and salvage in patients with STEMI and the association with exenatide treatment: results from a randomized study. Diabetes. 2014;63:2474–2485. doi: 10.2337/db13-1849. [DOI] [PubMed] [Google Scholar]
- 56.Abdelmoneim AS, Welsh RC, Eurich DT, Simpson SH. Sulfonylurea use is associated with larger infarct size in patients with diabetes and ST-elevation myocardial infarction. Int J Cardiol. 2016;202:126–130. doi: 10.1016/j.ijcard.2015.08.213. [DOI] [PubMed] [Google Scholar]
- 57.Biondi-Zoccai G, Pinto A, Versaci F, Procaccini E, Neri G, Sesti G, Uccioli L, Vetere M, Peruzzi M, Nudi F. Comparative impact of hypoglycemic agents on severity and extent of myocardial ischemia in patients with type 2 diabetes mellitus undergoing myocardial perfusion scintigraphy. J Cardiovasc Pharmacol. 2016;68:162–170. doi: 10.1097/FJC.0000000000000399. [DOI] [PubMed] [Google Scholar]
- 58.Engbersen R, Riksen NP, Mol MJ, Bravenboer B, Boerman OC, Meijer P, Oyen WJ, Tack C, Rongen GA, Smits P. Improved resistance to ischemia and reperfusion, but impaired protection by ischemic preconditioning in patients with type 1 diabetes mellitus: a pilot study. Cardiovasc Diabetol. 2012;11:124. doi: 10.1186/1475-2840-11-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishihara M, Inoue I, Kawagoe T, Shimatani Y, Kurisu S, Nishioka K, Kouno Y, Umemura T, Nakamura S, Sato H. Diabetes mellitus prevents ischemic preconditioning in patients with a first acute anterior wall myocardial infarction. J Am Coll Cardiol. 2001;38:1007–1011. doi: 10.1016/S0735-1097(01)01477-2. [DOI] [PubMed] [Google Scholar]
- 60.Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM. Preconditioning the diabetic human myocardium. J Cell Mol Med. 2010;14:1740–1746. doi: 10.1111/j.1582-4934.2009.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barua A, Standen NB, Galiñanes M. Modulation of the nitric oxide metabolism overcomes the unresponsiveness of the diabetic human myocardium to protection against ischemic injury. J Surg Res. 2011;171:452–456. doi: 10.1016/j.jss.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 62.Lemoine S, Durand C, Zhu L, Ivasceau C, Lepage O, Babatasi G, Massetti M, Gérard JL, Hanouz JL. Desflurane-induced postconditioning of diabetic human right atrial myocardium in vitro. Diabetes Metab. 2010;36:21–28. doi: 10.1016/j.diabet.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Lemoine S, Zhu L, Buléon C, Massetti M, Gérard JL, Galera P, Hanouz JL. Mechanisms involved in the desflurane-induced post-conditioning of isolated human right atria from patients with type 2 diabetes. Br J Anaesth. 2011;107:510–518. doi: 10.1093/bja/aer201. [DOI] [PubMed] [Google Scholar]
- 64.Rezende PC, Rahmi RM, Uchida AH, da Costa LM, Scudeler TL, Garzillo CL, Lima EG, Segre CA, Girardi P, Takiuti M, Silva MF, Hueb W, Ramires JA, Kalil Filho R. Type 2 diabetes mellitus and myocardial ischemic preconditioning in symptomatic coronary artery disease patients. Cardiovasc Diabetol. 2015;14:66. doi: 10.1186/s12933-015-0228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou C, Yao Y, Zheng Z, Gong J, Wang W, Hu S, Li L. Stenting technique, gender, and age are associated with cardioprotection by ischaemic postconditioning in primary coronary intervention: a systematic review of 10 randomized trials. Eur Heart J. 2012;33:3070–3077. doi: 10.1093/eurheartj/ehs265. [DOI] [PubMed] [Google Scholar]
- 66.Pichot S, Mewton N, Bejan-Angoulvant T, Roubille F, Rioufol G, Giraud C, Boussaha I, Lairez O, Elbaz M, Piot C, Angoulvant D, Ovize M. Influence of cardiovascular risk factors on infarct size and interaction with mechanical ischaemic postconditioning in ST-elevation myocardial infarction. Open Heart. 2015;2:e000175. doi: 10.1136/openhrt-2014-000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomai F, Danesi A, Ghini AS, Crea F, Perino M, Gaspardone A, Ruggeri G, Chiariello L, Gioffrè PA. Effects of KATP channel blockade by glibenclamide on the warm-up phenomenon. Eur Heart J. 1999;20:196–202. doi: 10.1053/euhj.1998.1311. [DOI] [PubMed] [Google Scholar]
- 68.Kottenberg E, Thielmann M, Kleinbongard P, Frey UH, Heine T, Jakob H, Heusch G, Peters J. Myocardial protection by remote ischaemic pre-conditioning is abolished in sulphonylurea-treated diabetics undergoing coronary revascularisation. Acta Anaesthesiol Scand. 2014;58:453–462. doi: 10.1111/aas.12278. [DOI] [PubMed] [Google Scholar]
- 69.Cleveland JC, Jr, Meldrum DR, Cain BS, Banerjee A, Harken AH. Oral sulfonylurea hypoglycemic agents prevent ischemic preconditioning in human myocardium. Two paradoxes revisited. Circulation. 1997;96:29–32. doi: 10.1161/01.CIR.96.1.29. [DOI] [PubMed] [Google Scholar]
- 70.Hueb W, Uchida AH, Gersh BJ, Betti RT, Lopes N, Moffa PJ, Ferreira BM, Ramires JA, Wajchenberg BL. Effect of a hypoglycemic agent on ischemic preconditioning in patients with type 2 diabetes and stable angina pectoris. Coron Artery Dis. 2007;18:55–59. doi: 10.1097/MCA.0b013e328011c0a9. [DOI] [PubMed] [Google Scholar]
- 71.Rahmi RM, Uchida AH, Rezende PC, Lima EG, Garzillo CL, Favarato D, Strunz CM, Takiuti M, Girardi P, Hueb W, Kalil Filho R, Ramires JA. Effect of hypoglycemic agents on ischemic preconditioning in patients with type 2 diabetes and symptomatic coronary artery disease. Diabetes Care. 2013;36:1654–1659. doi: 10.2337/dc12-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol. 2003;285:H921–H930. doi: 10.1152/ajpheart.00421.2003. [DOI] [PubMed] [Google Scholar]
- 73.Currie CJ, Peters JR, Tynan A, Evans M, Heine RJ, Bracco OL, Zagar T, Poole CD. Survival as a function of HbA1c in people with type 2 diabetes: a retrospective cohort study. Lancet. 2010;375:481–489. doi: 10.1016/S0140-6736(09)61969-3. [DOI] [PubMed] [Google Scholar]
- 74.Gamble JM, Simpson SH, Eurich DT, Majumdar SR, Johnson JA. Insulin use and increased risk of mortality in type 2 diabetes: a cohort study. Diabetes Obes Metab. 2010;12:47–53. doi: 10.1111/j.1463-1326.2009.01125.x. [DOI] [PubMed] [Google Scholar]
- 75.Margolis DJ, Hoffstad O, Strom BL. Association between serious ischemic cardiac outcomes and medications used to treat diabetes. Pharmacoepidemiol Drug Saf. 2008;17:753–759. doi: 10.1002/pds.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colayco DC, Niu F, McCombs JS, Cheetham TC. A1C and cardiovascular outcomes in type 2 diabetes: a nested case-control study. Diabetes Care. 2011;34:77–83. doi: 10.2337/dc10-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smooke S, Horwich TB, Fonarow GC. Insulin-treated diabetes is associated with a marked increase in mortality in persons with advanced heart failure. Am Heart J. 2005;149:168–174. doi: 10.1016/j.ahj.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 78.Wang MY, Yu X, Lee Y, McCorkle SK, Clark GO, Strowig S, Unger RH, Raskin P. Iatrogenic hyperinsulinemia in type 1 diabetes: its effect on atherogenic risk markers. J Diabetes Complicat. 2013;27:70–74. doi: 10.1016/j.jdiacomp.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 79.Vadde Ramakrishna V, Jailkhani R. Oxidative stress in non-insulin-dependent diabetes mellitus (NIDDM) patients. Acta Diabetol. 2008;45:41–46. doi: 10.1007/s00592-007-0018-3. [DOI] [PubMed] [Google Scholar]
- 80.Hashimoto J, Ito S. Central pulse pressure and aortic stiffness determine renal hemodynamics: pathophysiological implication for microalbuminuria in hypertension. Hypertension. 2011;58:839–846. doi: 10.1161/HYPERTENSIONAHA.111.177469. [DOI] [PubMed] [Google Scholar]
- 81.Paty BW. The role of hypoglycemia in cardiovascular outcomes in diabetes. Can J Diabetes. 2015;39(Suppl 5):S155–S159. doi: 10.1016/j.jcjd.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 82.Freemantle N, Danchin N, Calvi-Gries F, Vincent M, Home PD. Relationship of glycaemic control and hypoglycaemic episodes to 4-year cardiovascular outcomes in people with type 2 diabetes starting insulin. Diabetes Obes Metab. 2016;18:152–158. doi: 10.1111/dom.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Malfitano C, Barboza CA, Mostarda C, da Palma RK, dos Santos CP, Rodrigues B, Freitas SC, Belló-Klein A, Llesuy S, Irigoyen MC, De Angelis K. Diabetic hyperglycemia attenuates sympathetic dysfunction and oxidative stress after myocardial infarction in rats. Cardiovasc Diabetol. 2014;13:131. doi: 10.1186/s12933-014-0131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feuvray D, Lopaschuk GD. Controversies on the sensitivity of the diabetic heart to ischemic injury: the sensitivity of the diabetic heart to ischemic injury is decreased. Cardiovasc Res. 1997;34:113–120. doi: 10.1016/S0008-6363(97)00037-0. [DOI] [PubMed] [Google Scholar]
- 85.Balakumar P, Sharma NK. Healing the diabetic heart: does myocardial preconditioning work? Cell Signal. 2012;24:53–59. doi: 10.1016/j.cellsig.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 86.Liu Y, Thomton JD, Cohen MV, Downey JM, Schaffer SW. Streptozotocin-induced non-insulin-dependent diabetes protects the heart from infarction. Circulation. 1993;88:1273–1278. doi: 10.1161/01.CIR.88.3.1273. [DOI] [PubMed] [Google Scholar]
- 87.Rana A, Sharma S. Mechanism of sphingosine-1-phosphate induced cardioprotection against I–R injury in diabetic rat heart: possible involvement of glycogen synthase kinase 3β and mitochondrial permeability transition pore. Clin Exp Pharmacol Physiol. 2016;43:166–173. doi: 10.1111/1440-1681.12516. [DOI] [PubMed] [Google Scholar]
- 88.Nawata T, Takahashi N, Ooie T, Kaneda K, Saikawa T, Sakata T. Cardioprotection by streptozotocin-induced diabetes and insulin against ischemia/reperfusion injury in rats. J Cardiovasc Pharmacol. 2002;40:491–500. doi: 10.1097/00005344-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 89.Ooie T, Takahashi N, Nawata T, Arikawa M, Yamanaka K, Kajimoto M, Shinohara T, Shigematsu S, Hara M, Yoshimatsu H, Saikawa T. Ischemia-induced translocation of protein kinase C-epsilon mediates cardioprotection in the streptozotocin-induced diabetic rat. Circ J. 2003;67:955–961. doi: 10.1253/circj.67.955. [DOI] [PubMed] [Google Scholar]
- 90.Xu G, Takashi E, Kudo M, Ishiwata T, Naito Z. Contradictory effects of short- and long-term hyperglycemias on ischemic injury of myocardium via intracellular signaling pathway. Exp Mol Pathol. 2004;76:57–65. doi: 10.1016/j.yexmp.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 91.Kristiansen SB, Løfgren B, Støttrup NB, Khatir D, Nielsen-Kudsk JE, Nielsen TT, Bøtker HE, Flyvbjerg A. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia. 2004;47:1716–1721. doi: 10.1007/s00125-004-1514-4. [DOI] [PubMed] [Google Scholar]
- 92.Povlsen JA, Løfgren B, Dalgas C, Birkler RI, Johannsen M, Støttrup NB, Bøtker HE. Protection against myocardial ischemia-reperfusion injury at onset of type 2 diabetes in Zucker diabetic fatty rats is associated with altered glucose oxidation. PLoS ONE. 2013;8:e64093. doi: 10.1371/journal.pone.0064093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adameová A, Kuzelová M, Andelová E, Faberová V, Pancza D, Svec P, Ziegelhöffer A, Ravingerová T. Hypercholesterolemia abrogates an increased resistance of diabetic rat hearts to ischemia-reperfusion injury. Mol Cell Biochem. 2007;295:129–136. doi: 10.1007/s11010-006-9282-8. [DOI] [PubMed] [Google Scholar]
- 94.Ma G, Al-Shabrawey M, Johnson JA, Datar R, Tawfik HE, Guo D, Caldwell RB, Caldwell RW. Protection against myocardial ischemia/reperfusion injury by short-term diabetes: enhancement of VEGF formation, capillary density, and activation of cell survival signaling. Naunyn Schmiedebergs Arch Pharmacol. 2006;373:415–427. doi: 10.1007/s00210-006-0102-1. [DOI] [PubMed] [Google Scholar]
- 95.Korkmaz-Icöz S, Lehner A, Li S, Vater A, Radovits T, Hegedűs P, Ruppert M, Brlecic P, Zorn M, Karck M, Szabó G. Mild type 2 diabetes mellitus reduces the susceptibility of the heart to ischemia/reperfusion injury: identification of underlying gene expression changes. J Diabetes Res. 2015;2015:396414. doi: 10.1155/2015/396414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marfella R, D’Amico M, Di Filippo C, Piegari E, Nappo F, Esposito K, Berrino L, Rossi F, Giugliano D. Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia. 2002;45:1172–1181. doi: 10.1007/s00125-002-0882-x. [DOI] [PubMed] [Google Scholar]
- 97.Di Filippo C, Marfella R, Cuzzocrea S, Piegari E, Petronella P, Giugliano D, Rossi F, D’Amico M. Hyperglycemia in streptozotocin-induced diabetic rat increases infarct size associated with low levels of myocardial HO-1 during ischemia/reperfusion. Diabetes. 2005;54:803–810. doi: 10.2337/diabetes.54.3.803. [DOI] [PubMed] [Google Scholar]
- 98.Liu X, Wei J, Peng DH, Layne MD, Yet SF. Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes. 2005;54:778–784. doi: 10.2337/diabetes.54.3.778. [DOI] [PubMed] [Google Scholar]
- 99.Xue R, Lei S, Xia ZY, Wu Y, Meng Q, Zhan L, Su W, Liu H, Xu J, Liu Z, Zhou B, Xia Z. Selective inhibition of PTEN preserves ischaemic post-conditioning cardioprotection in STZ-induced Type 1 diabetic rats: role of the PI3K/Akt and JAK2/STAT3 pathways. Clin Sci (Lond) 2016;130:377–392. doi: 10.1042/CS20150496. [DOI] [PubMed] [Google Scholar]
- 100.Sharma NK, Mahadevan N, Balakumar P. Adenosine transport blockade restores attenuated cardioprotective effects of adenosine preconditioning in the isolated diabetic rat heart: potential crosstalk with opioid receptors. Cardiovasc Toxicol. 2013;13:22–32. doi: 10.1007/s12012-012-9182-y. [DOI] [PubMed] [Google Scholar]
- 101.Tanaka K, Kehl F, Gu W, Krolikowski JG, Pagel PS, Warltier DC, Kersten JR. Isoflurane-induced preconditioning is attenuated by diabetes. Am J Physiol Heart Circ Physiol. 2002;282:H2018–H2023. doi: 10.1152/ajpheart.01130.2001. [DOI] [PubMed] [Google Scholar]
- 102.Ebel D, Müllenheim J, Frässdorf J, Heinen A, Huhn R, Bohlen T, Ferrari J, Südkamp H, Preckel B, Schlack W, Thämer V. Effect of acute hyperglycaemia and diabetes mellitus with and without short-term insulin treatment on myocardial ischaemic late preconditioning in the rabbit heart in vivo. Pflugers Arch. 2003;446:175–182. doi: 10.1007/s00424-003-1051-x. [DOI] [PubMed] [Google Scholar]
- 103.Kim HS, Cho JE, Hwang KC, Shim YH, Lee JH, Kwak YL. Diabetes mellitus mitigates cardioprotective effects of remifentanil preconditioning in ischemia-reperfused rat heart in association with anti-apoptotic pathways of survival. Eur J Pharmacol. 2010;628:132–139. doi: 10.1016/j.ejphar.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 104.Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH. Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology. 2011;114:1364–1372. doi: 10.1097/ALN.0b013e31820efafd. [DOI] [PubMed] [Google Scholar]
- 105.Lacerda L, Opie LH, Lecour S. Influence of tumour necrosis factor alpha on the outcome of ischaemic postconditioning in the presence of obesity and diabetes. Exp Diabetes Res. 2012;2012:502654. doi: 10.1155/2012/502654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tai W, Shi E, Yan L, Jiang X, Ma H, Ai C. Diabetes abolishes the cardioprotection induced by sevoflurane postconditioning in the rat heart in vivo: roles of glycogen synthase kinase-3β and its upstream pathways. J Surg Res. 2012;178:96–104. doi: 10.1016/j.jss.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 107.Chen QL, Gu EW, Zhang L, Cao YY, Zhu Y, Fang WP. Diabetes mellitus abrogates the cardioprotection of sufentanil against ischaemia/reperfusion injury by altering glycogen synthase kinase-3β. Acta Anaesthesiol Scand. 2013;57:236–242. doi: 10.1111/j.1399-6576.2012.02748.x. [DOI] [PubMed] [Google Scholar]
- 108.Potier L, Waeckel L, Vincent MP, Chollet C, Gobeil F, Jr, Marre M, Bruneval P, Richer C, Roussel R, Alhenc-Gelas F, Bouby N. Selective kinin receptor agonists as cardioprotective agents in myocardial ischemia and diabetes. J Pharmacol Exp Ther. 2013;346:23–30. doi: 10.1124/jpet.113.203927. [DOI] [PubMed] [Google Scholar]
- 109.Han Z, Cao J, Song D, Tian L, Chen K, Wang Y, Gao L, Yin Z, Fan Y, Wang C. Autophagy is involved in the cardioprotection effect of remote limb ischemic postconditioning on myocardial ischemia/reperfusion injury in normal mice, but not diabetic mice. PLoS ONE. 2014;9:e86838. doi: 10.1371/journal.pone.0086838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jamwal S, Kumar K, Reddy BV. Beneficial effect of zinc chloride and zinc ionophore pyrithione on attenuated cardioprotective potential of preconditioning phenomenon in STZ-induced diabetic rat heart. Perfusion. 2016;31:334–342. doi: 10.1177/0267659115608935. [DOI] [PubMed] [Google Scholar]
- 111.Lin J, Wang T, Li Y, Wang M, Li H, Irwin MG, Xia Z. N-Acetylcysteine restores sevoflurane postconditioning cardioprotection against myocardial ischemia-reperfusion injury in diabetic rats. J Diabetes Res. 2016;2016:9213034. doi: 10.1155/2016/9213034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang Y, Zhang L, Gu E, Zhu B, Zhao X, Chen J. Long-term insulin treatment restores cardioprotection induced by sufentanil postconditioning in diabetic rat heart. Exp Biol Med (Maywood) 2016;241:650–657. doi: 10.1177/1535370215622706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pourkhalili K, Hajizadeh S, Akbari Z, Dehaj ME, Akbarzadeh S, Alizadeh A. Hyperoxic preconditioning fails to confer additional protection against ischemia-reperfusion injury in acute diabetic rat heart. EXCLI J. 2012;11:263–273. [PMC free article] [PubMed] [Google Scholar]
- 114.Ravingerová T, Stetka R, Volkovova K, Pancza D, Dzurba A, Ziegelhöffer A, Styk J. Acute diabetes modulates response to ischemia in isolated rat heart. Mol Cell Biochem. 2000;210:143–151. doi: 10.1023/A:1007129708262. [DOI] [PubMed] [Google Scholar]
- 115.Ravingerová T, Neckár J, Kolár F. Ischemic tolerance of rat hearts in acute and chronic phases of experimental diabetes. Mol Cell Biochem. 2003;249:167–174. doi: 10.1023/A:1024751109196. [DOI] [PubMed] [Google Scholar]
- 116.Tosaki A, Engelman DT, Engelman RM, Das DK. The evolution of diabetic response to ischemia/reperfusion and preconditioning in isolated working rat hearts. Cardiovasc Res. 1996;31:526–536. doi: 10.1016/S0008-6363(95)00217-0. [DOI] [PubMed] [Google Scholar]
- 117.Kersten JR, Schmeling TJ, Orth KG, Pagel PS, Warltier DC. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am J Physiol. 1998;275:H721–H725. doi: 10.1152/ajpheart.1998.275.2.H721. [DOI] [PubMed] [Google Scholar]
- 118.Su H, Sun X, Ma H, Zhang HF, Yu QJ, Huang C, Wang XM, Luan RH, Jia GL, Wang HC, Gao F. Acute hyperglycemia exacerbates myocardial ischemia/reperfusion injury and blunts cardioprotective effect of GIK. Am J Physiol Endocrinol Metab. 2007;293:E629–E635. doi: 10.1152/ajpendo.00221.2007. [DOI] [PubMed] [Google Scholar]
- 119.Weber NC, Goletz C, Huhn R, Grueber Y, Preckel B, Schlack W, Ebel D. Blockade of anaesthetic-induced preconditioning in the hyperglycaemic myocardium: the regulation of different mitogen-activated protein kinases. Eur J Pharmacol. 2008;592:48–54. doi: 10.1016/j.ejphar.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 120.Yang Z, Tian Y, Liu Y, Hennessy S, Kron IL, French BA. Acute hyperglycemia abolishes ischemic preconditioning by inhibiting Akt phosphorylation: normalizing blood glucose before ischemia restores ischemic preconditioning. Oxid Med Cell Longev. 2013;2013:329183. doi: 10.1155/2013/329183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu M, Zhou B, Xia ZY, Zhao B, Lei SQ, Yang QJ, Xue R, Leng Y, Xu JJ, Xia Z. Hyperglycemia-induced inhibition of DJ-1 expression compromised the effectiveness of ischemic postconditioning cardioprotection in rats. Oxid Med Cell Longev. 2013;2013:564902. doi: 10.1155/2013/564902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Su H, Ji L, Xing W, Zhang W, Zhou H, Qian X, Wang X, Gao F, Sun X, Zhang H. Acute hyperglycaemia enhances oxidative stress and aggravates myocardial ischaemia/reperfusion injury: role of thioredoxin-interacting protein. J Cell Mol Med. 2013;17:181–191. doi: 10.1111/j.1582-4934.2012.01661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yu Q, Zhou N, Nan Y, Zhang L, Li Y, Hao X, Xiong L, Lau WB, Ma XL, Wang H, Gao F. Effective glycaemic control critically determines insulin cardioprotection against ischaemia/reperfusion injury in anaesthetized dogs. Cardiovasc Res. 2014;103:238–247. doi: 10.1093/cvr/cvu132. [DOI] [PubMed] [Google Scholar]
- 124.Mapanga RF, Joseph D, Symington B, Garson KL, Kimar C, Kelly-Laubscher R, Essop MF. Detrimental effects of acute hyperglycaemia on the rat heart. Acta Physiol (Oxf) 2014;210:546–564. doi: 10.1111/apha.12184. [DOI] [PubMed] [Google Scholar]
- 125.Kehl F, Krolikowski JG, Mraovic B, Pagel PS, Warltier DC, Kersten JR. Hyperglycemia prevents isoflurane-induced preconditioning against myocardial infarction. Anesthesiology. 2002;96:183–188. doi: 10.1097/00000542-200201000-00032. [DOI] [PubMed] [Google Scholar]
- 126.Keszler A, Brandal G, Baumgardt S, Ge ZD, Pratt PF, Riess ML, Bienengraeber M. Far red/near infrared light-induced protection against cardiac ischemia and reperfusion injury remains intact under diabetic conditions and is independent of nitric oxide synthase. Front Physiol. 2014;5:305. doi: 10.3389/fphys.2014.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Baranyai T, Nagy CT, Koncsos G, Onódi Z, Károlyi-Szabó M, Makkos A, Varga ZV, Ferdinandy P, Giricz Z. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc Diabetol. 2015;14:151. doi: 10.1186/s12933-015-0313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Raphael J, Gozal Y, Navot N, Zuo Z. Activation of adenosine triphosphate-regulated potassium channels during reperfusion restores isoflurane postconditioning-induced cardiac protection in acutely hyperglycemic rabbits. Anesthesiology. 2015;122:1299–1311. doi: 10.1097/ALN.0000000000000648. [DOI] [PubMed] [Google Scholar]
- 129.Chu LM, Osipov RM, Robich MP, Feng J, Oyamada S, Bianchi C. Sellke FWIs hyperglycemia bad for the heart during acute ischemia? J Thorac Cardiovasc Surg. 2010;140:1345–1352. doi: 10.1016/j.jtcvs.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kersten JR, Toller WG, Gross ER, Pagel PS, Warltier DC. Diabetes abolishes ischemic preconditioning: role of glucose, insulin, and osmolality. Am J Physiol Heart Circ Physiol. 2000;278:H1218–H1224. doi: 10.1152/ajpheart.2000.278.4.H1218. [DOI] [PubMed] [Google Scholar]
- 131.Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the diabetic heart. The importance of Akt phosphorylation. Diabetes. 2005;54:2360–2364. doi: 10.2337/diabetes.54.8.2360. [DOI] [PubMed] [Google Scholar]
- 132.Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, Terashima Y, Takada A, Ishikawa S, Shimamoto K. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes. 2009;58:2863–2872. doi: 10.2337/db09-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yadav HN, Singh M, Sharma PL. Involvement of GSK-3β in attenuation of the cardioprotective effect of ischemic preconditioning in diabetic rat heart. Mol Cell Biochem. 2010;343:75–81. doi: 10.1007/s11010-010-0500-z. [DOI] [PubMed] [Google Scholar]
- 134.Przyklenk K, Maynard M, Greiner DL, Whittaker P. Cardioprotection with postconditioning: loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxid Redox Signal. 2011;14:781–790. doi: 10.1089/ars.2010.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li H, Yao W, Liu Z, Xu A, Huang Y, Ma XL, Irwin MG, Xia Z. Hyperglycemia abrogates ischemic postconditioning cardioprotection by impairing AdipoR1/caveolin-3/STAT3 signaling in diabetic rats. Diabetes. 2016;65:942–955. doi: 10.2337/db15-0782. [DOI] [PubMed] [Google Scholar]
- 136.Broderick TL, Poirier P, Gillis M. Exercise training restores abnormal myocardial glucose utilization and cardiac function in diabetes. Diabetes Metab Res Rev. 2005;21:44–50. doi: 10.1002/dmrr.479. [DOI] [PubMed] [Google Scholar]
- 137.Chen L, Cai P, Cheng Z, Zhang Z, Fang J. Pharmacological postconditioning with atorvastatin calcium attenuates myocardial ischemia/reperfusion injury in diabetic rats by phosphorylating GSK3β. Exp Ther Med. 2017;14:25–34. doi: 10.3892/etm.2017.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Görbe A, Varga ZV, Kupai K, Bencsik P, Kocsis GF, Csont T, Boengler K, Schulz R, Ferdinandy P. Cholesterol diet leads to attenuation of ischemic preconditioning-induced cardiac protection: the role of connexin 43. Am J Physiol Heart Circ Physiol. 2011;300:H1907–H1913. doi: 10.1152/ajpheart.01242.2010. [DOI] [PubMed] [Google Scholar]
- 139.Csonka C, Kupai K, Bencsik P, Görbe A, Pálóczi J, Zvara A, Puskás LG, Csont T, Ferdinandy P. Cholesterol-enriched diet inhibits cardioprotection by ATP-sensitive K+ channel activators cromakalim and diazoxide. Am J Physiol Heart Circ Physiol. 2014;306:H405–H413. doi: 10.1152/ajpheart.00257.2013. [DOI] [PubMed] [Google Scholar]
- 140.Wu N, Zhang X, Jia P, Jia D. Hypercholesterolemia abrogates the protective effect of ischemic postconditioning by induction of apoptosis and impairment of activation of reperfusion injury salvage kinase pathway. Biochem Biophys Res Commun. 2015;458:148–153. doi: 10.1016/j.bbrc.2015.01.084. [DOI] [PubMed] [Google Scholar]
- 141.Katakam PV, Jordan JE, Snipes JA, Tulbert CD, Miller AW, Busija DW. Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;292:R920–R926. doi: 10.1152/ajpregu.00520.2006. [DOI] [PubMed] [Google Scholar]
- 142.Bouhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B. Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol Heart Circ Physiol. 2008;295:H1580–H1586. doi: 10.1152/ajpheart.00379.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xie N, Zhang W, Li J, Liang H, Zhou H, Duan W, Xu X, Yu S, Zhang H, Yi D. α-Linolenic acid intake attenuates myocardial ischemia/reperfusion injury through anti-inflammatory and anti-oxidative stress effects in diabetic but not normal rats. Arch Med Res. 2011;42:171–181. doi: 10.1016/j.arcmed.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 144.Van der Mieren G, Nevelsteen I, Vanderper A, Oosterlinck W, Flameng W, Herijgers P. Angiotensin-converting enzyme inhibition and food restriction restore delayed preconditioning in diabetic mice. Cardiovasc Diabetol. 2013;12:36. doi: 10.1186/1475-2840-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhu SG, Xi L, Kukreja RC. Type 2 diabetic obese db/db mice are refractory to myocardial ischaemic post-conditioning in vivo: potential role for Hsp 20, F1-ATPase δ and Echs1. J Cell Mol Med. 2012;16:950–958. doi: 10.1111/j.1582-4934.2011.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kleindienst A, Battault S, Belaidi E, Tanguy S, Rosselin M, Boulghobra D, Meyer G, Gayrard S, Walther G, Geny B, Durand G, Cazorla O, Reboul C. Exercise does not activate the β3 adrenergic receptor-eNOS pathway, but reduces inducible NOS expression to protect the heart of obese diabetic mice. Basic Res Cardiol. 2016;111:40. doi: 10.1007/s00395-016-0559-0. [DOI] [PubMed] [Google Scholar]
- 147.Huhn R, Heinen A, Weber NC, Kerindongo RP, Oei GT, Hollmann MW, Schlack W, Preckel B. Helium-induced early preconditioning and postconditioning are abolished in obese Zucker rats in vivo. J Pharmacol Exp Ther. 2009;329:600–607. doi: 10.1124/jpet.108.149971. [DOI] [PubMed] [Google Scholar]
- 148.Gundewar S, Calvert JW, Elrod JW, Lefer DJ. Cytoprotective effects of N,N,N-trimethylsphingosine during ischemia- reperfusion injury are lost in the setting of obesity and diabetes. Am J Physiol Heart Circ Physiol. 2007;293:H2462–H2471. doi: 10.1152/ajpheart.00392.2007. [DOI] [PubMed] [Google Scholar]
- 149.Muravyeva M, Baotic I, Bienengraeber M, Lazar J, Bosnjak ZJ, Sedlic F, Warltier DC, Kersten JR. Cardioprotection during diabetes: the role of mitochondrial DNA. Anesthesiology. 2014;120:870–879. doi: 10.1097/ALN.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bulhak AA, Jung C, Ostenson CG, Lundberg JO, Sjöquist PO, Pernow J. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-kinase/Akt and NO pathway. Am J Physiol Heart Circ Physiol. 2009;296:H719–H727. doi: 10.1152/ajpheart.00394.2008. [DOI] [PubMed] [Google Scholar]
- 151.Yi W, Sun Y, Gao E, et al. Reduced cardioprotective action of adiponectin in high-fat diet-induced type II diabetic mice and its underlying mechanisms. Antioxid Redox Signal. 2011;15:1779–1788. doi: 10.1089/ars.2010.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Poe AJ, Knowlton AA. Exosomes as agents of change in the cardiovascular system. J Mol Cell Cardiol. 2017;111:40–50. doi: 10.1016/j.yjmcc.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi-Doku C, Arjun S, Zheng Y, Riquelme JA, Kearney J, Sharma V, Multhoff G, Hall AR, Davidson SM. Plasma exosomes protect the myocardium from ischemia–reperfusion injury. J Am Coll Cardiol. 2015;65:1525–1536. doi: 10.1016/j.jacc.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 154.Povlsen JA, Løfgren B, Rasmussen LE, Nielsen JM, Nørregaard R, Kristiansen SB, Bøtker HE, Nielsen TT. Cardioprotective effect of l-glutamate in obese type 2 diabetic Zucker fatty rats. Clin Exp Pharmacol Physiol. 2009;36:892–898. doi: 10.1111/j.1440-1681.2009.05166.x. [DOI] [PubMed] [Google Scholar]
- 155.Peake BF, Nicholson CK, Lambert JP, Hood RL, Amin H, Amin S, Calvert JW. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am J Physiol Heart Circ Physiol. 2013;304:H1215–H1224. doi: 10.1152/ajpheart.00796.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kupai K, Szabó R, Veszelka M, Awar AA, Török S, Csonka A, Baráth Z, Pósa A, Varga C. Consequences of exercising on ischemia-reperfusion injury in type 2 diabetic Goto-Kakizaki rat hearts: role of the HO/NOS system. Diabetol Metab Syndr. 2015;7:85. doi: 10.1186/s13098-015-0080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Boardman NT, Hafstad AD, Lund J, Rossvoll L, Aasum E. Exercise of obese mice induces cardioprotection and oxygen-sparing in hearts exposed to high fat-load. Am J Physiol Heart Circ Physiol. 2017 doi: 10.1152/ajpheart.00382.2017. [DOI] [PubMed] [Google Scholar]
- 158.Stralfors P. Caveolins and caveolae, roles in insulin signalling and diabetes. Adv Exp Med Biol. 2012;729:111–126. doi: 10.1007/978-1-4614-1222-9_8. [DOI] [PubMed] [Google Scholar]
- 159.Murfitt L, Whiteley G, Iqbal MM, Kitmitto A. Targeting caveolin-3 for the treatment of diabetic cardiomyopathy. Pharmacol Ther. 2015;151:50–71. doi: 10.1016/j.pharmthera.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 160.McGrath KF, Yuki A, Manaka Y, Tamaki H, Saito K, Takekura H. Morphological characteristics of cardiac calcium release units in animals with metabolic and circulatory disorders. J Muscle Res Cell Motil. 2009;30:225–231. doi: 10.1007/s10974-009-9191-z. [DOI] [PubMed] [Google Scholar]
- 161.Stølen TO, Høydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, Wisløff U. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. 2009;105:527–536. doi: 10.1161/CIRCRESAHA.109.199810. [DOI] [PubMed] [Google Scholar]
- 162.Tse G, Lai ET, Tse V, Yeo JM. Molecular and electrophysiological mechanisms underlying cardiac arrhythmogenesis in diabetes mellitus. J Diabetes Res. 2016;2016:2848759. doi: 10.1155/2016/2848759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cohen AW, Combs TP, Scherer PE, Lisanti MP. Role of caveolin and caveolae in insulin signaling and diabetes. Am J Physiol Endocrinol Metab. 2003;285:E1151–E1160. doi: 10.1152/ajpendo.00324.2003. [DOI] [PubMed] [Google Scholar]
- 164.Quest AF, Leyton L, Párraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol. 2004;82:129–144. doi: 10.1139/o03-071. [DOI] [PubMed] [Google Scholar]
- 165.Insel PA, Head BP, Ostrom RS, Patel HH, Swaney JS, Tang CM, Roth DM. Caveolae and lipid rafts: g protein-coupled receptor signaling microdomains in cardiac myocytes. Ann N Y Acad Sci. 2005;1047:166–172. doi: 10.1196/annals.1341.015. [DOI] [PubMed] [Google Scholar]
- 166.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 168.Roth DM, Patel HH. Role of caveolae in cardiac protection. Pediatr Cardiol. 2011;32:329–333. doi: 10.1007/s00246-010-9881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schilling JM, Roth DM, Patel HH. Caveolins in cardioprotection—translatability and mechanisms. Br J Pharmacol. 2015;172:2114–2125. doi: 10.1111/bph.13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang Y, Ma Z, Hu W, Wang D, Jiang S, Fan C, Di S, Liu D, Sun Y, Yi W. Caveolin-1/-3: therapeutic targets for myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2016;111:45. doi: 10.1007/s00395-016-0561-6. [DOI] [PubMed] [Google Scholar]
- 171.Jin S, Zhou F, Katirai F, Li PL. Lipid raft redox signaling: molecular mechanisms in health and disease. Antioxid Redox Signal. 2011;15:1043–1083. doi: 10.1089/ars.2010.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Patel HH, Insel PA. Lipid rafts and caveolae and their role in compartmentation of redox signaling. Antioxid Redox Signal. 2009;11:1357–1372. doi: 10.1089/ars.2008.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ziegelhöffer A, Waczulíková I, Ferko M, Šikurová L, Mujkošová J, Ravingerová T. Involvement of membrane fluidity in endogenous protective processes running on subcellular membrane systems of the rat heart. Physiol Res. 2012;61(SUPPL 2):11–21. doi: 10.33549/physiolres.932361. [DOI] [PubMed] [Google Scholar]
- 174.Elmendorf JS. Fluidity of insulin action. Mol Biotechnol. 2004;27:127–138. doi: 10.1385/MB:27:2:127. [DOI] [PubMed] [Google Scholar]
- 175.Czech MP. Insulin action and the regulation of hexose transport. Diabetes. 1980;29:399–409. doi: 10.2337/diab.29.5.399. [DOI] [PubMed] [Google Scholar]
- 176.Pilch PF, Thompson PA, Czech MP. Coordinate modulation of d-glucose transport activity and bilayer fluidity in plasma membranes derived from control and insulin-treated adipocytes. Proc Natl Acad Sci USA. 1980;77:915–918. doi: 10.1073/pnas.77.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Whitesell RR, Regen DM, Beth AH, Pelletier DK, Abumrad NA. Activation energy of the slowest step in the glucose carrier cycle: break at 23 degrees C and correlation with membrane lipid fluidity. Biochemistry. 1989;28:5618–5625. doi: 10.1021/bi00439a042. [DOI] [PubMed] [Google Scholar]
- 178.Mitsutake S, Zama K, Yokota H, Yoshida T, Tanaka M, Mitsui M, Ikawa M, Okabe M, Tanaka Y, Yamashita T, Takemoto H, Okazaki T, Watanabe K, Igarashi Y. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J Biol Chem. 2011;286:28544–28555. doi: 10.1074/jbc.M111.255646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Calder PC. Polyunsaturated fatty acids and inflammatory processes: new twists in an old tale. Biochimie. 2009;91:791–795. doi: 10.1016/j.biochi.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 180.Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, Jung ME, Ford DA, Tontonoz P. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, Ory DS, Semenkovich CF. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–298. doi: 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lucas E, Vila-Bedmar R, Arcones AC, Cruces-Sande M, Cachofeiro V, Mayor F, Jr, Murga C. Obesity-induced cardiac lipid accumulation in adult mice is modulated by G protein-coupled receptor kinase 2 levels. Cardiovasc Diabetol. 2016;15(1):155. doi: 10.1186/s12933-016-0474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen X, Zhao S, Xia Y, Xiong Z, Li Y, Tao L, Zhang F, Wang X. G protein coupled receptor kinase-2 upregulation causes κ-opioid receptor desensitization in diabetic heart. Biochem Biophys Res Commun. 2017;482:658–664. doi: 10.1016/j.bbrc.2016.11.090. [DOI] [PubMed] [Google Scholar]
- 184.Otsuji S, Baba Y, Kamada T. Erythrocyte membrane microviscosity in diabetes. Horm Metab Res Suppl. 1981;11:97–102. [PubMed] [Google Scholar]
- 185.Bryszewska M, Watała C, Torzecka W. Changes in fluidity and composition of erythrocyte membranes and in composition of plasma lipids in type I diabetes. Br J Haematol. 1986;62:111–116. doi: 10.1111/j.1365-2141.1986.tb02906.x. [DOI] [PubMed] [Google Scholar]
- 186.Kamada T, McMillan DE, Yamashita T, Otsuji S. Lowered membrane fluidity of younger erythrocytes in diabetes. Diabetes Res Clin Pract. 1992;16:1–6. doi: 10.1016/0168-8227(92)90128-E. [DOI] [PubMed] [Google Scholar]
- 187.Bakan E, Yildirim A, Kurtul N, Polat MF, Dursun H. Cayir K Effects of type 2 diabetes mellitus on plasma fatty acid composition and cholesterol content of erythrocyte and leukocyte membranes. Acta Diabetol. 2006;43:109–113. doi: 10.1007/s00592-007-0224-4. [DOI] [PubMed] [Google Scholar]
- 188.Kröger J, Jacobs S, Jansen EH, Fritsche A, Boeing H, Schulze MB. Erythrocyte membrane fatty acid fluidity and risk of type 2 diabetes in the EPIC-Potsdam study. Diabetologia. 2015;58:282–289. doi: 10.1007/s00125-014-3421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ziegelhöffer-Mihalovicová B, Waczulíková I, Sikurová L, Styk J, Cársky J, Ziegelhöffer A. Remodelling of the sarcolemma in diabetic rat hearts: the role of membrane fluidity. Mol Cell Biochem. 2003;249:175–182. doi: 10.1023/A:1024703226034. [DOI] [PubMed] [Google Scholar]
- 190.Prisco D, Paniccia R, Coppo M, Vanni D, Rogasi PG, Tramontana M, Abbate R, Gensini GF. Red blood cell lipid alterations in type II diabetes mellitus. Thromb Res. 1989;54:751–758. doi: 10.1016/0049-3848(89)90139-4. [DOI] [PubMed] [Google Scholar]
- 191.Denton RM, Randle PJ. Concentrations of glycerides and phospholipids in rat heart and gastrocnemius muscles. Effects of alloxan-diabetes and perfusion. Biochem J. 1967;104:416–422. doi: 10.1042/bj1040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Paulson DJ, Crass MF., 3rd Endogenous triacylglycerol metabolism in diabetic heart. Am J Physiol. 1982;242:H1084–H1094. doi: 10.1152/ajpheart.1982.242.6.H1084. [DOI] [PubMed] [Google Scholar]
- 193.Gudbjarnason S, El-Hage AN, Whitehurst VE, Simental F, Balazs T. Reduced arachidonic acid levels in major phospholipids of heart muscle in the diabetic rat. J Mol Cell Cardiol. 1987;19:1141–1146. doi: 10.1016/S0022-2828(87)80357-7. [DOI] [PubMed] [Google Scholar]
- 194.Hao S, Xu R, Li D, Zhu Z, Wang T, Liu K. Attenuation of streptozotocin-induced lipid profile anomalies in the heart, brain, and mRNA expression of HMG-CoA reductase by diosgenin in rats. Cell Biochem Biophys. 2015;72:741–749. doi: 10.1007/s12013-015-0525-8. [DOI] [PubMed] [Google Scholar]
- 195.Kamat SG, Roy R. Evaluation of the effect of n-3 PUFA-rich dietary fish oils on lipid profile and membrane fluidity in alloxan-induced diabetic mice (Mus musculus) Mol Cell Biochem. 2016;416:117–129. doi: 10.1007/s11010-016-2701-6. [DOI] [PubMed] [Google Scholar]
- 196.Pierce GN, Kutryk MJ, Dhalla NS. Alterations in Ca2+ binding by and composition of the cardiac sarcolemmal membrane in chronic diabetes. Proc Natl Acad Sci USA. 1983;80:5412–5416. doi: 10.1073/pnas.80.17.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Holman RT, Johnson SB, Gerrard JM, Mauer SM, Kupcho-Sandberg S, Brown DM. Arachidonic acid deficiency in streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1983;80:2375–2379. doi: 10.1073/pnas.80.8.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huang YS, Horrobin DF, Manku MS, Mitchell J, Ryan MA. Tissue phospholipid fatty acid composition in the diabetic rat. Lipids. 1984;19:367–370. doi: 10.1007/BF02534790. [DOI] [PubMed] [Google Scholar]
- 199.Vecchini A, Del Rosso F, Binaglia L, Dhalla NS, Panagia V. Molecular defects in sarcolemmal glycerophospholipid subclasses in diabetic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1061–1074. doi: 10.1006/jmcc.2000.1140. [DOI] [PubMed] [Google Scholar]
- 200.Hu Q, Ishii E, Nakagawa Y. Differential changes in relative levels of arachidonic acid in major phospholipids from rat tissues during the progression of diabetes. J Biochem. 1994;115:405–408. doi: 10.1093/oxfordjournals.jbchem.a124351. [DOI] [PubMed] [Google Scholar]
- 201.Black SC, Katz S, McNeill JH. Influence of omega-3 fatty acid treatment on cardiac phospholipid composition and coronary flow of streptozocin-diabetic rats. Metabolism. 1993;42:320–326. doi: 10.1016/0026-0495(93)90081-X. [DOI] [PubMed] [Google Scholar]
- 202.Han X, Abendschein DR, Kelley JG, Gross RW. Diabetes-induced changes in specific lipid molecular species in rat myocardium. Biochem J. 2000;352:79–89. doi: 10.1042/bj3520079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Waczulíková I, Cagalinec M, Uličná O, Slezák P, Ziegelhöffer A. Biophysical investigation on left ventricular myocytes in rats with experimentally induced diabetes. Physiol Res. 2010;59(SUPPL 1):9–17. doi: 10.33549/physiolres.932011. [DOI] [PubMed] [Google Scholar]
- 204.Saini HK, Arneja AS, Dhalla NS. Role of cholesterol in cardiovascular dysfunction. Can J Cardiol. 2004;20:333–346. [PubMed] [Google Scholar]
- 205.Wang W, Yang L, Huang HW. Evidence of cholesterol accumulated in high curvature regions: implication to the curvature elastic energy for lipid mixtures. Biophys J. 2007;92:2819–2830. doi: 10.1529/biophysj.106.097923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Busija AR, Patel HH, Insel PA. Caveolins and cavins in the trafficking, maturation, and degradation of caveolae: implications for cell physiology. Am J Physiol Cell Physiol. 2017;312:C459–C477. doi: 10.1152/ajpcell.00355.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Brenner RR. Effects of unsaturated acids on membrane structure and enzyme kinetics. Progr Lipid Res. 1984;23:69–96. doi: 10.1016/0163-7827(84)90008-0. [DOI] [PubMed] [Google Scholar]
- 208.Kuwahara Y, Yanagishita T, Konno N, Katagiri T. Changes in microsomal membrane phospholipids and fatty acids and in activities of membrane- bound enzyme in diabetic rat heart. Basic Res Cardiol. 1997;92:214–222. doi: 10.1007/BF00788516. [DOI] [PubMed] [Google Scholar]
- 209.Muderhwa JM, Brockman HL. Lateral lipid distribution is a major regulator of lipase activity. Implications for lipid-mediated signal transduction. J Biol Chem. 1992;267:24184–24192. [PubMed] [Google Scholar]
- 210.Barthel A, Nakatani K, Dandekar AA, Roth RA. Protein kinase C modulates the insulin-stimulated increase in Akt1 and Akt3 activity in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 1998;243:509–513. doi: 10.1006/bbrc.1998.8134. [DOI] [PubMed] [Google Scholar]
- 211.Glaser PE, Gross RW. Plasmenylethanolamine facilitates rapid membrane fusion: a stopped-flow kinetic investigation correlating the propensity of a major plasma membrane constituent to adopt an HII phase with its ability to promote membrane fusion. Biochemistry. 1994;33:5805–5812. doi: 10.1021/bi00185a019. [DOI] [PubMed] [Google Scholar]
- 212.Exton JH. Signaling through phosphatidylcholine breakdown. J Biol Chem. 1990;265:1–4. [PubMed] [Google Scholar]
- 213.Panagia V, Taira Y, Ganguly PK, Tung S, Dhalla NS. Alterations in phospholipid N-methylation of cardiac subcellular membranes due to experimentally induced diabetes in rats. J Clin Invest. 1990;86:777–784. doi: 10.1172/JCI114774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Makino N, Dhalla KS, Elimban V, Dhalla NS. Sarcolemmal Ca2+ transport in streptozotocin-induced diabetic cardiomyopathy in rats. Am J Physiol. 1987;253:E202–E207. doi: 10.1152/ajpendo.1987.253.2.E202. [DOI] [PubMed] [Google Scholar]
- 215.Panagia V, Tappia PS, Dhalla NS. Phospholipid N-methylation as a signal transduction mechanism in normal and failing hearts. Heart Fail Rev. 1997;2(1):43–53. doi: 10.1023/A:1009767320693. [DOI] [Google Scholar]
- 216.Shaikh SR, Kinnun JJ, Leng X, Williams JA, Wassall SR. How polyunsaturated fatty acids modify molecular organization in membranes: insight from NMR studies of model systems. Biochim Biophys Acta. 2015;1848:211–219. doi: 10.1016/j.bbamem.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 217.Zhao S, Jia L, Gao P, Li Q, Lu X, Li J, Xu G. Study on the effect of eicosapentaenoic acid on phospholipids composition in membrane microdomains of tight junctions of epithelial cells by liquid chromatography/electrospray mass spectrometry. J Pharm Biomed Anal. 2008;47:343–350. doi: 10.1016/j.jpba.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 218.Rockett BD, Teague H, Harris M, Melton M, Williams J, Wassall SR, Shaikh SR. Fish oil increases raft size and membrane order of B cells accompanied by differential effects on function. J Lipid Res. 2012;53:674–685. doi: 10.1194/jlr.M021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Fan YY, Ly LH, Barhoumi R, McMurray DN, Chapkin RS. Dietary docosahexaenoic acid suppresses T cell protein kinase C theta lipid raft recruitment and IL-2 production. J Immunol. 2004;173:6151–6160. doi: 10.4049/jimmunol.173.10.6151. [DOI] [PubMed] [Google Scholar]
- 220.Fan YY, McMurray DN, Ly LH, Chapkin RS. Dietary (n-3) polyunsaturated fatty acids remodel mouse T-cell lipid rafts. J Nutr. 2003;133:1913–1920. doi: 10.1093/jn/133.6.1913. [DOI] [PubMed] [Google Scholar]
- 221.Grimm MO, Kuchenbecker J, Grösgen S, Burg VK, Hundsdörfer B, Rothhaar TL, Friess P, de Wilde MC, Broersen LM, Penke B, Péter M, Vígh L, Grimm HS, Hartmann T. Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. J Biol Chem. 2011;286:14028–14039. doi: 10.1074/jbc.M110.182329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hu J, Popp R, Frömel T, Ehling M, Awwad K, Adams RH, Hammes HP, Fleming I. Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J Exp Med. 2014;211:281–295. doi: 10.1084/jem.20131494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Brzustowicz MR, Cherezov V, Caffrey M, Stillwell W, Wassall SR. Molecular organization of cholesterol in polyunsaturated membranes: microdomain formation. Biophys J. 2002;82:285–298. doi: 10.1016/S0006-3495(02)75394-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Schley PD, Brindley DN, Field CJ. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J Nutr. 2007;137:548–553. doi: 10.1093/jn/137.3.548. [DOI] [PubMed] [Google Scholar]
- 225.Lee EJ, Yun UJ, Koo KH, Sung JY, Shim J, Ye SK, Hong KM, Kim YN. Down-regulation of lipid raft-associated onco-proteins via cholesterol-dependent lipid raft internalization in docosahexaenoic acid-induced apoptosis. Biochim Biophys Acta. 2014;1841:190–203. doi: 10.1016/j.bbalip.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 226.Moreno C, de la Cruz A, Valenzuela C. In-depth study of the interaction, sensitivity, and gating modulation by PUFAs on K+ channels; interaction and new targets. Front Physiol. 2016;7:578. doi: 10.3389/fphys.2016.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Elinder F, Liin SI. Actions and mechanisms of polyunsaturated fatty acids on voltage-gated ion channels. Front Physiol. 2017;8:43. doi: 10.3389/fphys.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 229.de la Serna JB, Schütz GJ, Eggeling C, Cebecauer M. There is no simple model of the plasma membrane organization. Front Cell Dev Biol. 2016;4:106. doi: 10.3389/fcell.2016.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Pani B, Singh BB. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium. 2009;45:625–633. doi: 10.1016/j.ceca.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Best JM, Kamp TJ. Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol. 2012;52:376–387. doi: 10.1016/j.yjmcc.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Harvey RD, Calaghan SC. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J Mol Cell Cardiol. 2012;52:366–375. doi: 10.1016/j.yjmcc.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Pilarczyk M, Mateuszuk L, Rygula A, Kepczynski M, Chlopicki S, Baranska M, Kaczor A. Endothelium in spots–high-content imaging of lipid rafts clusters in db/db mice. PLoS ONE. 2014;9:e106065. doi: 10.1371/journal.pone.0106065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Calaghan S, Kozera L, White E. Compartmentalisation of cAMP-dependent signalling by caveolae in the adult cardiac myocyte. J Mol Cell Cardiol. 2008;45:88–92. doi: 10.1016/j.yjmcc.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 235.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for beta2-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Makarewich CA, Correll RN, Gao H, Zhang H, Yang B, Berretta RM, Rizzo V, Molkentin JD, Houser SR. A caveolae-targeted L-type Ca2+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res. 2012;110:669–674. doi: 10.1161/CIRCRESAHA.111.264028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Glukhov AV, Balycheva M, Sanchez-Alonso JL, Ilkan Z, Alvarez-Laviada A, Bhogal N, Diakonov I, Schobesberger S, Sikkel MB, Bhargava A, Faggian G, Punjabi PP, Houser SR, Gorelik J. Direct evidence for microdomain-specific localization and remodeling of functional L-type calcium channels in rat and human atrial myocytes. Circulation. 2015;132:2372–2384. doi: 10.1161/CIRCULATIONAHA.115.018131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Correll RN, Makarewich CA, Zhang H, Zhang C, Sargent MA, York AJ, Berretta RM, Chen X, Houser SR, Molkentin JD. Caveolae-localized L-type Ca2+ channels do not contribute to function or hypertrophic signalling in the mouse heart. Cardiovasc Res. 2017;113:749–759. doi: 10.1093/cvr/cvx046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cavalli A, Eghbali M, Minosyan TY, Stefani E, Philipson KD. Localization of sarcolemmal proteins to lipid rafts in the myocardium. Cell Calcium. 2007;42:313–322. doi: 10.1016/j.ceca.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bossuyt J, Taylor BE, James-Kracke M, Hale CC. Evidence for cardiac sodium-calcium exchanger association with caveolin-3. FEBS Lett. 2002;511:113–117. doi: 10.1016/S0014-5793(01)03323-3. [DOI] [PubMed] [Google Scholar]
- 241.Liu L, Askari A. Beta-subunit of cardiac Na+-K+-ATPase dictates the concentration of the functional enzyme in caveolae. Am J Physiol Cell Physiol. 2006;291:C569–C578. doi: 10.1152/ajpcell.00002.2006. [DOI] [PubMed] [Google Scholar]
- 242.Bai Y, Wu J, Li D, Morgan EE, Liu J, Zhao X, Walsh A, Saikumar J, Tinkel J, Joe B, Gupta R, Liu L. Differential roles of caveolin-1 in ouabain-induced Na+/K+-ATPase cardiac signaling and contractility. Physiol Genom. 2016;48:739–748. doi: 10.1152/physiolgenomics.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Golfman L, Dixon IM, Takeda N, Lukas A, Dakshinamurti K, Dhalla NS. Cardiac sarcolemmal Na+-Ca2+ exchange and Na+-K+ ATPase activities and gene expression in alloxan-induced diabetes in rats. Mol Cell Biochem. 1998;188:91–101. doi: 10.1023/A:1006824623496. [DOI] [PubMed] [Google Scholar]
- 244.Pierce GN, Dhalla NS. Sarcolemmal Na-K-ATPase activity in diabetic rat heart. Am J Physiol Cell Physiol. 1983;245:C241–C247. doi: 10.1152/ajpcell.1983.245.3.C241. [DOI] [PubMed] [Google Scholar]
- 245.Papahadjopoulos D, Cowden M, Kimelberg H. Role of cholesterol in membranes effects on phospholipid-protein interactions, membrane permeability and enzymatic activity. BBA-Biomembr. 1973;330:8–26. doi: 10.1016/0005-2736(73)90280-0. [DOI] [PubMed] [Google Scholar]
- 246.Moffat MP, Dhalla NS. Heart sarcolemmal ATPase and calcium binding activities in rats fed a high cholesterol diet. Can J Cardiol. 1985;1:194–200. [PubMed] [Google Scholar]
- 247.Bilginoglu A, Kandilci HB, Turan B. Intracellular levels of Na+ and TTX-sensitive Na+ channel current in diabetic rat ventricular cardiomyocytes. Cardiovasc Toxicol. 2013;13:138–147. doi: 10.1007/s12012-012-9192-9. [DOI] [PubMed] [Google Scholar]
- 248.Camors E, Charue D, Trouvé P, Monceau V, Loyer X, Russo-Marie F, Charlemagne D. Association of annexin A5 with Na+/Ca2+ exchanger and caveolin-3 in non-failing and failing human heart. J Mol Cell Cardiol. 2006;40:47–55. doi: 10.1016/j.yjmcc.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 249.Kutryk MJ, Pierce GN. Stimulation of sodium-calcium exchange by cholesterol incorporation into isolated cardiac sarcolemmal vesicles. J Biol Chem. 1988;263:13167–13172. [PubMed] [Google Scholar]
- 250.Lei S, Li H, Xu J, Liu Y, Gao X, Wang J, Ng KF, Lau WB, Ma XL, Rodrigues B, Irwin MG, Xia Z. Hyperglycemia-induced PKCβ2 activation induces diastolic dysfunction in diabetic rats by impairing caveolin-3 expression and Akt/eNOS signaling. Diabetes. 2013;62:2318–2328. doi: 10.2337/db12-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Liu Y, Jin J, Qiao S, Lei S, Liao S, Ge ZD, Li H, Wong GT, Irwin MG, Xia Z. Inhibition of PKCβ2 overexpression ameliorates myocardial ischaemia/reperfusion injury in diabetic rats via restoring caveolin-3/Akt signaling. Clin Sci (Lond) 2015;129:331–344. doi: 10.1042/CS20140789. [DOI] [PubMed] [Google Scholar]
- 252.Penumathsa SV, Thirunavukkarasu M, Zhan L, Maulik G, Menon VP, Bagchi D, Maulik N. Resveratrol enhances GLUT-4 translocation to the caveolar lipid raft fractions through AMPK/Akt/eNOS signalling pathway in diabetic myocardium. J Cell Mol Med. 2008;12(6A):2350–2361. doi: 10.1111/j.1582-4934.2008.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sharma V, Sharma A, Saran V, Bernatchez PN, Allard MF, McNeill JH. β-receptor antagonist treatment prevents activation of cell death signaling in the diabetic heart independent of its metabolic actions. Eur J Pharmacol. 2011;657:117–125. doi: 10.1016/j.ejphar.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 254.Ha H, Pak Y. Modulation of the caveolin-3 and Akt status in caveolae by insulin resistance in H9c2 cardiomyoblasts. Exp Mol Med. 2005;37:169–178. doi: 10.1038/emm.2005.23. [DOI] [PubMed] [Google Scholar]
- 255.Mineo C, Shaul PW. Regulation of eNOS in caveolae. Adv Exp Med Biol. 2012;729:51–62. doi: 10.1007/978-1-4614-1222-9_4. [DOI] [PubMed] [Google Scholar]
- 256.Fridolfsson HN, Roth DM, Insel PA, Patel HH. Regulation of intracellular signaling and function by caveolin. FASEB J. 2014;28:3823–3831. doi: 10.1096/fj.14-252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Pilch PF, Liu L. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol Metab. 2011;22:318–324. doi: 10.1016/j.tem.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Qin L, Zhu N, Ao BX, Liu C, Shi YN, Du K, Chen JX, Zheng XL, Liao DF. Caveolae and caveolin-1 integrate reverse cholesterol transport and inflammation in atherosclerosis. Int J Mol Sci. 2016;17:429. doi: 10.3390/ijms17030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Schilling JM, Patel HH. Non-canonical roles for caveolin in regulation of membrane repair and mitochondria: implications for stress adaptation with age. J Physiol. 2016;594:4581–4589. doi: 10.1113/JP270591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Pelkmans, et al. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. 2004;118:767–780. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 261.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-Z. [DOI] [PubMed] [Google Scholar]
- 262.Breen MR, Camps M, Carvalho-Simoes F, Zorzano A, Pilch PF. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of cavin-2 in a switch-like fashion. PLoS ONE. 2012;7:e34516. doi: 10.1371/journal.pone.0034516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pike LJ, Miller JM. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J Biol Chem. 1998;273:22298–22304. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- 264.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 265.Horikawa YT, Patel HH, Tsutsumi YM, Jennings MM, Kidd MW, Hagiwara Y, Ishikawa Y, Insel PA, Roth DM. Caveolin-3 expression and caveolae are required for isoflurane- induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol. 2008;44:123–130. doi: 10.1016/j.yjmcc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.See Hoe LE, Schilling JM, Tarbit E, Kiessling CJ, Busija AR, Niesman IR, et al. Sarcolemmal cholesterol and caveolin-3 dependence of cardiac function, ischemic tolerance, and opioidergic cardioprotection. Am J Physiol Hear Circ Physiol. 2014;307:H895–H903. doi: 10.1152/ajpheart.00081.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hissa B, Oakes PW, Pontes B, Ramírez-San Juan G, Gardel ML. Cholesterol depletion impairs contractile machinery in neonatal rat cardiomyocytes. Sci Rep. 2017;7:43764. doi: 10.1038/srep43764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Pike LJ, Casey L. Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J Biol Chem. 1996;271:26453–26456. doi: 10.1074/jbc.271.43.26453. [DOI] [PubMed] [Google Scholar]
- 269.Fujita A, Cheng J, Tauchi-Sato K, Takenawa T, Fujimoto T. A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc Natl Acad Sci USA. 2009;106:9256–9261. doi: 10.1073/pnas.0900216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, Grinstein S, Parton RG. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol. 2011;194:257–275. doi: 10.1083/jcb.201012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ariotti N, Fernández-Rojo MA, Zhou Y, Hill MM, Rodkey TL, Inder KL, Tanner LB, Wenk MR, Hancock JF, Parton RG. Caveolae regulate the nanoscale organization of the plasma membrane to remotely control Ras signaling. J Cell Biol. 2014;204:777–792. doi: 10.1083/jcb.201307055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Fernández-Rojo MA, Gongora M, Fitzsimmons RL, Martel N, Martin SD, Nixon SJ, Brooks AJ, Ikonomopoulou MP, Martin S, Lo HP, Myers SA, Restall C, Ferguson C, Pilch PF, McGee SL, Anderson RL, Waters MJ, Hancock JF, Grimmond SM, Muscat GE, Parton RG. Caveolin-1 is necessary for hepatic oxidative lipid metabolism: evidence for crosstalk between caveolin-1 and bile acid signaling. Cell Rep. 2013;4:238–247. doi: 10.1016/j.celrep.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 273.Wanaski SP, Ng BK, Glaser M. Caveolin scaffolding region and the membrane binding region of SRC form lateral membrane domains. Biochemistry. 2003;42:42–56. doi: 10.1021/bi012097n. [DOI] [PubMed] [Google Scholar]
- 274.Kovtun O, Tillu VA, Ariotti N, Parton RG, Collins BM. Cavin family proteins and the assembly of caveolae. J Cell Sci. 2015;128:1269–1278. doi: 10.1242/jcs.167866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Su W, Zhang Y, Zhang Q, Xu J, Zhan L, Zhu Q, Lian Q, Liu H, Xia ZY, Xia Z, Lei S. N-acetylcysteine attenuates myocardial dysfunction and postischemic injury by restoring caveolin-3/eNOS signaling in diabetic rats. Cardiovasc Diabetol. 2016;15:146. doi: 10.1186/s12933-016-0460-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Knowles CJ, Cebova M, Pinz IM. Palmitate diet-induced loss of cardiac caveolin-3: a novel mechanism for lipid-induced contractile dysfunction. PLoS ONE. 2013;8:e61369. doi: 10.1371/journal.pone.0061369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Peart JN, Pepe S, Reichelt ME, Beckett N, See Hoe L, Ozberk V, Niesman IR, Patel HH, Headrick JP. Dysfunctional survival-signaling and stress-intolerance in aged murine and human myocardium. Exp Gerontol. 2014;50:72–81. 10.1016/j.exger.2013.11.015. [DOI] [PMC free article] [PubMed]
- 278.Carotenuto F, Minieri M, Monego G, Fiaccavento R, Bertoni A, Sinigaglia F, Vecchini A, Carosella L, Di Nardo P. A diet supplemented with ALA-rich flaxseed prevents cardiomyocyte apoptosis by regulating caveolin-3 expression. Cardiovasc Res. 2013;100:422–431. doi: 10.1093/cvr/cvt211. [DOI] [PubMed] [Google Scholar]
- 279.Bucci M, Roviezzo F, Brancaleone V, Lin MI, Lorenzo AD, Cicala C, Pinto A, Sessa WC, Farneti S, Fiorucci S, Cirino G. Diabetic mouse angiopathy is linked to progressive sympathetic receptor deletion coupled to an enhanced caveolin-1 expression. Arterioscler Thromb Vasc Biol. 2004;24:721–726. doi: 10.1161/01.ATV.0000122362.44628.09. [DOI] [PubMed] [Google Scholar]
- 280.Collins BM, Davis MJ, Hancock JF, Parton RG. Structure-based reassessment of the caveolin signaling model: do caveolae regulate signaling through caveolin-protein interactions? Dev Cell. 2012;23:11–20. doi: 10.1016/j.devcel.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Fridolfsson HN, Kawaraguchi Y, Ali SS, Panneerselvam M, Niesman IR, Finley JC, Kellerhals SE, Migita MY, Okada H, Moreno AL, Jennings M, Kidd MW, Bonds JA, Balijepalli RC, Ross RS, Patel PM, Miyanohara A, Chen Q, Lesnefsky EJ, Head BP, Roth DM, Insel PA, Patel HH. Mitochondria-localized caveolin in adaptation to cellular stress and injury. FASEB J. 2012;26:4637–4649. doi: 10.1096/fj.12-215798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Smart EJ, Ying YS, Donzell WC, Anderson RGW. A role for caveolin in transport of cholesterol from endoplasmic reticulum to plasma membrane. J Biol Chem. 1996;271:29427–29435. doi: 10.1074/jbc.271.46.29427. [DOI] [PubMed] [Google Scholar]
- 283.O’Connell KMS, Martens JR, Tamkun MM. Localization of ion channels to lipid raft domains within the cardiovascular system. Trends Cardiovasc Med. 2004;14:37–42. doi: 10.1016/j.tcm.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 284.Balse E, Steele DF, Abriel H, Coulombe A, Fedida D, Hatem SN. Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol Rev. 2012;92:1317–1358. doi: 10.1152/physrev.00041.2011. [DOI] [PubMed] [Google Scholar]
- 285.Fecchi K, Volonte D, Hezel MP, Schmeck K, Galbiati F. Spatial and temporal regulation of GLUT4 translocation by flotillin-1 and caveolin-3 in skeletal muscle cells. FASEB J. 2006;20:705–707. doi: 10.1096/fj.05-4661fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Talukder MA, Preda M, Ryzhova L, Prudovsky I, Pinz IM. Heterozygous caveolin-3 mice show increased susceptibility to palmitate-induced insulin resistance. Physiol Rep. 2016;4:e12736. doi: 10.14814/phy2.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, et al. Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol. 2002;160:2207–2217. doi: 10.1016/S0002-9440(10)61168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Augustus AS, Buchanan J, Addya S, Rengo G, Pestell RG, Fortina P, Koch WJ, Bensadoun A, Abel ED, Lisanti MP. Substrate uptake and metabolism are preserved in hypertrophic caveolin-3 knockout hearts. Am J Physiol Heart Circ Physiol. 2008;295:H657–H666. doi: 10.1152/ajpheart.00387.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Krawczyk KK, Yao Mattisson I, Ekman M, Oskolkov N, Grantinge R, Kotowska D, Olde B, Hansson O, Albinsson S, Miano JM, Rippe C, Swärd K. Myocardin family members drive formation of caveolae. PLoS ONE. 2015;10:e013393. doi: 10.1371/journal.pone.0133931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Madonna R, Geng YJ, Bolli R, Rokosh G, Ferdinandy P, Patterson C, De Caterina R. Co-activation of nuclear factor-κB and myocardin/serum response factor conveys the hypertrophy signal of high insulin levels in cardiac myoblasts. J Biol Chem. 2014;289:19585–19598. doi: 10.1074/jbc.M113.540559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG, Gupte SA. Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol. 2014;306:H214–H224. doi: 10.1152/ajpheart.00310.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Swärd K, Stenkula KG, Rippe C, Alajbegovic A, Gomez MF, Albinsson S. Emerging roles of the myocardin family of proteins in lipid and glucose metabolism. J Physiol. 2016;594:4741–4752. doi: 10.1113/JP271913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Paulson DJ. The diabetic heart is more sensitive to ischemic injury. Cardiovasc Res. 1997;34:104–112. doi: 10.1016/S0008-6363(97)00018-7. [DOI] [PubMed] [Google Scholar]
- 294.Salem KA, Qureshi MA, Sydorenko V, Parekh K, Jayaprakash P, Iqbal T, Singh J, Oz M, Adrian TE, Howarth FC. Effects of exercise training on excitation-contraction coupling and related mRNA expression in hearts of Goto-Kakizaki type 2 diabetic rats. Mol Cell Biochem. 2013;380:83–96. doi: 10.1007/s11010-013-1662-2. [DOI] [PubMed] [Google Scholar]
- 295.Knowles CJ, Dionne M, Cebova M, Pinz IM. Palmitate-induced translocation of caveolin-3 and endothelial nitric oxide synthase in cardiomyocytes. Online J Biol Sci. 2011;11(2):27–36. doi: 10.3844/ojbsci.2011.27.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 297.Feron O, Dessy C, Opel DJ, Arstall MA, Kelly RA, Michel T. Modulation of the endothelial nitric-oxide synthase-caveolin interaction in cardiac myocytes. Implications for the autonomic regulation of heart rate. J Biol Chem. 1998;273:30249–30254. doi: 10.1074/jbc.273.46.30249. [DOI] [PubMed] [Google Scholar]
- 298.García-Cardeña G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- 299.Horikawa YT, Panneerselvam M, Kawaraguchi Y, Tsutsumi YM, Ali SS, Balijepalli RC, Murray F, Head BP, Niesman IR, Rieg T, Vallon V, Insel PA, Patel HH, Roth DM. Cardiac-specific overexpression of caveolin-3 attenuates cardiac hypertrophy and increases natriuretic peptide expression and signaling. J Am Coll Cardiol. 2011;57:2273–2283. doi: 10.1016/j.jacc.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Ajmani P, Yadav HN, Singh M, Sharma PL. Possible involvement of caveolin in attenuation of cardioprotective effect of ischemic preconditioning in diabetic rat heart. BMC Cardiovasc Disord. 2011;11:43. doi: 10.1186/1471-2261-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Zuluaga S, Alvarez-Barrientos A, Gutiérrez-Uzquiza A, Benito M, Nebreda AR, Porras A. Negative regulation of Akt activity by p38alpha MAP kinase in cardiomyocytes involves membrane localization of PP2A through interaction with caveolin-1. Cell Signal. 2007;19:62–74. doi: 10.1016/j.cellsig.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 302.Nassar ZD, Parat MO. Cavin family: new players in the biology of caveolae. Int Rev Cell Mol Biol. 2015;320:235–305. doi: 10.1016/bs.ircmb.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 303.Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kaakinen M, Reichelt ME, Ma Z, Ferguson C, Martel N, Porrello ER, Hudson JE, Thomas WG, Parton RG, Headrick JP. Cavin-1 deficiency modifies myocardial and coronary function, stretch responses and ischaemic tolerance: roles of NOS over-activity. Basic Res Cardiol. 2017;112:24. doi: 10.1007/s00395-017-0613-6. [DOI] [PubMed] [Google Scholar]
- 305.Patel HH, Tsutsumi YM, Head BP, Niesman IR, Jennings M, Horikawa Y, Huang D, Moreno AL, Patel PM, Insel PA, Roth DM. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J. 2007;21:1565–1574. doi: 10.1096/fj.06-7719com. [DOI] [PubMed] [Google Scholar]
- 306.Jasmin JF, Rengo G, Lymperopoulos A, Gupta R, Eaton GJ, Quann K, Gonzales DM, Mercier I, Koch WJ, Lisanti MP. Caveolin-1 deficiency exacerbates cardiac dysfunction and reduces survival in mice with myocardial infarction. Am J Physiol Heart Circ Physiol. 2011;300:H1274–H1281. doi: 10.1152/ajpheart.01173.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wunderlich C, Schober K, Kasper M, Heerwagen C, Marquetant R, Ebner B, Forkmann M, Schoen S, Braun-Dullaeus RC, Schmeisser A, Strasser RH. Nitric oxide synthases are crucially involved in the development of the severe cardiomyopathy of caveolin-1 knockout mice. Biochem Biophys Res Com. 2008;377:769–774. doi: 10.1016/j.bbrc.2008.10.068. [DOI] [PubMed] [Google Scholar]
- 308.Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, Lisanti MP. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol. 2003;285:C222–C235. doi: 10.1152/ajpcell.00006.2003. [DOI] [PubMed] [Google Scholar]
- 309.Gustavsson J, Parpal S, Karlsson M, Ramsing C, Thorn H, Borg M, Lindroth M, Peterson KH, Magnusson KE, Strâlfors P. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999;13:1961–1971. [PubMed] [Google Scholar]
- 310.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 311.Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 312.Yang J, Holman GD. Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem. 2005;280:4070–4078. doi: 10.1074/jbc.M410213200. [DOI] [PubMed] [Google Scholar]
- 313.Antonescu CN, Díaz M, Femia G, Planas JV, Klip A. Clathrin-dependent and independent endocytosis of glucose transporter 4 (GLUT4) in myoblasts: regulation by mitochondrial uncoupling. Traffic. 2008;9:1173–1190. doi: 10.1111/j.1600-0854.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- 314.Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG, Jr, Ishikawa Y. Caveolin is an activator of insulin receptor signaling. J Biol Chem. 1998;273:26962–26968. doi: 10.1074/jbc.273.41.26962. [DOI] [PubMed] [Google Scholar]
- 315.Otsu K, Toya Y, Oshikawa J, Kurotani R, Yazawa T, Sato M, Yokoyama U, Umemura S, Minamisawa S, Okumura S, Ishikawa Y. Caveolin gene transfer improves glucose metabolism in diabetic mice. Am J Physiol Cell Physiol. 2010;298:C450–C456. doi: 10.1152/ajpcell.00077.2009. [DOI] [PubMed] [Google Scholar]
- 316.Augustus AS, Buchanan J, Gutman E, Rengo G, Pestell RG, Fortina P, Koch WJ, Bensadoun A, Abel ED, Lisanti MP. Hearts lacking caveolin-1 develop hypertrophy with normal cardiac substrate metabolism. Cell Cycle. 2008;7:2509–2518. doi: 10.4161/cc.7.16.6421. [DOI] [PubMed] [Google Scholar]
- 317.Oshikawa J, Otsu K, Toya Y, Tsunematsu T, Hankins R, Kawabe J, Minamisawa S, Umemura S, Hagiwara Y, Ishikawa Y. Insulin resistance in skeletal muscles of caveolin-3-null mice. Proc Natl Acad Sci USA. 2004;101:12670–12675. doi: 10.1073/pnas.0402053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- 319.Gomez-Ruiz A, Milagro FI, Campion J, Martinez JA, de Miguel C. Caveolin expression and activation in retroperitoneal and subcutaneous adipocytes: influence of a high-fat diet. J Cell Physiol. 2010;225:206–213. doi: 10.1002/jcp.22241. [DOI] [PubMed] [Google Scholar]
- 320.Ehehalt R, Sparla R, Herrmann T, Kulaksiz H, Fullekrug J, Stremmel W. Uptake of long chain fatty acids is regulated by dynamic interaction of FAT/CD36 with cholesterol/sphingolipid enriched microdomains (lipid rafts) BMC Cell Biol. 2008;9:45. doi: 10.1186/1471-2121-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Coort SL, Bonen A, van der Vusse GJ, Glatz JF, Luiken JJ. Cardiac substrate uptake and metabolism in obesity and type-2 diabetes: role of sarcolemmal substrate transporters. Mol Cell Biochem. 2007;299:5–18. doi: 10.1007/s11010-005-9030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc Res. 2008;79:249–258. doi: 10.1093/cvr/cvn116. [DOI] [PubMed] [Google Scholar]
- 323.Steinbusch LK, Schwenk RW, Ouwens DM, Diamant M, Glatz JF, Luiken JJ. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell Mol Life Sci. 2011;68:2525–2538. doi: 10.1007/s00018-011-0690-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R, Abumrad NA. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem. 1999;274:26761–26766. doi: 10.1074/jbc.274.38.26761. [DOI] [PubMed] [Google Scholar]
- 325.Kuang M, Febbraio M, Wagg C, Lopaschuk GD, Dyck JR. Fatty acid translocase/CD36 deficiency does not energetically or functionally compromise hearts before or after ischemia. Circulation. 2004;109:1550–1557. doi: 10.1161/01.CIR.0000121730.41801.12. [DOI] [PubMed] [Google Scholar]
- 326.Bezaire V, Bruce CR, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Identification of fatty acid translocase on human skeletal muscle mitochondrial membranes: essential role in fatty acid oxidation. Am J Physiol Endocrinol Metab. 2006;290:E509–E515. doi: 10.1152/ajpendo.00312.2005. [DOI] [PubMed] [Google Scholar]
- 327.Abumrad NA, Goldberg IJ. CD36 actions in the heart: lipids, calcium, inflammation, repair and more? Biochim Biophys Acta. 2016;1860:1442–1449. doi: 10.1016/j.bbalip.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Ring A, Le Lay S, Pohl J, Verkade P, Stremmel W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim Biophys Acta. 2006;1761:416–423. doi: 10.1016/j.bbalip.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 329.Wang L, Ko KW, Lucchinetti E, Zhang L, Troxler H, Hersberger M, Omar MA, Posse de Chaves EI, Lopaschuk GD, Clanachan AS, Zaugg M. Metabolic profiling of hearts exposed to sevoflurane and propofol reveals distinct regulation of fatty acid and glucose oxidation: CD36 and pyruvate dehydrogenase as key regulators in anesthetic-induced fuel shift. Anesthesiology. 2010;113:541–551. doi: 10.1097/ALN.0b013e3181e2c1a1. [DOI] [PubMed] [Google Scholar]
- 330.Zhu Y, Zhang C, Chen B, Chen R, Guo A, Hong J, Song LS. Cholesterol is required for maintaining T-tubule integrity and intercellular connections at intercalated discs in cardiomyocytes. J Mol Cell Cardiol. 2016;97:204–212. doi: 10.1016/j.yjmcc.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Barrientos G, Sánchez-Aguilera P, Jaimovich E, Hidalgo C, Llanos P. Membrane cholesterol in skeletal muscle: a novel player in excitation-contraction coupling and insulin resistance. J Diabetes Res. 2017;2017:3941898. doi: 10.1155/2017/3941898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Haque MZ, McIntosh VJ, Abou Samra AB, Mohammad RM, Lasley RD. Cholesterol depletion alters cardiomyocyte subcellular signaling and increases contractility. PLoS ONE. 2016;11:e0154151. doi: 10.1371/journal.pone.0154151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Liu P, Leffler BJ, Weeks LK, Chen G, Bouchard CM, Strawbridge AB, Elmendorf JS. Sphingomyelinase activates GLUT4 translocation via a cholesterol-dependent mechanism. Am J Physiol Cell Physiol. 2004;286:C317–C329. doi: 10.1152/ajpcell.00073.2003. [DOI] [PubMed] [Google Scholar]
- 334.Habegger KM, Penque BA, Sealls W, Tackett L, Bell LN, Blue EK, Gallagher PJ, Sturek M, Alloosh MA, Steinberg HO, Considine RV, Elmendorf JS. Fat-induced membrane cholesterol accrual provokes cortical filamentous actin destabilisation and glucose transport dysfunction in skeletal muscle. Diabetologia. 2012;55:457–467. doi: 10.1007/s00125-011-2334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Habegger KM, Hoffman NJ, Ridenour CM, Brozinick JT, Elmendorf JS. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology. 2012;153:2130–2141. doi: 10.1210/en.2011-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Llanos P, Contreras-Ferrat A, Georgiev T, Osorio-Fuentealba C, Espinosa A, Hidalgo J, Hidalgo C, Jaimovich E. The cholesterol-lowering agent methyl-β-cyclodextrin promotes glucose uptake via GLUT4 in adult muscle fibers and reduces insulin resistance in obese mice. Am J Physiol Endocrinol Metab. 2015;308:E294–E305. doi: 10.1152/ajpendo.00189.2014. [DOI] [PubMed] [Google Scholar]
- 337.Fantini J, Barrantes FJ. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol. 2013;4:31. doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Levitan I, Singh DK, Rosenhouse-Dantsker A. Cholesterol binding to ion channels. Front Physiol. 2014;5:65. doi: 10.3389/fphys.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Sengupta D, Chattopadhyay A. Molecular dynamics simulations of GPCR-cholesterol interaction: an emerging paradigm. Biochim Biophys Acta. 2015;1848:1775–1782. doi: 10.1016/j.bbamem.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 340.Ueda Y, Kitakaze M, Komamura K, Minamino T, Asanuma H, Sato H, Kuzuya T, Takeda H, Hori M. Pravastatin restored the infarct size-limiting effect of ischemic preconditioning blunted by hypercholesterolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol. 1999;34:2120–2125. doi: 10.1016/S0735-1097(99)00440-4. [DOI] [PubMed] [Google Scholar]
- 341.Balk EM, Tatsioni A. Lichten Habegger stein AH, Lau J, Pittas AG. Effect of chromium supplementation on glucose metabolism and lipids: a systematic review of randomized controlled trials. Diabetes Care. 2007;30:2154–2163. doi: 10.2337/dc06-0996. [DOI] [PubMed] [Google Scholar]
- 342.Hoffman NJ, Penque BA, Habegger KM, Sealls W, Tackett L, Elmendorf JS. Chromium enhances insulin responsiveness via AMPK. J Nutr Biochem. 2014;25:565–572. doi: 10.1016/j.jnutbio.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Baier CJ, Fantini J, Barrantes FJ. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep. 2011;1:69. doi: 10.1038/srep00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Gimpl G. Interaction of G protein coupled receptors and cholesterol. Chem Phys Lipids. 2016;199:61–73. doi: 10.1016/j.chemphyslip.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 345.Legler DF, Matti C, Laufer JM, Jakobs BD, Purvanov V, Uetz-von Allmen E, Thelen M. Modulation of chemokine receptor function by cholesterol: new prospects for pharmacological intervention. Mol Pharmacol. 2017;91:331–338. doi: 10.1124/mol.116.107151. [DOI] [PubMed] [Google Scholar]
- 346.Tappia PS, Dhalla NS. Mechanisms for the defects in phospholipid signal transduction in diabetic cardiomyopathy. Indian J Biochem Biophys. 2014;51:431–440. [PubMed] [Google Scholar]
- 347.Prasad MR, Popescu LM, Moraru II, Liu XK, Maity S, Engelman RM, Das DK. Role of phospholipases A2 and C in myocardial ischemic reperfusion injury. Am J Physiol. 1991;260:H877–H883. doi: 10.1152/ajpheart.1991.260.3.H877. [DOI] [PubMed] [Google Scholar]
- 348.Gysembergh A, Lemaire S, Piot C, Sportouch C, Richard S, Kloner RA, Przyklenk K. Pharmacological manipulation of Ins(1,4,5)P3 signaling mimics preconditioning in rabbit heart. Am J Physiol. 1999;277:H2458–H2469. doi: 10.1152/ajpheart.1999.277.6.H2458. [DOI] [PubMed] [Google Scholar]
- 349.Tappia PS, Asemu G, Rodriguez-Leyva D. Phospholipase C as a potential target for cardioprotection during oxidative stress. Can J Physiol Pharmacol. 2010;88:249–263. doi: 10.1139/Y10-019. [DOI] [PubMed] [Google Scholar]
- 350.Tappia PS, Maddaford TG, Hurtado C, Dibrov E, Austria JA, Sahi N, Panagia V, Pierce GN. Defective phosphatidic acid-phospholipase C signaling in diabetic cardiomyopathy. Biochem Biophys Res Commun. 2004;316:280–289. doi: 10.1016/j.bbrc.2004.02.045. [DOI] [PubMed] [Google Scholar]
- 351.Wald M, Borda ES, Sterin-Borda L. Alpha-adrenergic supersensitivity and decreased number of alpha-adrenoceptors in heart from acute diabetic rats. Can J Physiol Pharmacol. 1988;66:1154–1160. doi: 10.1139/y88-190. [DOI] [PubMed] [Google Scholar]
- 352.Heyliger CE, Pierce GN, Singal PK, Beamish RE, Dhalla NS. Cardiac alpha- and beta-adrenergic receptor alterations in diabetic cardiomyopathy. Basic Res Cardiol. 1982;77:610–618. doi: 10.1007/BF01908314. [DOI] [PubMed] [Google Scholar]
- 353.Puceat M, Vassort G. Purinergic stimulation of rat cardiomyocytes induces tyrosine phosphorylation and membrane association of phospholipase C gamma: a major mechanism for InsP3 generation. Biochem J. 1996;318:723–728. doi: 10.1042/bj3180723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.RES.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 355.Okumura K, Akiyama N, Hashimoto H, Ogawa K, Satake T. Alteration of 1,2-diacylglycerol content in myocardium from diabetic rats. Diabetes. 1988;37:1168–1172. doi: 10.2337/diab.37.9.1168. [DOI] [PubMed] [Google Scholar]
- 356.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Das S, Rand RP. Diacylglycerol causes major structural transitions in phospholipid bilayer membranes. Biochem Biophys Res Commun. 1984;124:491–496. doi: 10.1016/0006-291X(84)91580-8. [DOI] [PubMed] [Google Scholar]
- 358.Das S, Rand RP. Modification by diacylglycerol of the structure and interaction of various phospholipid bilayer membranes. Biochemistry. 1986;25:2882–2889. doi: 10.1021/bi00358a022. [DOI] [PubMed] [Google Scholar]
- 359.Williams SA, Tappia PS, Yu CH, Bibeau M, Panagia V. Impairment of the sarcolemmal phospholipase D-phosphatidate phosphohydrolase pathway in diabetic cardiomyopathy. J Mol Cell Cardiol. 1998;30:109–118. doi: 10.1006/jmcc.1997.0576. [DOI] [PubMed] [Google Scholar]
- 360.McHowat J, Creer MH, Hicks KK, Jones JH, McCrory R, Kennedy RH. Induction of Ca-independent PLA(2) and conservation of plasmalogen polyunsaturated fatty acids in diabetic heart. Am J Physiol Endocrinol Metab. 2000;279:E25–E32. doi: 10.1152/ajpendo.2000.279.1.E25. [DOI] [PubMed] [Google Scholar]
- 361.Hope HR, Pike LJ. Phosphoinositides and phosphoinositide-utilizing enzymes in detergent- insoluble lipid domains. Mol Biol Cell. 1996;7:843–851. doi: 10.1091/mbc.7.6.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Liu Y, Casey L, Pike LJ. Compartmentalization of phosphatidylinositol 4,5- bisphosphate in low-density membrane domains in the absence of caveolin. Biochem Biophys Res Commun. 1998;245:684–690. doi: 10.1006/bbrc.1998.8329. [DOI] [PubMed] [Google Scholar]
- 363.Fujita T, Toya Y, Iwatsubo K, Onda T, Kimura K, Umemura S, et al. Accumulation of molecules involved in alpha1-adrenergic signal within caveolae: caveolin expression and the development of cardiac hypertrophy. Cardiovasc Res. 2001;51:709–716. doi: 10.1016/S0008-6363(01)00348-0. [DOI] [PubMed] [Google Scholar]
- 364.Alday A, Urrutia J, Gallego M, Casis O. alpha1-adrenoceptors regulate only the caveolae-located subpopulation of cardiac K(V)4 channels. Channels (Austin) 2010;4:168–178. doi: 10.4161/chan.4.3.11479. [DOI] [PubMed] [Google Scholar]
- 365.Morris JB, Huynh H, Vasilevski O, Woodcock EA. Alpha1-adrenergic receptor signaling is localized to caveolae in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2006;41:17–25. doi: 10.1016/j.yjmcc.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 366.Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Beech DJ. Integration of transient receptor potential canonical channels with lipids. Acta Physiol (Oxf) 2012;204:227–237. doi: 10.1111/j.1748-1716.2011.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Blaskovic S, Blanc M. van der Goot FG What does S-palmitoylation do to membrane proteins? FEBS J. 2013;280:2766–2774. doi: 10.1111/febs.12263. [DOI] [PubMed] [Google Scholar]
- 369.Howie J, Tulloch LB, Shattock MJ, Fuller W. Regulation of the cardiac Na+ pump by palmitoylation of its catalytic and regulatory subunits. Biochem Soc Trans. 2013;41:95–100. doi: 10.1042/BST20120269. [DOI] [PubMed] [Google Scholar]
- 370.Reilly L, Howie J, Wypijewski K, Ashford ML, Hilgemann DW, Fuller W. Palmitoylation of the Na/Ca exchanger cytoplasmic loop controls its inactivation and internalization during stress signaling. FASEB J. 2015;29:4532–4543. doi: 10.1096/fj.15-276493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Thorne RF, Ralston KJ, de Bock CE, Mhaidat NM, Zhang XD, Boyd AW, Burns GF. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim Biophys Acta. 2010;1803:1298–1307. doi: 10.1016/j.bbamcr.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 372.Wei X, Song H, Semenkovich CF. Insulin-regulated protein palmitoylation impacts endothelial cell function. Arterioscler Thromb Vasc Biol. 2014;34:346–354. doi: 10.1161/ATVBAHA.113.302848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, Burrell LM. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. 2003;92:785–792. doi: 10.1161/01.RES.0000065620.39919.20. [DOI] [PubMed] [Google Scholar]
- 374.Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko M, Song F, Sell DR, Strauch C, Monnier VM, Yan SF, Schmidt AM, Ramasamy R. RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes. 2008;57:1941–1951. doi: 10.2337/db07-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Suchal K, Malik S, Khan SI, Malhotra RK, Goyal SN, Bhatia J, Kumari S, Ojha S, Arya DS. Protective effect of mangiferin on myocardial ischemia-reperfusion injury in streptozotocin-induced diabetic rats: role of AGE-RAGE/MAPK pathways. Sci Rep. 2017;7:42027. doi: 10.1038/srep42027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Suchal K, Malik S, Khan SI, Malhotra RK, Goyal SN, Bhatia J, Ojha S, Arya DS. Molecular pathways involved in the amelioration of myocardial injury in diabetic rats by kaempferol. Int J Mol Sci. 2017;18(5):E1001. doi: 10.3390/ijms18051001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Khodeer DM, Zaitone SA, Farag NE, Moustafa YM. Cardioprotective effect of pioglitazone in diabetic and non-diabetic rats subjected to acute myocardial infarction involves suppression of AGE-RAGE axis and inhibition of apoptosis. Can J Physiol Pharmacol. 2016;94:463–476. doi: 10.1139/cjpp-2015-0135. [DOI] [PubMed] [Google Scholar]
- 378.Marsh SA, Chatham JC. The paradoxical world of protein O-GlcNAcylation: a novel effector of cardiovascular (dys)function. Cardiovasc Res. 2011;89:487–488. doi: 10.1093/cvr/cvq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 380.Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am J Physiol Heart Circ Physiol. 2010;299:H1715–H1727. doi: 10.1152/ajpheart.00337.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Liu J, Marchase RB, Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol. 2007;42:177–185. doi: 10.1016/j.yjmcc.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Jensen RV, Zachara NE, Nielsen PH, Kimose HH, Kristiansen SB, Bøtker HE. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc Res. 2013;97:369–378. doi: 10.1093/cvr/cvs337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Rajamani U, Essop MF. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am J Physiol Cell Physiol. 2010;299:C139–C147. doi: 10.1152/ajpcell.00020.2010. [DOI] [PubMed] [Google Scholar]
- 384.Marsh SA, Powell PC, Dell’italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci. 2013;92:648–656. doi: 10.1016/j.lfs.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Ramirez-Correa GA, Ma J, Slawson C, Zeidan Q, Lugo-Fagundo NS, Xu M, Shen X, Gao WD, Caceres V, Chakir K, DeVine L, Cole RN, Marchionni L, Paolocci N, Hart GW, Murphy AM. Removal of abnormal myofilament O-GlcNAcylation restores Ca2+ sensitivity in diabetic cardiac muscle. Diabetes. 2015;64:3573–3587. doi: 10.2337/db14-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Dassanayaka S, Readnower RD, Salabei JK, Long BW, Aird AL, Zheng YT, Muthusamy S, Facundo HT, Hill BG, Jones SP. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem J. 2015;467:115–126. doi: 10.1042/BJ20141018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Makino A, Dai A, Han Y, Youssef KD, Wang W, Donthamsetty R, Scott BT, Wang H, Dillmann WH. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am J Physiol Cell Physiol. 2015;309:C593–C599. doi: 10.1152/ajpcell.00069.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Liu J, Pang Y, Chang T, Bounelis P, Chatham JC, Marchase RB. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40:303–312. doi: 10.1016/j.yjmcc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 389.Greaves J, Prescott GR, Gorleku OA, Chamberlain LH. The fat controller: roles of palmitoylation in intracellular protein trafficking and targeting to membrane microdomains. Mol Membr Biol. 2009;26:67–79. doi: 10.1080/09687680802620351. [DOI] [PubMed] [Google Scholar]
- 390.Shahinian S, Silvius J. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry. 1995;34:3813–3822. doi: 10.1021/bi00011a039. [DOI] [PubMed] [Google Scholar]
- 391.Greaves J, Chamberlain LH. Dual role of the cysteine-string domain in membrane binding and palmitoylation-dependent sorting of the molecular chaperone cysteine-string protein. Mol Biol Cell. 2006;17:4748–4759. doi: 10.1091/mbc.E06-03-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Melkonian KA, Ostermeyer AG, Chen JZ, Roth MG, Brown DA. Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J Biol Chem. 1999;274:3910–3917. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- 393.Brown DA. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology. 2006;21:430–439. doi: 10.1152/physiol.00032.2006. [DOI] [PubMed] [Google Scholar]
- 394.Escribá PV, Wedegaertner PB, Goñi FM, Vögler O. Lipid-protein interactions in GPCR-associated signaling. Biochim Biophys Acta. 2007;1768:836–852. doi: 10.1016/j.bbamem.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 395.Karnik SS, Ridge KD, Bhattacharya S, Khorana HG. Palmitoylation of bovine opsin and its cysteine mutants in COS cells. Proc Natl Acad Sci USA. 1993;90:40–44. doi: 10.1073/pnas.90.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Fukushima Y, Saitoh T, Anai M, Ogihara T, Inukai K, Funaki M, Sakoda H, Onishi Y, Ono H, Fujishiro M, Ishikawa T, Takata K, Nagai R, Omata M, Asano T. Palmitoylation of the canine histamine H2 receptor occurs at Cys305 and is important for cell surface targeting. Biochim Biophys Acta. 2001;1539:181–191. doi: 10.1016/S0167-4889(01)00104-5. [DOI] [PubMed] [Google Scholar]
- 397.Hilgemann DW, Fine M, Linder ME, Jennings BC, Lin MJ. Massive endocytosis triggered by surface membrane palmitoylation under mitochondrial control in BHK fibroblasts. eLife. 2013;2:e01293. doi: 10.7554/eLife.01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Lin MJ, Fine M, Lu JY, Hofmann SL, Frazier G, Hilgemann DW. Massive palmitoylation-dependent endocytosis during reoxygenation of anoxic cardiac muscle. eLife. 2013;2:e01295. doi: 10.7554/eLife.01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Levental I, Lingwood D, Grzybek M, Coskun U, Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci USA. 2010;107:22050–22054. doi: 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Larsen JB, et al. Membrane curvature enables N-Ras lipid anchor sorting to liquid-ordered membrane phases. Nat Chem Biol. 2015;11:192–194. doi: 10.1038/nchembio.1733. [DOI] [PubMed] [Google Scholar]
- 401.Wypijewski KJ, Tinti M, Chen W, Lamont D, Ashford ML, Calaghan SC, Fuller W. Identification of caveolar resident proteins in ventricular myocytes using a quantitative proteomic approach: dynamic changes in caveolar composition following adrenoceptor activation. Mol Cell Proteomics. 2015;14:596–608. doi: 10.1074/mcp.M114.038570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Fuller W, Reilly L, Hilgemann DW. S-palmitoylation and the regulation of NCX1. Channels (Austin) 2016;10:75–77. doi: 10.1080/19336950.2015.1099329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Maritim AC, Sanders RA, Watkins JB. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17:24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- 404.Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, Suzuki H, Toyama K, Spin JM, Tsao PS. Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci. 2015;16(10):25234–25263. doi: 10.3390/ijms161025234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Dombrowski L, Roy D, Marette A. Selective impairment in GLUT4 translocation to transverse tubules in skeletal muscle of streptozotocin-induced diabetic rats. Diabetes. 1998;47:5–12. doi: 10.2337/diab.47.1.5. [DOI] [PubMed] [Google Scholar]
- 406.Lauritzen HP, Ploug T, Prats C, Tavaré JM, Galbo H. Imaging of insulin signaling in skeletal muscle of living mice shows major role of T-tubules. Diabetes. 2006;55:1300–1306. doi: 10.2337/db05-1216. [DOI] [PubMed] [Google Scholar]
- 407.Louch WE, Mørk HK, Sexton J, Strømme TA, Laake P, Sjaastad I, Sejersted OM. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D’Hooge J, Sipido K. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–346. doi: 10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- 409.Øyehaug L, Loose KØ, Jølle GF, Røe ÅT, Sjaastad I, Christensen G, Sejersted OM, Louch WE. Synchrony of cardiomyocyte Ca2+ release is controlled by T-tubule organization, SR Ca2+ content, and ryanodine receptor Ca2+ sensitivity. Biophys J. 2013;104:1685–1697. doi: 10.1016/j.bpj.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Shao CH, Rozanski GJ, Patel KP, Bidasee KR. Dyssynchronous (non-uniform) Ca2+ release in myocytes from streptozotocin-induced diabetic rats. J Mol Cell Cardiol. 2007;42:234–246. doi: 10.1016/j.yjmcc.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 411.Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 412.Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Uphues I, Kolter T, Goud B, Eckel J. Failure of insulin-regulated recruitment of the glucose transporter GLUT4 in cardiac muscle of obese Zucker rats is associated with alterations of small-molecular-mass GTP-binding proteins. Biochem J. 1995;311:161–166. doi: 10.1042/bj3110161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Maria Z, Campolo AR, Lacombe VA. Diabetes alters the expression and translocation of the insulin-sensitive glucose transporters 4 and 8 in the atria. PLoS ONE. 2015;10:e0146033. doi: 10.1371/journal.pone.0146033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Wang W, Hansen PA, Marshall BA, Holloszy JO, Mueckler M. Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J Cell Biol. 1996;135:415–430. doi: 10.1083/jcb.135.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Ploug T, vanDeurs B, Ai H, Cushman SW, Ralston E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol. 1998;142:1429–1446. doi: 10.1083/jcb.142.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Lau YH, Caswell AH, Brunschwig JP, Baerwald R, Garcia M. Lipid analysis and freeze-fracture studies on isolated transverse tubules and sarcoplasmic reticulum subfractions of skeletal muscle. J Biol Chem. 1979;254:540–546. [PubMed] [Google Scholar]
- 418.Rosemblatt M, Hidalgo C, Vergara C, Ikemoto N. Immunological and biochemical properties of transverse tubule membranes isolated from rabbit skeletal muscle. J Biol Chem. 1981;256:8140–8148. [PubMed] [Google Scholar]
- 419.Sumnicht GE, Sabbadini RA. Lipid composition of transverse tubular membranes from normal and dystrophic skeletal muscle. Arch Biochem Biophys. 1982;215:628–637. doi: 10.1016/0003-9861(82)90124-2. [DOI] [PubMed] [Google Scholar]
- 420.Hidalgo C. Lipid phase of transverse tubule membranes from skeletal muscle. An electron paramagnetic resonance study. Biophys J. 1985;47:757–764. doi: 10.1016/S0006-3495(85)83978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Parton RG, Way M, Zorzi N, Stang E. Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol. 1997;136:137–154. doi: 10.1083/jcb.136.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. doi: 10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 423.Bakth S, Arena J, Lee W, Torres R, Haider B, Patel BC, Lyons MM, Regan TJ. Arrhythmia susceptibility and myocardial composition in diabetes. Influence of physical conditioning. J Clin Invest. 1986;77:382–395. doi: 10.1172/JCI112316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Yang Q, Kiyoshige K, Fujimoto T, Katayama M, Fujino K, Saito K, Nakaya Y, Mori H. Signal-averaging electrocardiogram in patients with diabetic mellitus. Jpn Heart J. 1990;31:25–33. doi: 10.1536/ihj.31.25. [DOI] [PubMed] [Google Scholar]
- 425.Hekimian G, Khandoudi N, Feuvary D, Beigelman PM. Abnormal cardiac rhythm in diabetic rats. Life Sci. 1985;37:547–555. doi: 10.1016/0024-3205(85)90467-9. [DOI] [PubMed] [Google Scholar]
- 426.Olsen KB, Axelsen LN, Braunstein TH, Sørensen CM, Andersen CB, Ploug T, Holstein-Rathlou NH, Nielsen MS. Myocardial impulse propagation is impaired in right ventricular tissue of Zucker diabetic fatty (ZDF) rats. Cardiovasc Diabetol. 2013;12:19. doi: 10.1186/1475-2840-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Axelsen LN, Calloe K, Braunstein TH, Riemann M, Hofgaard JP, Liang B, Jensen CF, Olsen KB, Bartels ED, Baandrup U, Jespersen T, Nielsen LB, Holstein-Rathlou NH, Nielsen MS. Diet-induced pre-diabetes slows cardiac conductance and promotes arrhythmogenesis. Cardiovasc Diabetol. 2015;14:87. doi: 10.1186/s12933-015-0246-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Schulz R, Görge PM, Görbe A, Ferdinandy P, Lampe PD, Leybaert L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol Ther. 2015;153:90–106. doi: 10.1016/j.pharmthera.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Münster PN, Weingart R. Effects of phorbol ester on gap junctions of neonatal rat heart cells. Pflugers Arch. 1993;423:181–188. doi: 10.1007/BF00374392. [DOI] [PubMed] [Google Scholar]
- 430.Malhotra A, Kang BPS, Hashmi S, Meggs LG. PKCε inhibits the hyperglycemia-induced apoptosis signal in adult rat ventricular myocytes. Mol Cell Biochem. 2005;268:169–173. doi: 10.1007/s11010-005-3858-6. [DOI] [PubMed] [Google Scholar]
- 431.Doble BW, Ping P, Kardami E. The εsubtype of protein kinase C is required for cardiomyocytes connexin 43 phosphorylation. Circ Res. 2000;86:293–301. doi: 10.1161/01.RES.86.3.293. [DOI] [PubMed] [Google Scholar]
- 432.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kléber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexion-43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.RES.87.8.656. [DOI] [PubMed] [Google Scholar]
- 434.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. J Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- 435.Lin H, Ogawa K, Imanaga I, Tribulova N. Remodeling of connexin 43 in the diabetic rat heart. Mol Cell Biochem. 2006;290:69–78. doi: 10.1007/s11010-006-9166-y. [DOI] [PubMed] [Google Scholar]
- 436.Yu L, Zhao Y, Fan Y, Wang M, Xu S, Fu G. Epigallocatechin-3 gallate, a green tea catechin, attenuated the downregulation of the cardiac gap junction induced by high glucose in neonatal rat cardiomyocytes. Cell Physiol Biochem. 2010;26:403–412. doi: 10.1159/000320564. [DOI] [PubMed] [Google Scholar]
- 437.Yu L, Yu H, Li X, Jin C, Zhao Y, Xu S, Sheng X. P38 MAPK/miR-1 are involved in the protective effect of EGCG in high glucose-induced Cx43 downregulation in neonatal rat cardiomyocytes. Cell Biol Int. 2016;40:934–942. doi: 10.1002/cbin.10637. [DOI] [PubMed] [Google Scholar]
- 438.Yu L, Zhao Y, Xu S, Ding F, Jin C, Fu G, Weng S. Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) system upregulated connexin43 expression in rat cardiomyocytes via PKC and ERK MAPK pathways. Int J Mol Sci. 2013;14:2242–2257. doi: 10.3390/ijms14022242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Howarth FC, Nowotny N, Zilahi E, El Haj MA, Lei M. Altered expression of gap junction connexin proteins may partly underlie heart rhythm disturbances in the streptozotocin-induced diabetic rat heart. Mol Cell Biochem. 2007;305:145–151. doi: 10.1007/s11010-007-9537-z. [DOI] [PubMed] [Google Scholar]
- 440.Mitasíková M, Lin H, Soukup T, Imanaga I, Tribulová N. Diabetes and thyroid hormones affect connexin-43 and PKC-epsilon expression in rat heart atria. Physiol Res. 2009;58:211–217. doi: 10.33549/physiolres.931425. [DOI] [PubMed] [Google Scholar]
- 441.Howarth FC, Chandler NJ, Kharche S, Tellez JO, Greener ID, Yamanushi TT, Billeter R, Boyett MR, Zhang H, Dobrzynski H. Effects of streptozotocin-induced diabetes on connexin43 mRNA and protein expression in ventricular muscle. Mol Cell Biochem. 2008;319:105–114. doi: 10.1007/s11010-008-9883-5. [DOI] [PubMed] [Google Scholar]
- 442.Radosinska J, Kurahara LH, Hiraishi K, Viczenczova C, Egan Benova T, Szeiffova Bacova B, Dosenko V, Navarova J, Obsitnik B, Imanaga I, Soukup T, Tribulova N. Modulation of cardiac connexin-43 by omega-3 fatty acid ethyl-ester supplementation demonstrated in spontaneously diabetic rats. Physiol Res. 2015;64:795–806. doi: 10.33549/physiolres.933075. [DOI] [PubMed] [Google Scholar]
- 443.Joshi MS, Mihm MJ, Cook AC, Schanbacher BL, Bauer JA. Alterations in connexin 43 during diabetic cardiomyopathy: competition of tyrosine nitration versus phosphorylation. J Diabetes. 2015;7:250–259. doi: 10.1111/1753-0407.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Lin H, Mitasikova M, Dlugosova K, Okruhlicova L, Imanaga I, Ogawa K, Weismann P, Tribulova N. Thyroid hormones suppress epsilon-PKC signalling, down-regulate connexin-43 and increase lethal arrhythmia susceptibility in non-diabetic and diabetic rat hearts. J Physiol Pharmacol. 2008;59:271–285. [PubMed] [Google Scholar]
- 445.Veeranki S, Givvimani S, Kundu S, Metreveli N, Pushpakumar S, Tyagi SC. Moderate intensity exercise prevents diabetic cardiomyopathy associated contractile dysfunction through restoration of mitochondrial function and connexin 43 levels in db/db mice. J Mol Cell Cardiol. 2016;92:163–173. doi: 10.1016/j.yjmcc.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Hesari FS, Khajehnasiri N, Khojasteh SM, Soufi FG, Dastranj A. Attenuation of phosphorylated connexin-43 protein levels in diabetic rat heart by regular moderate exercise. Arch Iran Med. 2014;17:569–573. [PubMed] [Google Scholar]
- 447.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, Gómez AM. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–615. doi: 10.2337/diabetes.55.03.06.db05-1284. [DOI] [PubMed] [Google Scholar]
- 448.Howarth FC, Qureshi MA, Hassan Z, Al Kury LT, Isaev D, Parekh K, Yammahi SR, Oz M, Adrian TE, Adeghate E. Changing pattern of gene expression is associated with ventricular myocyte dysfunction and altered mechanisms of Ca2+ signalling in young type 2 Zucker diabetic fatty rat heart. Exp Physiol. 2011;96:325–337. doi: 10.1113/expphysiol.2010.055574. [DOI] [PubMed] [Google Scholar]
- 449.Salem KA, Adrian TE, Qureshi MA, Parekh K, Oz M, Howarth FC. Shortening and intracellular Ca2+ in ventricular myocytes and expression of genes encoding cardiac muscle proteins in early onset type 2 diabetic Goto-Kakizaki rats. Exp Physiol. 2012;97:1281–1291. doi: 10.1113/expphysiol.2012.066639. [DOI] [PubMed] [Google Scholar]
- 450.Gaber EM, Jayaprakash P, Qureshi MA, Parekh K, Oz M, Adrian TE, Howarth FC. Effects of a sucrose-enriched diet on the pattern of gene expression, contraction and Ca2+ transport in Goto -Kakizaki type 2 diabetic rat heart. Exp Physiol. 2014;99:881–893. doi: 10.1113/expphysiol.2013.077594. [DOI] [PubMed] [Google Scholar]
- 451.da Silva MF, Natali AJ, da Silva E, Gomes GJ, Teodoro BG, Cunha DN, Drummond LR, Drummond FR, Moura AG, Belfort FG, de Oliveira A, Maldonado IR, Alberici LC. Attenuation of Ca2+ homeostasis, oxidative stress, and mitochondrial dysfunctions in diabetic rat heart: insulin therapy or aerobic exercise? J Appl Physiol. 1985;2015(119):148–156. doi: 10.1152/japplphysiol.00915.2014. [DOI] [PubMed] [Google Scholar]
- 452.Lengyel C, Virág L, Bíró T, Jost N, Magyar J, Biliczki P, Kocsis E, Skoumal R, Nánási PP, Tóth M, Kecskeméti V, Papp JG, Varró A. Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res. 2007;73:512–520. doi: 10.1016/j.cardiores.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 453.Lopez-Izquierdo A, Pereira RO, Wende AR, Punske BB, Abel ED, Tristani-Firouzi M. The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am J Physiol Heart Circ Physiol. 2014;306:H747–H754. doi: 10.1152/ajpheart.00849.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Sato T, Kobayashi T, Kuno A, Miki T, Tanno M, Kouzu H, Itoh T, Ishikawa S, Kojima T, Miura T, Tohse N. Type 2 diabetes induces subendocardium-predominant reduction in transient outward K+ current with downregulation of Kv4.2 and KChIP2. Am J Physiol Heart Circ Physiol. 2014;306:H1054–H1065. doi: 10.1152/ajpheart.00414.2013. [DOI] [PubMed] [Google Scholar]
- 455.Hansen PS, Clarke RJ, Buhagiar KA, Hamilton E, Garcia A, White C, Rasmussen HH. Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol. 2007;292:C1070–C1077. doi: 10.1152/ajpcell.00288.2006. [DOI] [PubMed] [Google Scholar]
- 456.Lu Z, Jiang YP, Xu XH, Ballou LM, Cohen IS, Lin RZ. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes. 2007;56:2780–2789. doi: 10.2337/db06-1629. [DOI] [PubMed] [Google Scholar]
- 457.Markandeya YS, Phelan LJ, Woon MT, Keefe AM, Reynolds CR, August BK, Hacker TA, Roth DM, Patel HH, Balijepalli RC. Caveolin-3 overexpression attenuates cardiac hypertrophy via inhibition of T-type Ca2+ current modulated by protein kinase Cα in cardiomyocytes. J Biol Chem. 2015;290:22085–22100. doi: 10.1074/jbc.M115.674945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Duzhyy DE, Viatchenko-Karpinski VY, Khomula EV, Voitenko NV, Belan PV. Upregulation of T-type Ca2+ channels in long-term diabetes determines increased excitability of a specific type of capsaicin-insensitive DRG neurons. Mol Pain. 2015;11:29. doi: 10.1186/s12990-015-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Torres-Jacome J, Gallego M, Rodríguez-Robledo JM, Sanchez-Chapula JA, Casis O. Improvement of the metabolic status recovers cardiac potassium channel synthesis in experimental diabetes. Acta Physiol (Oxf) 2013;207:447–459. doi: 10.1111/apha.12043. [DOI] [PubMed] [Google Scholar]
- 460.Chen ZC, Cheng YZ, Chen LJ, Cheng KC, Li Y, Cheng J. Increase of ATP-sensitive potassium (KATP) channels in the heart of type-1 diabetic rats. Cardiovasc Diabetol. 2012;11:8. doi: 10.1186/1475-2840-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Carotenuto F, Costa A, Albertini MC, Rocchi MBL, Rudov A, Coletti D, Minieri M, Di Nardo P, Teodori L. Dietary flaxseed mitigates impaired skeletal muscle regeneration: in vivo, in vitro and in silico studies. Int J Med Sci. 2016;13:206–219. doi: 10.7150/ijms.13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Xia Z, Kuo KH, Nagareddy PR, Wang F, Guo Z, Guo T, et al. N-acetylcysteine attenuates PKCbeta2 overexpression and myocardial hypertrophy in streptozotocin-induced diabetic rats. Cardiovasc Res. 2007;73:770–782. doi: 10.1016/j.cardiores.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 463.Yamamoto M, Okumura S, Oka N, Schwencke C, Ishikawa Y. Downregulation of caveolin expression by cAMP signal. Life Sci. 1999;64:1349–1357. doi: 10.1016/S0024-3205(99)00070-3. [DOI] [PubMed] [Google Scholar]
- 464.Quach NL, Biressi S, Reichardt LF, Keller C, Rando TA. Focal adhesion kinase signaling regulates the expression of caveolin 3 and beta1 integrin, genes essential for normal myoblast fusion. Mol Biol Cell. 2009;20:3422–3435. doi: 10.1091/mbc.E09-02-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Mori S, Takemoto M, Yokote K, Asaumi S, Saito Y. Hyperglycemia-induced alteration of vascular smooth muscle phenotype. J Diabetes Complicat. 2002;16:65–68. doi: 10.1016/S1056-8727(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 466.Fortes MA, Pinheiro CH, Guimarães-Ferreira L, Vitzel KF, Vasconcelos DA, Curi R. Overload-induced skeletal muscle hypertrophy is not impaired in STZ-diabetic rats. Physiol Rep. 2015;3:e12457. doi: 10.14814/phy2.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Huang D, Khoe M, Ilic D, Bryer-Ash M. Reduced expression of focal adhesion kinase disrupts insulin action in skeletal muscle cells. Endocrinology. 2006;147:3333–3343. doi: 10.1210/en.2005-0382. [DOI] [PubMed] [Google Scholar]
- 468.Wichelhaus A, Russ M, Petersen S, Eckel J. G protein expression and adenylate cyclase regulation in ventricular cardiomyocytes from STZ-diabetic rats. Am J Physiol. 1994;267:H548–H555. doi: 10.1152/ajpheart.1994.267.2.H548. [DOI] [PubMed] [Google Scholar]
- 469.Matsumoto T, Wakabayashi K, Kobayashi T, Kamata K. Functional changes in adenylyl cyclases and associated decreases in relaxation responses in mesenteric arteries from diabetic rats. Am J Physiol Heart Circ Physiol. 2005;289:H2234–H2243. doi: 10.1152/ajpheart.00971.2004. [DOI] [PubMed] [Google Scholar]
- 470.Vatner SF, Park M, Yan L, Lee GJ, Lai L, Iwatsubo K, Ishikawa Y, Pessin J, Vatner DE. Adenylyl cyclase type 5 in cardiac disease, metabolism, and aging. Am J Physiol Heart Circ Physiol. 2013;305:H1–H8. doi: 10.1152/ajpheart.00080.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Colhoun HM. Lipid goals in metabolic syndrome and diabetes. Curr Atheroscler Rep. 2007;9:286–295. doi: 10.1007/s11883-007-0035-2. [DOI] [PubMed] [Google Scholar]
- 472.Van de Ree M, Huisman M, Princen H, Meinders A, Kluft C. Strong decrease of high sensitivity C-reactive protein with highdose atorvastatin in patients with type 2 diabetes mellitus. Atherosclerosis. 2003;166:129–135. doi: 10.1016/S0021-9150(02)00316-7. [DOI] [PubMed] [Google Scholar]
- 473.Betteridge DJ, Carmena R. The diabetogenic action of statins—mechanisms and clinical implications. Nat Rev Endocrinol. 2016;12:99–110. doi: 10.1038/nrendo.2015.194. [DOI] [PubMed] [Google Scholar]
- 474.Parida S, Swain TR, Routray SN, Maiti R. Effect of atorvastatin on glycaemic parameters in normoglycaemic and prediabetic subjects: a prospective, panel study. J Clin Diagn Res. 2017;11:FC04–FC09. doi: 10.7860/JCDR/2017/23741.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Maki KC, Dicklin MR, Baum SJ. Statins and diabetes. Endocrinol Metab Clin North Am. 2016;45:87–100. doi: 10.1016/j.ecl.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 476.Adameová A, Harcarova A, Matejikova J, Pancza D, Kuzelova M, Carnicka S, Svec P, Bartekova M, Styk J, Ravingerová T. Simvastatin alleviates myocardial contractile dysfunction and lethal ischemic injury in rat heart independent of cholesterol-lowering effects. Physiol Res. 2009;58:449–454. doi: 10.33549/physiolres.931751. [DOI] [PubMed] [Google Scholar]
- 477.Rajtík T, Čarnická S, Szobi A, Mesárošová L, Máťuš M, Švec P, Ravingerová T, Adameová A. Pleiotropic effects of simvastatin are associated with mitigation of apoptotic component of cell death upon lethal myocardial reperfusion-induced injury. Physiol Res. 2012;61(Suppl 2):S33–S41. doi: 10.33549/physiolres.932420. [DOI] [PubMed] [Google Scholar]
- 478.Verkerk AO, van Ginneken AC, Berecki G, den Ruijter HM, Schumacher CA, Veldkamp MW, Baartscheer A, Casini S, Opthof T, Hovenier R, Fiolet JW, Zock PL, Coronel R. Incorporated sarcolemmal fish oil fatty acids shorten pig ventricular action potentials. Cardiovasc Res. 2006;70:509–520. doi: 10.1016/j.cardiores.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 479.Benediktsdottir V, Gudbjarnason S. Modification of the fatty acid composition of rat heart sarcolemma with dietary cod liver oil, corn oil or butter. J Mol Cell Cardiol. 1988;20:141–147. doi: 10.1016/S0022-2828(88)80027-0. [DOI] [PubMed] [Google Scholar]
- 480.Al Makdessi S, Sweidan H, Jacob R. n-3 versus n-6 fatty acid incorporation into the phospholipids of rat heart sarcolemma. A comparative study of four different oil diets. J Mol cell cardiol. 1994;26:23–29. doi: 10.1006/jmcc.1994.1004. [DOI] [PubMed] [Google Scholar]
- 481.Creus A, Ferreira M, Oliva M, Lombardo Y. Mechanisms involved in the improvement of lipotoxicity and impaired lipid metabolism by dietary α-linolenic acid rich Salvia hispanica l (salba) seed in the heart of dyslipemic insulin-resistant rats. J Clin Med. 2016;5:E18. doi: 10.3390/jcm5020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Franekova V, Angin Y, Hoebers NTH, Coumans WA, Simons PJ, Glatz JFC, et al. Marine omega-3 fatty acids prevent myocardial insulin resistance and metabolic remodeling as induced experimentally by high insulin exposure. Am J Physiol Cell Physiol. 2015;308:C297–C307. doi: 10.1152/ajpcell.00073.2014. [DOI] [PubMed] [Google Scholar]
- 483.Kahleova H, Matoulek M, Bratova M, Malinska H, Kazdova L, Hill M, Pelikanova T. Vegetarian diet-induced increase in linoleic acid in serum phospholipids is associated with improved insulin sensitivity in subjects with type 2 diabetes. Nutr Diabetes. 2013;3:e75. doi: 10.1038/nutd.2013.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Field CJ, Ryan EA, Thomson AB, Clandinin MT. Diet fat composition alters membrane phospholipid composition, insulin binding, and glucose metabolism in adipocytes from control and diabetic animals. J Biol Chem. 1990;265:11143–11150. [PubMed] [Google Scholar]
- 485.Den Ruijter HM, Verkerk AO, Coronel R. Incorporated fish oil fatty acids prevent action potential shortening induced by circulating fish oil fatty acids. Front Physiol. 2010;1:149. doi: 10.3389/fphys.2010.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Bogdanov KY, Spurgeon HA, Vinogradova TM, Lakatta EG. Modulation of the transient outward current in adult rat ventricular myocytes by polyunsaturated fatty acids. Am J Physiol. 1998;274:H571–H579. doi: 10.1152/ajpheart.1998.274.2.H571. [DOI] [PubMed] [Google Scholar]
- 487.Xiao YF, Gomez AM, Morgan JP, Lederer WJ, Leaf A. Suppression of voltage-gated L-type Ca2+ currents by polyunsaturated fatty acids in adult and neonatal rat ventricular myocytes. Proc Natl Acad Sci USA. 1997;94:4182–4187. doi: 10.1073/pnas.94.8.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Goel DP, Maddaford TG, Pierce GN. Effects of omega-3 polyunsaturated fatty acids on cardiac sarcolemmal Na+/H+ exchange. Am J Physiol Heart Circ Physiol. 2002;283:H1688–H1694. doi: 10.1152/ajpheart.00664.2001. [DOI] [PubMed] [Google Scholar]
- 489.Han X, Cheng H, Mancuso DJ, Gross RW. Caloric restriction results in phospholipid depletion, membrane remodeling, and triacylglycerol accumulation in murine myocardium. Biochemistry. 2004;43:15584–15594. doi: 10.1021/bi048307o. [DOI] [PubMed] [Google Scholar]
- 490.Yamagishi T, Bessho M, Yanagida S, Nishizawa K, Kusuhara M, Ohsuzu F, Tamai S. Severe, short-term food restriction improves cardiac function following ischemia/reperfusion in perfused rat hearts. Heart Vessels. 2010;25:417–425. doi: 10.1007/s00380-009-1222-6. [DOI] [PubMed] [Google Scholar]
- 491.Marina Prendes MG, González MS, Torresín ME, Hermann R, Pascale NG, del Mar Jaitovich M, Savino EA, Varela A. Involvement of mitochondrial permeability transition, glutathione status, pentose phosphate pathway and oxidative damage in the protective effect of fasting on the ischaemic-reperfused rat heart. Clin Exp Pharmacol Physiol. 2009;36:637–642. doi: 10.1111/j.1440-1681.2008.05122.x. [DOI] [PubMed] [Google Scholar]
- 492.Snorek M, Hodyc D, Sedivý V, Durišová J, Skoumalová A, Wilhelm J, Neckář J, Kolář F, Herget J. Short-term fasting reduces the extent of myocardial infarction and incidence of reperfusion arrhythmias in rats. Physiol Res. 2012;61:567–574. doi: 10.33549/physiolres.932338. [DOI] [PubMed] [Google Scholar]
- 493.Park SY, Choi GH, Choi HI, Ryu J, Jung CY, Lee W. Calorie restriction improves whole-body glucose disposal and insulin resistance in association with the increased adipocyte-specific GLUT4 expression in Otsuka Long-Evans Tokushima fatty rats. Arch Biochem Biophys. 2005;436:276–284. doi: 10.1016/j.abb.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 494.Minamiyama Y, Bito Y, Takemura S, Takahashi Y, Kodai S, Mizuguchi S, Nishikawa Y, Suehiro S, Okada S. Calorie restriction improves cardiovascular risk factors via reduction of mitochondrial reactive oxygen species in type II diabetic rats. J Pharmacol Exp Ther. 2007;320:535–543. doi: 10.1124/jpet.106.110460. [DOI] [PubMed] [Google Scholar]
- 495.Liepinsh E, Makrecka M, Kuka J, Makarova E, Vilskersts R, Cirule H, Sevostjanovs E, Grinberga S, Pugovics O, Dambrova M. The heart is better protected against myocardial infarction in the fed state compared to the fasted state. Metabolism. 2014;63:127–136. doi: 10.1016/j.metabol.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 496.van den Brom CE, Boer C, van den Akker RF, Loer SA, Bouwman RA. Reducing caloric intake prevents ischemic injury and myocardial dysfunction and affects anesthetic cardioprotection in type 2 diabetic rats. J Diabetes Res. 2017;2017:4126820. doi: 10.1155/2017/4126820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Jamieson HA, Hilmer SN, Cogger VC, Warren A, Abernethy DR, Everitt AV, Fraser R, de Cabo R, Le Couteur DG. Caloric restriction reduces age-related pseudocapillarization of the hepatic sinusoid. Exp Gerontol. 2007;42:374–378. doi: 10.1016/j.exger.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Ørom UA, Lim MK, Savage JE, Jin L, Saleh AD, Lisanti MP, Simone NL. MicroRNA-203 regulates caveolin-1 in breast tissue during caloric restriction. Cell Cycle. 2012;11:1291–1295. doi: 10.4161/cc.19704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Melkani GC, Panda S. Time-restricted feeding for prevention and treatment of cardiometabolic disorders. J Physiol. 2017;595:3691–3700. doi: 10.1113/JP273094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JA, Ellisman MH, Panda S. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Yasumoto Y, Hashimoto C, Nakao R, Yamazaki H, Hiroyama H, Nemoto T, Yamamoto S, Sakurai M, Oike H, Wada N, Yoshida-Noro C, Oishi K. Short-term feeding at the wrong time is sufficient to desynchronize peripheral clocks and induce obesity with hyperphagia, physical inactivity and metabolic disorders in mice. Metabolism. 2016;65:714–727. doi: 10.1016/j.metabol.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 502.Gill S, Le HD, Melkani GC, Panda S. Time-restricted feeding attenuates age-related cardiac decline in Drosophila. Science. 2015;347:1265–1269. doi: 10.1126/science.1256682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Booth FW, Roberts CK, Laye MJ. Lack of exercise is a major cause of chronic diseases. Compr Physiol. 2012;2:1143–1211. doi: 10.1002/cphy.c110025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Colditz GA, Cannuscio CC, Frazier AL. Physical activity and reduced risk of colon cancer: implications for prevention. Cancer Causes Control. 1997;8:649–667. doi: 10.1023/A:1018458700185. [DOI] [PubMed] [Google Scholar]
- 505.Manson JE, Hu FB, Rich-Edwards JW, Colditz GA, Stampfer MJ, Willett WC, Speizer FE, Hennekens CH. A prospective study of walking as compared with vigorous exercise in the prevention of coronary heart disease in women. N Engl J Med. 1999;341:650–658. doi: 10.1056/NEJM199908263410904. [DOI] [PubMed] [Google Scholar]
- 506.Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, Willett WC. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med. 2001;345:790–797. doi: 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- 507.Roberts CK, Won D, Pruthi S, Kurtovic S, Sindhu RK, Vaziri ND, Barnard RJ. Effect of a short-term diet and exercise intervention on oxidative stress, inflammation, MMP-9, and monocyte chemotactic activity in men with metabolic syndrome factors. J Appl Physiol. 2006;100:1657–1665. doi: 10.1152/japplphysiol.01292.2005. [DOI] [PubMed] [Google Scholar]
- 508.Brown JC, Winters-Stone K, Lee A, Schmitz KH. Cancer, physical activity, and exercise. Compr Physiol. 2012;2:2775–2809. doi: 10.1002/cphy.c120005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Eyre HA, Papps E, Baune BT. Treating depression and depression-like behavior with physical activity: an immune perspective. Front Psychiatry. 2013;4:3. doi: 10.3389/fpsyt.2013.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Lavie CJ, Arena R, Swift DL, Johannsen NM, Sui X, Lee DC, Earnest CP, Church TS, O’Keefe JH, Milani RV, Blair SN. Exercise and the cardiovascular system: clinical science and cardiovascular outcomes. Circ Res. 2015;117:207–219. doi: 10.1161/CIRCRESAHA.117.305205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Lancaster GI, Febbraio MA. The immunomodulating role of exercise in metabolic disease. Trends Immunol. 2014;35:262–269. doi: 10.1016/j.it.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 512.Pareja-Galeano H, Garatachea N, Lucia A. Exercise as a polypill for chronic diseases. Prog Mol Biol Transl Sci. 2015;135:497–526. doi: 10.1016/bs.pmbts.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 513.Lumb A. Diabetes and exercise. Clin Med (Lond) 2014;14:673–676. doi: 10.7861/clinmedicine.14-6-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Staimez LR, Weber MB, Gregg EW. The role of lifestyle change for prevention of cardiovascular disease in diabetes. Curr Atheroscler Rep. 2014;16:460. doi: 10.1007/s11883-014-0460-y. [DOI] [PubMed] [Google Scholar]
- 515.Powers SK, Smuder AJ, Kavazis AN, Quindry JC. Mechanisms of exercise-induced cardioprotection. Physiology (Bethesda) 2014;29:27–38. doi: 10.1152/physiol.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Wang W, Zhang H, Xue G, Zhang L, Zhang W, Wang L, Lu F, Li H, Bai S, Lin Y, Lou Y, Xu C, Zhao Y. Exercise training preserves ischemic preconditioning in aged rat hearts by restoring the myocardial polyamine pool. Oxid Med Cell Longev. 2014;2014:457429. doi: 10.1155/2014/457429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Mataix J, Quiles JL, Huertas JR, Battino M, Mañas M. Tissue specific interactions of exercise, dietary fatty acids, and vitamin E in lipid peroxidation. Free Radic Biol Med. 1998;24:511–521. doi: 10.1016/S0891-5849(97)00288-8. [DOI] [PubMed] [Google Scholar]
- 518.Nikolaidis MG, Petridou A, Matsakas A, Schulz T, Michna H, Mougios V. Effect of chronic wheel running on the fatty acid composition of phospholipids and triacylglycerols in rat serum, skeletal muscle and heart. Acta Physiol Scand. 2004;181:199–208. doi: 10.1111/j.1365-201X.2004.01277.x. [DOI] [PubMed] [Google Scholar]
- 519.Marini M, Abruzzo PM, Bolotta A, Veicsteinas A, Ferreri C. Aerobic training affects fatty acid composition of erythrocyte membranes. Lipids Health Dis. 2011;10:188. doi: 10.1186/1476-511X-10-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Pons S, Martin V, Portal L, Zini R, Morin D, Berdeaux A, Ghaleh B. Regular treadmill exercise restores cardioprotective signaling pathways in obese mice independently from improvement in associated co-morbidities. J Mol Cell Cardiol. 2013;54:82–89. doi: 10.1016/j.yjmcc.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 521.Wang H, Bei Y, Lu Y, Sun W, Liu Q, Wang Y, Cao Y, Chen P, Xiao J, Kong X. Exercise prevents cardiac injury and improves mitochondrial biogenesis in advanced diabetic cardiomyopathy with PGC-1α and Akt activation. Cell Physiol Biochem. 2015;35:2159–2168. doi: 10.1159/000374021. [DOI] [PubMed] [Google Scholar]
- 522.Schrauwen-Hinderling VB, Meex RC, Hesselink MK, van de Weijer T, Leiner T, Schär M, Lamb HJ, Wildberger JE, Glatz JF, Schrauwen P, Kooi ME. Cardiac lipid content is unresponsive to a physical activity training intervention in type 2 diabetic patients, despite improved ejection fraction. Cardiovasc Diabetol. 2011;10:47. doi: 10.1186/1475-2840-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Bennett CE, Johnsen VL, Shearer J, Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci. 2013;92:657–663. doi: 10.1016/j.lfs.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 524.Belke DD. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J Appl Physiol. 1985;2011(111):157–162. doi: 10.1152/japplphysiol.00147.2011. [DOI] [PubMed] [Google Scholar]
- 525.Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013;305:H114–H123. doi: 10.1152/ajpheart.00135.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Giusti B, Marini M, Rossi L, Lapini I, Magi A, Capalbo A, Lapalombella R, di Tullio S, Samaja M, Esposito F, Margonato V, Boddi M, Abbate R, Veicsteinas A. Gene expression profile of rat left ventricles reveals persisting changes following chronic mild exercise protocol: implications for cardioprotection. BMC Genom. 2009;10:342. doi: 10.1186/1471-2164-10-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.