

Abstract
The genetic abnormalities underlying multiple myeloma (MM) are notoriously complex and intraclonal heterogeneity is a common disease feature. In the current study, we describe the establishment of a monoclonal IgA kappa (κ) MM cell line, designated MC-B11/14. Cytogenetic and FISH analyses of the original and relapse patient samples revealed the MM clone was non-hyperdiploid and possessed an 11;14 chromosomal translocation. The MC-B11/14 cell line, established from the relapse sample, is tetraploid and houses the t(11;14) abnormality. Given our long standing interest in immunoglobulin function and secretion, we next used CRISPR technology to knock out IgA heavy chain expression in the MC-B11/14 cells to assess the biological consequences of converting this cell line to one only expressing κ light chains. As expected, secretion of intact IgA was undetectable from MC-B11/14IgA− cells. Sensitivity to pomalidomide treatment was similar between the MC-B11/14WT and MC-B11/14IgA− cells, however, MC-B11/14IgA− cells were found to be significantly more resistant to bortezomib treatment. This study describes the establishment of a new human MM cell line tool to study disease biology and use of CRISPR technology to create a potentially useful model to study MM light chain escape.
Keywords: multiple myeloma, bortezomib, light chain escape
INTRODUCTION
Multiple myeloma (MM), a devastating and often fatal plasma cell (PC) malignancy, is the second most common hematological malignancy in the USA [1]. On the basis of immunoglobulin gene rearrangement and somatic mutation, MM is considered a clonal disease. However, additional genetic hits acquired during disease progression results in considerable intratumor genetic heterogeneity and selection of MM subclones with growth and survival advantages [2–4] that compete for resources within the BM micro-environment [5].
The development and use of novel therapeutic strategies for treatment of MM has considerably improved quality of life for MM patients and has substantially extended life expectancy. However, a patient’s response to treatment can be significantly impacted by intraclonal heterogeneity and the diverse sensitivity of individual MM clones to various chemotherapeutic agents [6]. In fact, changes in MM clonal dominance have been observed following MM treatment regimens [3]. Indeed, one of the first observations of clonal divergence was made by Hobbs in 1969 during the first medical research council trial when he observed that 5% of MM patients, who were originally determined to have abnormal levels of intact Ig, had relapsed with increased levels of free light chains (FLC) in the absence of intact Ig [3, 7]. This phenomenon, now referred to as light chain escape (LCE), is specifically defined as an increase in monoclonal FLC in the absence of a corresponding increase in monoclonal intact Ig [8].
Although the mechanisms underlying LCE remain to be precisely determined, clonal evolution/selection is believed to play an important role. Patients who relapse with LCE typically have a poorer prognosis [9]. In the current study, we established a new human IgAκ secreting MM cell line (HMCL) and exploited this new HMCL tool and CRISPR technology to investigate the biological consequences following loss of IgA heavy chain (HC) expression.
MATERIALS and METHODS
Immunoglobulin gene analysis, immunophenotypic analysis, annexin V binding, western blotting, and enzyme-linked immunosorbent assays (ELISA) are described in Supplementary Methods.
Cell line establishment
This study was performed with the approval of the Mayo Clinic Institutional Review Board, and informed consent was obtained in accordance with the Declaration of Helsinki. The MC-B11/14 HMCL was established from the BM aspirate obtained following the patient’s second relapse. The BM aspirate was processed according to previously described procedures [10]. At onset, CD138+ cells were cultured in Iscove’s modified Dulbecco medium with GlutaMAX (Invitrogen, Carlsbad, CA), supplemented with 50 U/mL penicillin G, 50 μg/mL streptomycin (Invitrogen), 10% heat-inactivated fetal calf serum (Atlanta Biologicals, Norcross, GA), 1 ng/mL interleukin-6 (IL-6; kindly provided by Novartis, Basel, Switzerland) and 10 ng/mL insulin-like growth factor-I (IGF-I; Sigma-Aldrich, St. Louis, MO). The MC-B11/14 HMCL is routinely cultured under these same conditions. This HMCL is responsive to both IL-6 and IGF-I and addition of both cytokines has an additive effect on cell proliferation; thus, the cells are routinely cultured with both cytokines to maintain optimal in vitro cell line passaging. The MC-B11/14 HMCL has been tested for the presence of Epstein Barr virus (EBV) using PCR methodology and was determined to be negative (data not shown). All studies used MC-B11/14 cells that had been in continuous culture for less than three months.
Karyotype analysis
Chromosome banding was performed by the Medical Genomics Facility at Mayo Clinic. Standard methods for brightfield G-banding (GTL-banding) were utilized [11, 12].
Fluorescence in situ hybridization (FISH) analysis
FISH analysis was performed as previously described [13]. The 11;14 probe used was from Vysis (Abbott Laboratories, Abbott Park, Illinois).
Cell proliferation assay
Cell proliferation assays were performed as previously described [10]. Recombinant human IL-6 and IGF-1 were used at concentrations of 1 ng/mL and 10 ng/mL, respectively. Recombinant human IFN-α (Biosource; Camarillo, CA) was used at 250 U/mL; recombinant human IFN-γ (Peprotech; Rocky Hill, NJ, USA) was used at 1 ng/mL; and recombinant human OSM and TNF-α (R&D; Minneapolis, MN) were used at final concentrations of 10 ng/mL and 1 ng/mL, respectively. Statistical analysis was performed using a paired t-test and GraphPad (San Diego, CA) analytical software.
Immunofluorescence (IF) analysis
IF analysis was performed as previously described [14] using an anti-IgA-FITC (Biosource) or anti-κ-TRITC (Southern Biotech) antibody.
CRISPR design, construction and genome editing
To knockout IgA1 gene expression in IgA1 expressing MC-B11/14 cells, we designed a guide RNA specific to a sequence in the IgA1 CH1 region (5′-GAACGTGGTCATCGCCTGCC3′). Oligos of the IgA1 single-guide RNA (sgRNA) sequence were then annealed, phosphorylated, and cloned into the BssI/BssI sites of the pX458 backbone vector (Addgene plasmid 48138). Construct clones were sequenced to verify the presence of the correct IgA1-sgRNA sequence. The IgA1-sgRNA-pX458 construct was then transiently transfected into MC-B11/14 cells by electroporation. 24 hours later, single GFP expressing MC-B11/14 cells were sorted into a 96-well plate at 1 cell/well using a BD Aria II cell sorter and expanded in vitro. Subclones were genotyped using IgA1 specific primers and Sanger sequencing to assess disruption of the locus. The null IgA expression in the knockout clones was validated by Western blotting.
Results
Case Report
A 46-year-male patient was referred to Mayo Clinic after diagnosis with MM and initial treatment. At diagnosis, serum protein electrophoresis revealed the patient was biclonal for IgGκ and IgAκ. A skeletal survey and magnetic resonance imaging revealed multiple lytic lesions and extensive myelomatous bone involvement. Chromosome analysis of the malignant PCs revealed an abnormal tetraploid clone with an 11;14 translocation [t(11;14)(q13;q32)]. The patient was started on bortezomib with dexamethasone and lenalidomide and received radiation to his L3 vertebral body. After 4 rounds of chemotherapy, a BM biopsy revealed a significant BM response to treatment with less than 5% monoclonal kappa cells in the BM. However, a serum IgAκ monoclonal protein was still detectable and FISH analysis of the BM biopsy demonstrated a diploid t(11;14) clone and persistence of a small t(11;14) tetraploid population. The patient was also found to exhibit significant paraspinal mass lesions, compression fractures and soft tissue uptake around the right lung. The patient was considered to be in relapse with a significant amount of extramedullary disease prompting subsequent treatment with an autologous BM stem cell transplant. Two months later the right side pleural mass still persisted and an additional lesion was detected in the left abdominal wall. Biopsy and analysis of the BM and pleural mass revealed a monoclonal IgAκ plasma cell population and the patient was determined to be in relapse again. The MC-B11/14 HMCL was established from this BM aspirate. The patient was started on high-dose combination chemotherapy but succumbed to disease shortly thereafter.
MC-B11/14 cell morphology and immunophenotype
With regard to morphology, the patient sample obtained at second relapse and the MC-B11/14 HMCL both displayed classic PC morphology with eccentrically located nuclei, prominent nucleoli, and a perinuclear hof was present in some cells. Several multi-nucleated cells were observed in both the primary patient sample and the MC-B11/14 HMCL (data not shown). The immunophenotype of the MC-B11/14 HMCL was tested via flow cytometry with a panel of antibodies. As shown in Table I, MC-B11/14 cells were determined to express high levels of CD28, CD38, CD44, CD49d, CD56, CD63, CD81, and CD138. Low level expression was observed for CD45, CD130, PD-1, and the IGF-I receptor. The expression pattern of these molecules was found to be consistent over different testing time points.
Table I.
Immunophenotyping of the MC-B11/14 HMCL was performed via flow cytometry using a panel of antibodies. Data reflect the delta mean fluorescence intensity (MFI) as calculated as MFI of a specific antibody/MFI of the matched isotype control antibody.
| Marker | Delta MFI | Marker | Delta MFI |
|---|---|---|---|
| CD19 | 1.0 | CD56 | 6.3 |
| CD20 | 1.0 | CD69 | 1.0 |
| CD27 | 1.0 | CD63 | 7.7 |
| CD28 | 14.4 | CD81 | 9.3 |
| CD38 | 7.1 | CD126 | 1.0 |
| CD44 | 19.5 | CD130 | 2.1 |
| CD45 | 2.7 | CD138 | 3.1 |
| CD49d | 10.0 | IGF-1R | 1.4 |
| CD49e | 1.0 | PD-1 | 1.4 |
Karyotype analysis
As previously stated, the malignant clone was determined to be tetraploid for DNA content at diagnosis. After treatment and at the second relapse the clone was found to be primarily diploid with persistence of a small tetraploid clone. Table II displays the karyotype of the patient sample at diagnosis and at second relapse, and the resulting HMCL. All samples possessed complex numeric and structural abnormalities, including an IGH rearrangement and thus were genetically classified as non-hyperdiploid (NHD). Specifically, the IGH rearrangement was determined to be an 11;14 translocation [t(11;14)(q13;q32)]. Of note, the tetraploid subclone persisting in the relapse sample also possessed the t(11;14)(q13;q32)(data not shown). Likewise, FISH analysis of the MC-B11/14 HMCL (Figure 1A) demonstrated the presence of the t(11;14)(q13;q32) abnormality and suggested that the cell line was tetraploid for DNA content, which was confirmed by cell cycle analysis (data not shown). Thus, the MC-B11/14 HMCL originated from outgrowth of the tetraploid subclonal population of cells present from initial disease diagnosis as well as in the relapse patient sample. The t(11;14)(q13;q32) is a common translocation in MM and results in upregulation of cyclin D1. Indeed, the MC-B11/14 HMCL was shown to overexpress cyclin D1 by western blotting (Figure 1B) and was compared to other MM HMCLs lacking this translocation.
Table II.
Cytogenetic analysis of primary patient cells at diagnosis and relapse and the MC-B11/14 HMCL. (sl indicates stemline clone).
| Cells | Chromosome No. | Representative Karyotype |
|---|---|---|
| Patient at diagnosis | 92 | XXYY, add(1)(p13), −6, t(6;8)(q21;q24.1) x2, +der(6)t(6;8),der(11)t(11;14)(q13;q32)x2, der(14)add(14)(p11.2)t(11;14)(q13;q32)x2[2]/46,XY[19] |
| Patient at relapse | 46–49 | 49,XY,del(6)(q13q25),+7,add(8)(q22), der(11)t(11;14)(q13;q32),der(14)t(11;14)(q13;p11.2)t(11;14) (q13;q32),+19,+21[6]/48,sl,add(1)(p13),+add(22) (p11.2)[2]/80,slx2,+5mar[1]/46,XY[11] |
| MC-B11/14 HMCL | 91–94 | XX, −X, −X or −Y, Y, der(1;12) (p10;q10), add(1)(q31), −2, −4, der (5) t(5;12) (q35;q13), add (6) (p25)x2, add (6)(q13)x2, +add(7) (q11.2)x2, add(8)(q22)x2, add(8)(q24.1)x2, −9, −10, add(10)(p13), +11, der(11)t(11;14)(q13;q32)X2, der(11;14)(q10;10)t(11;14)x2, +12, −18, +19, +19, +21, +21, +0–6mar[cp20] |
Figure 1.

A) Multiple green/red fusions following FISH analysis demonstrate that the MC-B11/14 HMCL possesses an 11;14 translocation; B) Western blotting reveals that the MC-B11/14 HMCL overexpresses cyclin D1.
Immunoglobulin gene analysis
Sequence analysis of the rearranged light (data not shown) and heavy chain variable region genes in the primary patient sample and the MC-B11/14 HMCL demonstrated that the sequences were identical (Figure 2). The specific rearranged genes were determined to be IGKV4-1, IGKJ2, IGHV5-a, IGHD2-2 and IGHJ6b. The level of V region somatic mutation was 2.8% in the IGHV gene and 3.3% in the IGKV gene.
Figure 2.

Alignment of germline IgA heavy chain variable sequence with the cDNA sequence from the original patient sample (CD138+ cells) and the MC-B11/14 HMCL demonstrates that the sequence of the patient sample and the cell line are identical. The corresponding amino acid sequence is shown below. Amino acid changes resulting from somatic hypermutation are indicated in larger bold font with asterisks.
Cytokine responsiveness
The MC-B11/14 HMCL was found to proliferate in response to IL-6 and IGF-1 (Figure 3). Of note, an additive effect was observed when cells were stimulated with both IL-6 and IGF-1 (data not shown). MC-B11/14 cells did not proliferate in response to stimulation with TNF-α, IFN-α, IFN-γ, or OSM.
Figure 3.
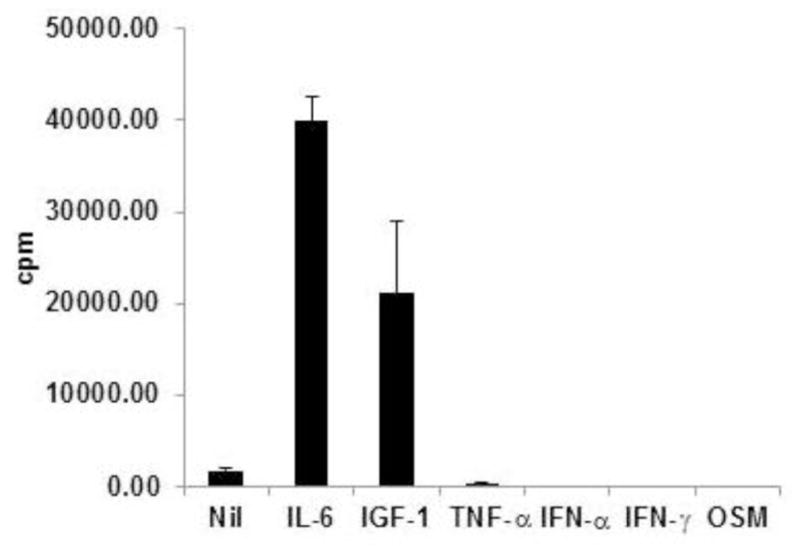
Proliferative response of MC-B11/14 HMCL to various cytokines. Proliferation was assessed by [3H]thymidine incorporation after 72 hours. Data reflect the mean +/− SD of triplicate experimental points and are reflective of multiple experiments.
Knockout of IgA heavy chain in MC-B11/14 cells
Figure 4 illustrates the region of the IgA gene we selected as our CRISPR target sequence and our overall CRISPR-mediated strategy to obtain MC-B11/14 cells that no longer express IgA HC. Once the MC-B11/14 cells were transfected and sorted for IgA expression or lack thereof, we stained the parental or wildtype (MC-B11/14WT) cells and the IgA knockout (MC-B11/14IgA−) cells for expression of IgA via IF. As shown in Figure 5A, the MC-B11/14IgA− knockout cells were completely negative for expression of IgA HC. Similar results were obtained by western blotting (Figure 5B) and flow cytometry (data not shown). Of note, IF staining of MC-B11/14IgA− knockout cells for κLC revealed much less cytoplasmic κLC than the WT cells (Figure 5A).
Figure 4.
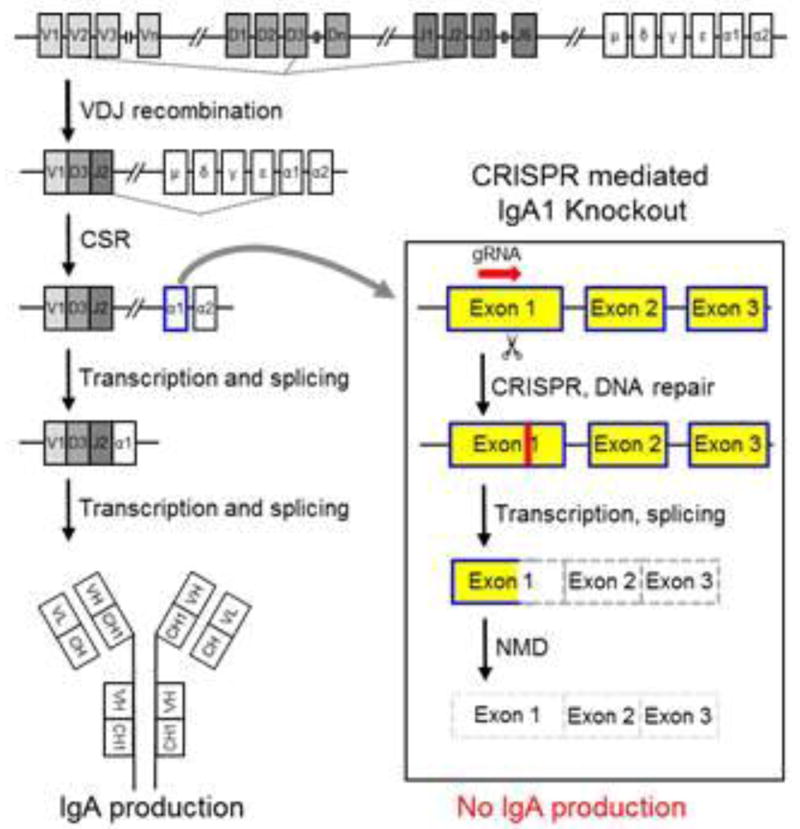
Experimental design of IgA1 gene knockout. Ig gene rearrangement and maturation scheme and IgA1 knockout design strategy. (NMD= nonsense-mediated decay).
Figure 5.

A) Immunofluorescent detection of IgA HC and κLC in MC-B11/14WT and MC-B11/14IgA− cells. B) Western blot demonstrating loss of IgA expression in MC-B11/14IgA− cells. ELISA was used to measure secretion of C) IgA; D) κLC; and E) κFLC from MC-B11-14WT cells and MC-B11/14IgA− cells following a 24 hr culture period. Data reflect the mean +/− SD and are reflective of multiple experiments (n=4).
Ig secretion
To further ensure that IgA expression was successfully knocked out, we next determined the secretion levels of intact IgA, total κLC (bound and free) and free κLC in the MC-B11/14WT and MC-B11/14IgA− knockout cells. As predicted, the MC-B11/14WT cells secreted a significant amount of IgA whereas IgA was undetectable in the supernatant collected from the MC-B11/14IgA− cells (Figure 5C). Of interest, the total level of κLC secretion (bound and free) was lower in the IgA knockout cells (Figure 5D) while the secretion of free κLC was observed to be higher in the IgA knockout cells (Figure 5E).
Pomalidomide and bortezomib mediated inhibition of cellular proliferation
Because LCE is most often observed in relapsing MM patients and that the malignant PCs of these patients generally become more resistant to therapy, we next wanted to test the sensitivity of MC-B11/14WT and MC-B11/14IgA− cells to pomalidomide and bortezomib. As shown in Figure 6A, no difference in sensitivity to pomalidomide was observed between the two HMCLs following a 5-day incubation. By contrast, in comparison to the MC-B11/14WTcells, the MC-B11/14IgA− cells were found to be more resistant to treatment with bortezomib following a 3-day incubation (Figure 6B). Moreover, MC-B11/14IgA− cells as compared to MC-B11/14WT cells, were found to have substantially fewer annexin-V+ cells following a 24 hr treatment with various concentrations of bortezomib (Figure 6C).
Figure 6.

Sensitivity of MC-B11/14WT cells and MC-B11/14IgA − cells to: A) Pomalidomide, 5-day treatment; and B) Bortezomib, 3-day treatment, was assessed by [3H]thymidine incorporation. Data are expressed as % growth response as compared to control. C) Annexin-V+ cells following 24 hr treatment with various concentrations of bortezomib. Assays were performed in the presence of IL-6 and data reflect the mean +/− SD of triplicate experimental points and are reflective of multiple experiments (n=4). Statistically significant differences (p<0.01) are indicated by an asterisk.
Discussion
In this study, we describe the establishment of a monoclonal IgAκ HMCL from a NHD-MM patient at relapse. FISH analysis determined that the abnormal PCs possessed an 11;14 translocation [t(11;14)(q13;q32)]. Overall, the frequency of t(11;14)(q13;q32) in MM patients is ~10–14% [15] and is the most common translocation in MM [16]. Notably, the t(11;14)(q13;q32) has also been shown be present at the MM precursor condition stage, i.e., monoclonal gammopathy of undetermined significance; and thus, is believed to be an early cytogenetic event [17]. Presence of the t(11;14)(q13;q32) results in upregulation of cyclin D1, which typically promotes cell cycle progression of cells in G1 to S phase [18]. The MC-B11/14 HMCL was found to overexpress cyclin D1 when compared to HMCLs lacking the t(11;14)(q13;q32) and thus, exemplifies the known obligate relationship between t(11;14)(q13;q32) and overexpression of cyclin D1 [17].
The t(11;14)(q13;q32) abnormality is the most common translocation in MM, however it is surprising that several of the more frequently used HMCLs lack this translocation. The MC-B11/14 HMCL, was established from a patient whom had received both bortezomib and lenalidomide prior to HMCL establishment, and thus, is a unique HMCL in the respect that it more adequately models MM t(11;14)(q13;q32) patients who have received novel therapeutics. In addition, given that most t(11;14)(q13;q32) HMCLs are near diploid, the near tetraploid MC-B11/14 HMCL is a unique entity representing a distinct group of MM patients.
Production of monoclonal Ig is a classic feature of MM and can consist of intact Ig, free light chain, both, or the clone can be non-secretory [3]. Our long standing interest in the biological function of both membrane bound and secreted Ig and our previous study using genome editing technologies [19] lead to our use of CRISPR technology to knockout IgA HC expression in the MC-B11/14 HMCL. Knockout of IgA HC resulted in a HMCL that only secreted κFLC. Although mature plasma cells are generally thought to lack transmembrane Ig and only express secreted Ig, ~35% of MM patients have been shown to express transmembrane Ig [20] and the MC-B11/14 HMCL was also determined to express transmembrane Ig. The significance of MM cell transmembrane Ig expression is not known. Because HCs uniquely possess a hydrophobic transmembrane domain whereas LCs do not, knockout of IgA HC in the MC-B11/14 HMCL also resulted in loss of transmembrane Ig. Our observations that IgA HC loss did not impact MC-B11/14IgA − cell viability suggests that transmembrane Ig is not required for sustained survival of this HMCL and provides distinction from B-cell development, where membrane Ig expression is essential for both B-cell survival and differentiation [21]. Indeed, although more than 80% of MM patients present with production of intact Ig, ~50% of these patients present with monoclonal FLC, in the urine and/or serum [22]. Selective outgrowth of an FLC secreting clone without an increase in the intact Ig secreting clone is exemplary of the intraclonal heterogeneity observed in MM. LCE occurs in ~2.5 to 8% of IgG and IgA MM patients following treatment [23] and is associated with MM progression and/or relapse. LCE is believed to result from combined selective pressure from the BM microenvironment and chemotherapeutic treatment [3, 24].
The use of novel therapeutic agents such as the IMiDs, lenalidomide and pomalidomide, and the highly selective proteasome inhibitor, bortezomib, has significantly improved the overall survival of MM patients [25]. Treatment of MC-B11/14IgA− cells with pomalidomide resulted in no significant difference in proliferation as compared to MC-B11/14WT cells whereas MC-B11/14IgA− cells were found to be significantly more resistant to treatment with bortezomib. In addition to induction of apoptosis of MM cells through blocking NF-κB activation [26–28], several studies have shown that bortezomib can induce additional effects on MM cells. Indeed, MM sensitivity to bortezomib has also been shown to be related to high levels of protein synthesis or more specifically, Ig synthesis [29]. In general, high protein synthesis leads to a higher degree of unfolded proteins in the endoplasmic reticulum (ER) and cytoplasm. Unfolded proteins are cleared by the proteasome dependent endoplasmic reticulum-associated degradation pathway [29]. Inhibition of the proteasome results in accumulation of unfolded proteins in the ER lumen, which subsequently triggers a signaling pathway called the unfolded protein response (UPR) [30]. In the event that the UPR pathway cannot eliminate the misfolded proteins, overwhelming ER stress ensues and a terminal UPR pathway involving apoptosis is activated [31]. Meister et al observed that decreased Ig synthesis conferred decreased sensitivity to bortezomib and thus, a decrease in bortezomib-induced apoptosis was observed. Likewise, MC-B11/14IgA− cells were found to be substantially more resistant to bortezomib-induced apoptosis. Accordingly, one mechanism of MM clone resistance to bortezomib may include decreased protein synthesis and more specifically, Ig synthesis [29] and/or clonal selection of a non-secretory or LC only producing clone. In this regard, in results not shown, we observed that several LC only HMCLs were also less sensitive to bortezomib when compared to HMCLs that produce both heavy and light chains.
Taken together, we describe the establishment of a new, biologically useful HMCL, MC-B11/14. We further demonstrate the utility of this line to model LCE using CRISPR technology. The latter findings suggest that MM patients producing high levels of Ig may be more sensitive to treatment with bortezomib patients whose myeloma cells are synthesizing lower levels of Ig or protein. These findings also underscore the importance of monitoring clonal evolution over the course of disease and that a clinical investigation into whether there is a correlation between bortezomib sensitivity and Ig production may be warranted in the future.
Supplementary Material
Highlights.
A novel human monoclonal IgA kappa myeloma cell line was established.
CRISPR mediated gene editing was used to disrupt IgA heavy chain expression.
Loss of IgA expression, significantly lessened sensitivity to bortezomib.
Acknowledgments
This work was supported in part by the National Institutes of Health (grant CA196831 awarded to DFJ). We would also like to thank Kimberly Henderson for purifying the primary MM cells and Greg Ahmann and Scott Van Weir for performing the FISH analysis of the MC-B11/14 HMCL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–113. doi: 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 2.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335–348. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 3.Kraj M, Kruk B, Endean K, Warzocha K, Budziszewska K, Dabrowska M. Light Chain Escape in 3 Cases: Evidence of Intraclonal Heterogeneity in Multiple Myeloma from a Single Institution in Poland. Case Rep Hematol. 2015;2015:809840. doi: 10.1155/2015/809840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brioli A, Melchor L, Walker BA, Davies FE, Morgan GJ. Biology and treatment of myeloma. Clin Lymphoma Myeloma Leuk. 2014;14(Suppl):S65–70. doi: 10.1016/j.clml.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Walker BA, Wardell CP, Melchor L, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood. 2012;120:1077–1086. doi: 10.1182/blood-2012-03-412981. [DOI] [PubMed] [Google Scholar]
- 6.Brioli A, Melchor L, Cavo M, Morgan GJ. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br J Haematol. 2014;165:441–454. doi: 10.1111/bjh.12805. [DOI] [PubMed] [Google Scholar]
- 7.Hobbs JR. Growth rates and responses to treatment in human myelomatosis. Br J Haematol. 1969;16:607–617. doi: 10.1111/j.1365-2141.1969.tb00441.x. [DOI] [PubMed] [Google Scholar]
- 8.Caldini A, Nozzoli C, Terreni A, et al. New patterns of relapse in multiple myeloma: a case of “light chain escape” in which FLC predicted relapse earlier than urine and serum immunofixation. Clin Chem Lab Med. 2016;54:991–995. doi: 10.1515/cclm-2015-0689. [DOI] [PubMed] [Google Scholar]
- 9.Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood. 2014;123:3414–3419. doi: 10.1182/blood-2013-12-542662. [DOI] [PubMed] [Google Scholar]
- 10.Arendt BK, Ramirez-Alvarado M, Sikkink LA, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood. 2008;112:1931–1941. doi: 10.1182/blood-2008-03-143040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971;2:971–972. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- 12.Spurbeck JL, Adams SA, Stupca PJ, Dewald GW. Primer on medical genomics. Part XI: Visualizing human chromosomes. Mayo Clin Proc. 2004;79:58–75. doi: 10.4065/79.1.58. [DOI] [PubMed] [Google Scholar]
- 13.Ahmann GJ, Jalal SM, Juneau AL, et al. A novel three-color, clone-specific fluorescence in situ hybridization procedure for monoclonal gammopathies. Cancer Genet Cytogenet. 1998;101:7–11. doi: 10.1016/s0165-4608(97)00058-7. [DOI] [PubMed] [Google Scholar]
- 14.Walters DK, Arendt BK, Jelinek DF. CD147 regulates the expression of MCT1 and lactate export in multiple myeloma cells. Cell Cycle. 2013;12:3175–3183. doi: 10.4161/cc.26193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Corral L, Gutierrez NC, Vidriales MB, et al. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin Cancer Res. 2011;17:1692–1700. doi: 10.1158/1078-0432.CCR-10-1066. [DOI] [PubMed] [Google Scholar]
- 16.Leiba M, Duek A, Amariglio N, et al. Translocation t(11;14) in newly diagnosed patients with multiple myeloma: Is it always favorable? Genes Chromosomes Cancer. 2016;55:710–718. doi: 10.1002/gcc.22372. [DOI] [PubMed] [Google Scholar]
- 17.Fonseca R, Blood EA, Oken MM, et al. Myeloma and the t(11;14)(q13;q32); evidence for a biologically defined unique subset of patients. Blood. 2002;99:3735–3741. doi: 10.1182/blood.v99.10.3735. [DOI] [PubMed] [Google Scholar]
- 18.Hoyer JD, Hanson CA, Fonseca R, Greipp PR, Dewald GW, Kurtin PJ. The (11;14)(q13;q32) translocation in multiple myeloma. A morphologic and immunohistochemical study. Am J Clin Pathol. 2000;113:831–837. doi: 10.1309/4W8E-8F4K-BHUP-UBE7. [DOI] [PubMed] [Google Scholar]
- 19.Wu X, Blackburn PR, Tschumper RC, Ekker SC, Jelinek DF. TALEN-mediated genetic tailoring as a tool to analyze the function of acquired mutations in multiple myeloma cells. Blood Cancer J. 2014;4:e210. doi: 10.1038/bcj.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ocqueteau M, San Miguel JF, Gonzalez M, Almeida J, Orfao A. Do myelomatous plasma cells really express surface immunoglobulins? Haematologica. 1996;81:460–463. [PubMed] [Google Scholar]
- 21.Corcos D, Osborn MJ, Matheson LS. B-cell receptors and heavy chain diseases: guilty by association? Blood. 2011;117:6991–6998. doi: 10.1182/blood-2011-02-336164. [DOI] [PubMed] [Google Scholar]
- 22.Ahn JS, Jung SH, Yang DH, et al. Patterns of relapse or progression after bortezomib-based salvage therapy in patients with relapsed/refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2014;14:389–394. doi: 10.1016/j.clml.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Farges C, Roussel M, Huynh A, Blancher A, Puissant-Lubrano B. Aggressive FLC Escape in a Patient with IgD Myeloma. Case Rep Hematol. 2015;2015:694730. doi: 10.1155/2015/694730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hobbs JA, Drayson MT, Sharp K, Harding S, Bradwell AR, Mead GP. Frequency of altered monoclonal protein production at relapse of multiple myeloma. Br J Haematol. 2010;148:659–661. doi: 10.1111/j.1365-2141.2009.07952.x. [DOI] [PubMed] [Google Scholar]
- 25.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitsiades N, Mitsiades CS, Poulaki V, et al. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99:4079–4086. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 27.Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 28.Hideshima T, Richardson PG, Anderson KC. Targeting proteasome inhibition in hematologic malignancies. Rev Clin Exp Hematol. 2003;7:191–204. [PubMed] [Google Scholar]
- 29.Meister S, Schubert U, Neubert K, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007;67:1783–1792. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 30.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 31.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.