

ABSTRACT
The overall health of the oral cavity is dependent on proper homeostasis between health-associated bacterial colonizers and bacteria known to promote dental caries. Streptococcus sanguinis is a health-associated commensal organism, a known early colonizer of the acquired tooth pellicle, and is naturally competent. We have shown that LytF, a competence-controlled murein hydrolase, is capable of inducing the release of extracellular DNA (eDNA) from oral bacteria. Precipitated LytF and purified LytF were used as treatments against planktonic cultures and biofilms. Larger amounts of eDNA were released from cultures treated with protein samples containing LytF. Additionally, LytF could affect biofilm formation and cellular morphology. Biofilm formation was significantly decreased in the lytF-complemented strain, in which increased amounts of LytF are present. The same strain also exhibited cell morphology defects in both planktonic cultures and biofilms. Furthermore, the LytF cell morphology phenotype was reproducible in wild-type cells using purified LytF protein. In sum, our findings demonstrate that LytF can induce the release of eDNA from oral bacteria, and they suggest that, without proper regulation of LytF, cells display morphological abnormalities that contribute to biofilm malformation. In the context of the oral biofilm, LytF may play important roles as part of the competence and biofilm development programs, as well as increasing the availability of eDNA.
IMPORTANCE Streptococcus sanguinis, a commensal organism in the oral cavity and one of the pioneer colonizers of the tooth surface, is associated with the overall health of the oral environment. Our laboratory showed previously that, under aerobic conditions, S. sanguinis can produce H2O2 to inhibit the growth of bacterial species that promote dental caries. This production of H2O2 by S. sanguinis also induces the release of eDNA, which is essential for proper biofilm formation. Under anaerobic conditions, S. sanguinis does not produce H2O2 but DNA is still released. Determining how S. sanguinis releases DNA is thus essential to understand biofilm formation in the oral cavity.
KEYWORDS: Streptococcus
INTRODUCTION
Within the oral cavity, more than 600 bacterial species have been identified and are known to persist through both cooperative and competitive means in a biofilm environment (1). Overall health in the oral cavity depends on the homeostasis between health-associated species and species known to contribute to dental caries and periodontitis (2, 3). For example, Streptococcus sanguinis and Streptococcus gordonii are considered health-associated pioneer colonizers within the oral biofilm. Both species are known to combat actively the growth of cariogenic and periodontal pathogens, such as Streptococcus mutans and Porphyromonas gingivalis, via the production of H2O2 (4, 5). Essential to competition, cooperation, and overall communication within the oral environment is the ability of certain bacterial species to develop natural competence. The competence pathway has been studied extensively in Streptococcus pneumoniae and is considered a stress-response pathway (6). Among several other stress-associated cellular functions, competence controls the uptake of exogenous or extracellular DNA (eDNA) (7). Interestingly, commensal species such as S. sanguinis and S. gordonii naturally develop competence under normal growth conditions, suggesting that genes regulated by the competence pathway play a role in establishing and maintaining biofilms (8, 9).
Competence is controlled by the competence-stimulating peptide (CSP). As an example, when CSP is secreted by Streptococcus pneumoniae cells, it subsequently is sensed and activates a two-component signal transduction system composed of the membrane-bound histidine kinase ComD and the corresponding response regulator ComE. CSP binds to ComD and allows the phosphorylation of ComE, which activates a number of so-called “early” competence genes (10, 11). Encoded by one of these early competence constituents, ComX is an alternative sigma factor that activates the transcription of late competence genes by binding to the cin box, which serves as a ComX-dependent promoter. The majority of late competence genes are involved in DNA repair, regulation, and uptake (12).
Extracellular DNA and its subsequent uptake are vital for oral biofilms. eDNA is a necessary component of the exopolysaccharide matrix of biofilms, as it provides structural stability and promotes cell-cell and cell-tooth adhesion (13 – 15). A lack of eDNA, such as that caused by treatment with DNase, causes biofilm destruction and inhibition of cell aggregation (16, 17). Since eDNA is present within the biofilm matrix, naturally competent bacteria readily import this eDNA to promote genetic exchange through horizontal gene transfer, especially under conditions that provide selective pressure (9, 18, 19). The release of this eDNA is considered one of the many functions of murein hydrolases, enzymes that act to digest the bacterial cell wall (20 – 22). In S. pneumoniae, murine hydrolase activity, competence development, and eDNA release are all coordinated processes. Specifically, when the competence-associated LytA is deleted, eDNA release from S. pneumoniae is decreased. This phenotype is exacerbated in a double deletion mutant with LytC (23). Although LytC is itself not associated with competence, further evidence suggests that the overall hydrolytic activity of LytA peaks when LytC and a third murein hydrolase, CbpD, all act together (24, 25).
Our group demonstrated previously that both S. sanguinis and S. gordonii are able to generate eDNA in an H2O2-dependent manner (17, 18). H2O2 is produced by and promotes eDNA release from S. sanguinis and S. gordonii during early biofilm formation, when there is an ample supply of environmental oxygen (26). As biofilms mature and thicken, oxygen availability and H2O2 production both decrease, although eDNA release is not completely inhibited (17, 18). This indicates that an additional mechanism for eDNA release must be utilized in oral streptococci. Since competence is known to play a role in eDNA release in S. pneumoniae, we focused on competence-regulated murein hydrolases (23).
Interestingly, of the competence-controlled genes in S. sanguinis and S. gordonii, we identified two potential murein hydrolases, encoded by sgo_2094 in S. gordonii and ssa_0036 in S. sanguinis. Both genes are predicted to be under the control of the alternative sigma factor ComX, indicating that they would be part of a late competence regulon. The murein hydrolase encoded by sgo_2094 was characterized previously and was named LytF (27). While the genes in both species lie among purine biosynthesis genes, S. gordonii lytF is also surrounded by the competence genes comA, comB, and comX (Fig. 1A). In fact, comX and sgo_2093 even overlap lytF (sgo_2094). S. sanguinis does not possess genes overlapping lytF (ssa_0036), and other competence genes are not present in the immediate vicinity, although S. sanguinis is not known to contain homologs of either comA or comB. Due to the similarities between the genes encoding LytF in S. gordonii and S. sanguinis, it is likely that the two gene products are orthologs. After a BLAST comparison, it is apparent that the cysteine histidine-dependent aminohydrolase/peptidase (CHAP) domains, the catalytic regions of the LytF protein responsible for hydrolyzing peptidoglycan via amidase and/or peptidase activities, share >75% identity (27). The differences thus lie in the number of Bsp (group B streptococcal secreted protein) domains. While S. sanguinis LytF possesses five Bsp domains, S. gordonii LytF has only three (Fig. 1B). Considering that Bsp domains are thought to give proteins their specificity for peptidoglycan structures, we were interested, in the current study, in examining the activities of LytF in S. sanguinis, specifically how these functions would relate to overall biofilm formation and the development of competence (27, 28).
FIG 1.

Genetic context of lytF in S. sanguinis and S. gordonii, with lytF colored dark gray for each species. Note that open reading frames are approximate and are not drawn to scale. (A) Top, S. sanguinis genetic context, with lytF (ssa_0036) present on the negative strand and surrounded by purine biosynthesis genes; ssa_0036 is 1,950 bp and produces a 659-amino-acid protein, LytF. Bottom, S. gordonii genetic context, with lytF (sgo_2094) present on the positive strand. Two genes overlapping sgo_2094 are present, along with purine biosynthesis genes upstream and the competence-associated comAB operon downstream; sgo_2094 is 1,650 bp and produces a 549-amino-acid LytF. (B) Comparison of protein domains within LytF of S. sanguinis and LytF of S. gordonii. Both proteins contain the hydrolytic CHAP domain (light gray rectangles) at the C terminus. S. sanguinis LytF contains five Bsp regions (dark gray diamonds), while S. gordonii LytF contains three, highlighting the main difference in the proteins between the two species. Small black rectangles upstream of LytF indicate the cin box.
RESULTS
LytF is part of the late competence regulon.
To first confirm that LytF is indeed part of the competence pathway, we induced competence by adding CSP to mid-log-phase cultures (optical density at 600 nm [OD600] of 0.4) growing in pH 6.8 medium (Fig. 2A). It is necessary to adjust the pH of the medium when examining the competence of S. sanguinis because the cells naturally induce competence at higher pH values, specifically pH 7.4 (as demonstrated in Fig. 2B), which is routinely the approximate pH of our autoclaved media (29, 30). By examining the expression of comD and comYA, well-characterized early and late competence genes, respectively, we concluded that at pH 6.8 we were able to induce the expression of the competence genes comD and comYA and our gene of interest, lytF, comparable to findings reported previously (29, 31). Specifically, the expression of comD was induced >4-fold, while comYA and lytF were induced >100-fold.
FIG 2.

Induction of competence-associated genes with CSP addition. Cultures were grown to mid-log phase (OD600 of ∼0.4), 0.5 μg/ml CSP was added to the appropriate samples, and the cultures were further incubated for 2 h. RT-PCR was performed with 16S rRNA as the housekeeping gene. Expression levels are presented relative to levels in noninduced, no-CSP-added controls, which were set to 1. Data presented are the means and standard deviations of three independent experiments performed in duplicate on different days. (A) Cultures grown in TH medium at pH 6.8. (B) Cultures grown in TH medium at pH 7.4.
Previous expression data indicated that lytF (ssa_0036) is regulated as a late competence gene (29). Late competence gene expression is regulated by the alternative sigma factor ComX. The association of ComX with genes possessing a cin box promoter region, predominantly DNA maintenance genes, activates their expression (12). To determine the promoter region and to identify the cin box for lytF, 5′ rapid amplification of cDNA ends (RACE) was performed. Accordingly, a likely cin box was identified 8 bases upstream of the identified transcription start site (Fig. 3).
FIG 3.

RACE results, showing the sequence of the identified lytF promoter. The agarose gel image shows the final RACE PCR product from the lytF promoter region that was ultimately sequenced, and the arrow indicates the band that correlates with the size of the lytF promoter with the 5′-RACE adapter.
In order to further examine the regulation of lytF and the activity of its gene product, it was necessary to create a lytF mutant and to complement this mutant. To do this, we created a lytF allelic replacement using an erythromycin resistance cassette, ermAM, and complemented this mutant by expressing lytF via its native promoter from the shuttle vector pDL278. We refer to the lytF mutant and its complemented strain as the ΔlytF and lytF-c strains, respectively. Considering that the competence pathway is highly regulated and responds to a number of different stimuli, it was necessary to confirm that the competence pathway was still properly expressed in the newly created strains. All examined competence genes were similarly expressed in the ΔlytF and lytF-c strains, compared to the wild-type SK36 strain. It is important to note that lytF expression is highly overexpressed in the lytF-c strain, likely due to the copy number of the vector (Fig. 4; also see Fig. 9 for increased amounts of LytF in the supernatant of the lytF-c strain).
FIG 4.

Induction of competence-associated genes in lytF mutant strains. The relative expression of competence-associated genes in the lytF mutant and complemented strains was calculated with the levels in S. sanguinis SK36 control samples set to 1. Data presented are the means and standard deviations of three independent experiments performed in duplicate on different days.
FIG 9.
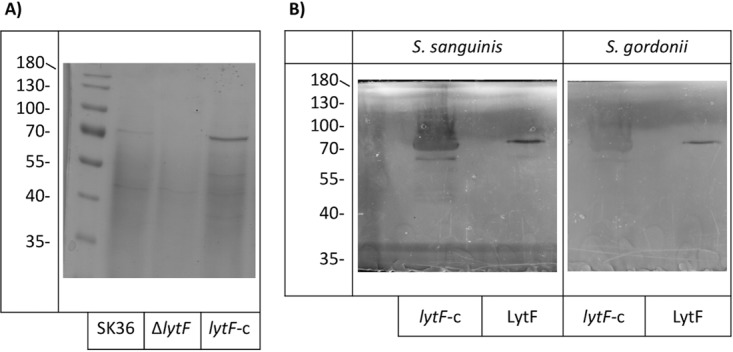
Precipitation and purification of LytF from S. sanguinis. (A) SDS-PAGE analysis of proteins precipitated from culture supernatants with 60% ammonium sulfate saturation. The image is representative of at least three experiments performed on different days. (B) Zymogram assays with S. sanguinis substrate cells (left) or S. gordonii substrate cells (right). Both lytF-c protein samples (left lanes) and purified LytF (right lanes) showed hydrolysis of both species. Zymograms are representative of three separate experiments performed on different days. Images were processed to increase the visibility of the bands.
Transformation efficiency and cell division are affected by LytF.
Given that LytF is a predicted murein hydrolase associated with competence, we wanted to determine whether the protein is involved in transformation and/or cell division. Transformation assays showed a 4-fold decrease in transformation in the ΔlytF strain, compared to the wild-type strain (Fig. 5A). The three strains also showed differences in their bacterial chain lengths. The ΔlytF strain had larger amounts of elongated chains than did the wild-type SK36 strain, while the lytF-c strain exhibited short chain lengths overall (Fig. 5B). We next treated planktonic cultures with the green fluorescent vancomycin-BODIPY-FL conjugate, which incorporates into the peptidoglycan and thus serves as an effective cell wall stain for Gram-positive organisms (32). Although there was no apparent difference in cell morphology between SK36 cells and ΔlytF cells, we did notice that lytF-c cells appeared more spherical and bloated (Fig. 6A). To examine this phenotype more closely, we grew the wild-type and mutant strains as biofilms in chemically defined medium (CDM) with added sucrose and examined the cell morphology with scanning electron microscopy (SEM). The observed phenotype was consistent for cells in biofilms; lytF-c biofilm cells appeared bloated and distended, whereas ΔlytF cells appeared similar to wild-type SK36 cells (Fig. 6B). Taken together, these results indicate that LytF may play a role in cell division as a murein hydrolase, which presumably also affects the ability of cells to take up exogenous DNA.
FIG 5.

LytF effects on transformation and cell division. (A) Cells were grown to an OD600 of ∼0.08 in TH medium with 2.5% heat-inactivated horse serum. Chromosomal DNA (kanamycin resistance) (37.6 ng/μl) and 0.5 μg/ml CSP were added, and cells were further incubated for 2 h. Cells were plated on selective and nonselective TH plates. Transformation efficiency was calculated as the number of kanamycin-resistant CFU relative to the CFU on nonselective agar. The decrease in ΔlytF transformation efficiency was statistically significant, compared to the wild-type value (**, P = 0.0099). Data represent the means and standard deviations of three independent experiments. (B) Cells were grown to mid-log phase, and images were obtained with oil immersion using an Olympus BX51 microscope, an Olympus DP72 digital camera, and cellSens 1.3 software. Images were adjusted for brightness and contrast and are representative of three independent experiments with similar outcomes. Chains from three images per strain were measured using ImageJ. Chain length differences between the strains were not statistically significant. Scale bars, 10 μm.
FIG 6.
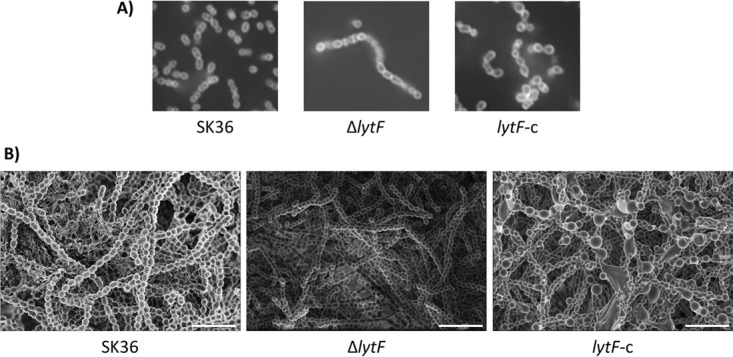
Altered cell morphologies of LytF mutant strains. (A) Cells were grown to mid-log phase and stained with 1 μg/ml vancomycin-BODIPY-FL conjugate. Images were obtained with oil immersion using an Olympus IX73 microscope, an Olympus DP72 digital camera, and cellSens standard software. Images were adjusted for brightness and contrast and are representative of multiple experiments with similar outcomes. (B) SEM was performed on 18-hour biofilms grown on Thermanox discs in CDM. Images are representative of at least three fields of view from two experiments performed on different days. Scale bars, 5 μm.
LytF affects biofilm formation.
As shown in Fig. 6, an excess of the LytF protein appears to be unhealthy for cells in both planktonic and biofilm cultures, as there was a multitude of enlarged and irregularly shaped cells with the lytF-c strain. Therefore, we wanted to examine the overall biofilm formation among the various strains. The ability of S. sanguinis wild-type and mutant strains to form biofilms was examined by quantifying biofilm formation in 96-well plate assays. Biofilms were stained with safranin and the absorbance at 490 nm was measured to assess differences in biofilm formation among the SK36, ΔlytF, and lytF-c strains. Although the absorbance was not largely changed for the lytF mutant, compared with the wild-type strain, the overexpression of lytF in the lytF-c strain resulted in a significant decrease in biofilm formation, compared to both the SK36 wild-type and ΔlytF strains (P < 0.0001) (Fig. 7).
FIG 7.
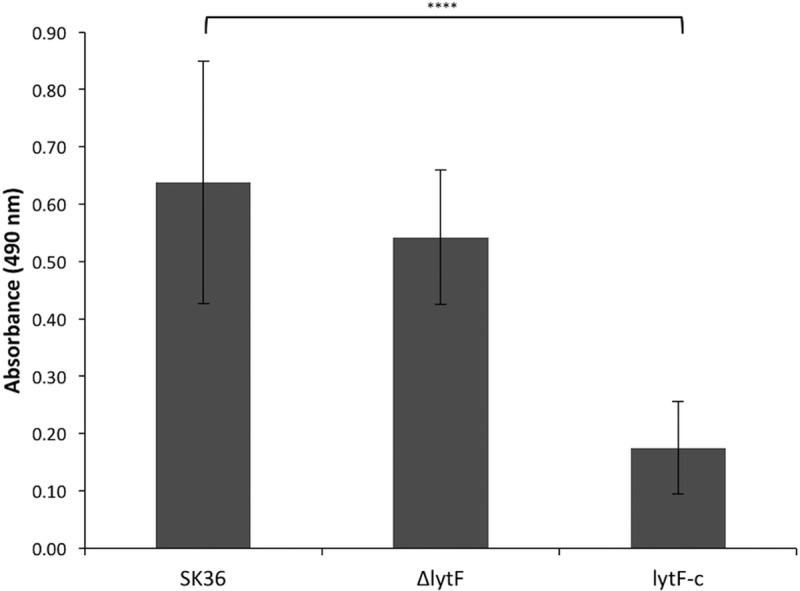
Inhibition of proper biofilm formation by excess LytF. Biofilms of S. sanguinis SK36, ΔlytF, and lytF-c strains were grown in CDM for 18 h, washed, and stained with 0.01% safranin. Biofilm formation was determined by measuring the absorbance of air-dried biofilms at 490 nm. The lytF-c biofilms showed a significant decrease in absorbance after safranin staining, indicating that biofilm formation was inhibited in this strain (****, P < 0.0001). Data represent averages and standard deviations from quadruplicate samples in three separate plate experiments.
LytF is a murein hydrolase.
To further examine the activities of LytF from S. sanguinis, we adapted a zymogram protocol. Zymogram assays were performed with whole-protein extracts of the wild-type, ΔlytF, and lytF-c strains. A clearance band at 70 kDa, the approximate size of LytF, was visible with the protein extracts of the wild-type and lytF-c strains, but no clearance was visible with proteins from the ΔlytF strain (Fig. 8). Interestingly, the lytF-c samples showed much greater clearance of the substrate than did the wild-type whole-protein samples, which correlates with the overexpression of lytF in this strain.
FIG 8.
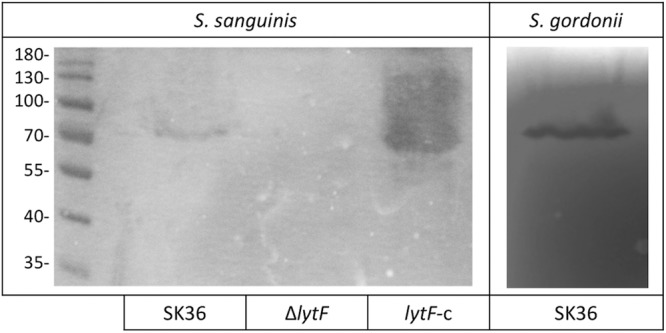
S. sanguinis LytF effects as a murein hydrolase. Zymograms with either S. sanguinis or S. gordonii substrate cells show clearance bands where hydrolytic proteins contained in whole-cell suspensions of the indicated strains are active. Zymograms were photographed on a black background and are representative of at least three separate experiments performed on different days. Entire pictures were processed to increase the visibility of the bands.
Since LytF of S. sanguinis and LytF of S. gordonii are orthologs, we determined, through similar zymogram assays, whether LytF of S. sanguinis could also digest S. gordonii substrate cells. We observed hydrolytic clearance in the S. gordonii substrate zymogram similar to that observed with S. sanguinis substrate cells; thus, S. sanguinis LytF is not strictly species specific (Fig. 8). Additionally, LytF of S. sanguinis could digest S. mutans substrate cells, albeit with lower efficiency (see Fig. S1 in the supplemental material).
LytF is secreted and retains activity in the environment.
Although we have confirmed LytF to be a murein hydrolase, the overall function or purpose of the protein is still unknown. Murein hydrolases, acting as autolysins, can be involved in cell division, programmed cell death, and fratricide, depending on environmental stimuli (20 – 22). To further characterize LytF, we attempted to overexpress and to purify LytF from Escherichia coli, but we were unsuccessful. Instead, since LytF is predicted to be a secreted protein, we precipitated proteins from the supernatants of actively growing cultures by using ammonium sulfate, in an attempt to collect LytF. In SDS-PAGE analysis, a band of approximately 70 kDa was present from the wild-type precipitation and a stronger band of the same size was visible from the lytF-c precipitation. The lytF mutant lacked a band of the same size (Fig. 9A). These proteins were excised from the polyacrylamide gel and submitted to the Oregon Health and Science University Proteomics Shared Resource for identification via mass spectrometry. This precipitated protein was confirmed to be LytF of S. sanguinis. Precipitated proteins were used in zymogram assays and remained active (data not shown). To confirm that any hydrolytic activity was due to LytF and not another protein present in the precipitated sample, proteins precipitated from the lytF-c strain were used for additional purification by fast protein liquid chromatography (FPLC). Protein samples were first dialyzed against 2-(N-morpholino)ethanesulfonic acid (MES) buffer and then purified over a cationic Sepharose column. After SDS-PAGE analysis of the fractions (Fig. S2), the purified protein was determined by zymogram assays to be still active (Fig. 9B).
LytF is able to cause eDNA release.
To determine whether LytF could induce the release of eDNA from S. sanguinis, SK36 cultures were treated with 100 μg ml−1 precipitated proteins from the three strains, and samples were taken over time. At 15 min posttreatment, significantly higher relative concentrations of eDNA could be detected from cultures treated with SK36 and lytF-c precipitated proteins than from ΔlytF protein-treated cultures (Fig. 10A). Since LytF of S. sanguinis could digest S. gordonii and S. mutans substrate cells in zymograms, we sought to determine whether the purified LytF could also induce the release of DNA from other oral streptococci. As shown in Fig. 10B, treating S. sanguinis SK36 and S. gordonii DL1 cultures with purified LytF (12.1 μg ml−1) induced larger amounts of eDNA release from both species, compared with the no-treatment samples. This release was significant for S. sanguinis (P = 0.0019). The same trend was evident in analyses of eDNA release from S. mutans and Streptococcus oralis after pure LytF treatment (Fig. S3).
FIG 10.

LytF induction of eDNA release. (A) Top, agarose gel showing precipitated eDNA samples. Bottom, relative amounts of eDNA released from S. sanguinis SK36 cultures after treatment for 15 min with precipitated proteins from the indicated samples. Compared to no treatment, cells treated with SK36 and lytF-c proteins released significantly more eDNA (*, P = 0.0211; **, P = 0.0060, respectively). Data represent the means and standard deviations of three independent experiments. (B) Relative amounts of eDNA released at 15 min from S. sanguinis (SS) or S. gordonii (SG) cultures, with or without pure LytF treatment (12.1 μg ml−1). LytF treatment significantly increased the amount of eDNA released from S. sanguinis cultures (**, P = 0.0019). n.s., not significant. Data represent the means and standard deviations of four independent experiments.
LytF causes morphological abnormalities.
As shown in Fig. 3, there were differences in cell division between the wild-type and lytF mutant strains in both planktonic and biofilm cultures. To further examine this phenotype, S. sanguinis SK36 biofilms were grown in the presence of precipitated proteins from the SK36, ΔlytF, and lytF-c strains. Cells within the biofilms treated with precipitated LytF (SK36 and lytF-c protein treatments) appeared misshapen, compared to biofilms grown without additional LytF (Fig. 11A). Furthermore, SK36 biofilms were grown in the presence of purified LytF, which yielded similar misshapen cells in addition to what appeared to be membrane blebs (Fig. 11B). To determine whether cells treated with protein samples containing excess LytF survived to the same extent as those without excess LytF, planktonic S. sanguinis SK36 cultures were grown in the presence of the various precipitated protein samples. A similar trend was apparent, with cells grown in the presence of excess LytF exhibiting lower CFU values than wild-type cells grown without any treatment (Fig. 11C). SEM was also used to examine S. gordonii DL1 biofilms grown in the presence of pure LytF. S. gordonii cells also displayed abnormalities, as they were more rounded at the center but pointed at the poles (Fig. S4).
FIG 11.

Excess LytF induction of morphological abnormalities. (A) S. sanguinis SK36 biofilms treated with precipitated proteins from the SK36 wild-type, lytF mutant, or lytF-complemented strains. Biofilms grown in the presence of LytF-containing protein samples had enlarged and irregular cells present within chains in the biofilms. Images were adjusted for brightness and contrast. Images are representative of at least three fields of view from two experiments performed on different days. Scale bars, 4 μm. (B) S. sanguinis SK36 biofilm grown in the presence of 12.1 μg ml−1 LytF. Cells within the biofilm again appeared enlarged and also displayed membrane blebs (white arrowheads). The dashed white box represents the area of the enlarged image to the right. Scale bar, 4 μm. The image is representative of multiple fields of view from one experiment. (C) Survival assay showing CFU of S. sanguinis SK36 grown in the presence of precipitated proteins from the indicated strains. Cultures were sonicated, serially diluted, and plated on BHI agar. Data represent the means and standard deviations of two individual experiments performed on different days.
DISCUSSION
This work provides documentation of eDNA release from S. sanguinis as a result of LytF activity. LytF is indeed a murein hydrolase, as evidenced in zymogram analysis, and not only is capable of digesting peptidoglycan corresponding to its own species but also can digest the peptidoglycans of S. gordonii and S. mutans. Although the target range of murein hydrolases is limited, we have confirmed, as have Berg et al. (27), that LytF is capable of digesting the peptidoglycans of multiple streptococcal species. This is particularly interesting because we have now shown that LytF can hydrolyze the peptidoglycan of the oral pathogen S. mutans. While Bsp domains are thought to provide binding specificity for the accompanying hydrolytic protein activity, it is likely that the close relationship between the streptococci examined allows such interspecies activity of LytF.
As discussed previously, the presence of DNA within the biofilm exopolysaccharide matrix is essential for maintaining stability (13 – 15). We originally hypothesized that LytF, being competence associated and a murein hydrolase, is a likely candidate to induce the release of eDNA from S. sanguinis. Although we were unable to overexpress and to purify LytF from E. coli, the protein extracts obtained from precipitated S. sanguinis supernatants proved to be relatively clean and were confirmed to contain active LytF (Fig. 9). Therefore, samples of LytF could be used as treatments against bacteria. Exogenously added LytF, in the case of precipitated protein samples from the wild-type SK36 and lytF-c strains, could significantly induce the release of eDNA from S. sanguinis (Fig. 10A). To confirm that these results were due to the activity of LytF and not an unidentified protein present in the precipitated samples, we also purified LytF using FPLC. Purified LytF could indeed induce the release of eDNA from S. sanguinis, as well as from S. gordonii (Fig. 10B). Furthermore, purified LytF induces the release of eDNA from S. oralis and S. mutans, indicating that the activity of LytF can affect bacteria present throughout the biofilm, likely contributing to overall biofilm stability (see Fig. S3 in the supplemental material).
Under aerobic conditions, S. sanguinis produces H2O2, which can induce the release of eDNA (17, 18). As S. sanguinis begins to colonize the tooth during early biofilm formation, oxygen tension in saliva is high enough to stimulate H2O2 production and thus eDNA release (26). As the oral biofilm grows, it becomes increasingly anaerobic, particularly for early colonizers such as S. sanguinis, which are present deeper within the community. Since H2O2 would no longer be efficiently produced by S. sanguinis, we predict that the competence stress pathway would become more active, since diffusion limitation would accumulate CSP, thus increasing the amount of LytF to compensate for the loss of H2O2-induced eDNA release. Throughout our experiments, we effectively controlled for the production of H2O2 by growing all bacteria statically. Without aeration, cells lacked the proper conditions to produce H2O2; therefore, any eDNA release cannot be attributed to induction by H2O2.
Upon treatment of S. sanguinis biofilms with either precipitated proteins or purified LytF, an excess of LytF appeared to dramatically influence cell morphology (Fig. 11). A similar phenotype was also observed with S. gordonii biofilms (data not shown and Fig. S4). Furthermore, when cell morphology in planktonic cultures was examined with the vancomycin-conjugated BODIPY-FL stain, lytF-complemented cells were engorged. This is similar to the phenotype observed for lytF-complemented biofilms Since lytF is actually overexpressed in this strain, and considering the induction of abnormal cell structure in the presence of excess LytF, we conclude that a specific range of LytF concentrations is essential for proper cell division and biofilm formation. It is thus likely that the decrease in biofilm mass of the lytF-c strain evidenced in Fig. 7 is associated with the abnormal morphology of the cells.
Straume et al. recently discussed a similar phenomenon in S. pneumoniae; with overexpression of the competence-associated protein ComM, which antagonizes the activity of the LytF ortholog CbpD, growth was inhibited and cells experienced morphological abnormalities (33). This abnormal regulation of competence controlling a murein hydrolase in S. pneumoniae leads us to think that changes in regulation patterns would also affect streptococci within the oral cavity. S. sanguinis, S. gordonii, S. mutans, and even Streptococcus parasanguinis all possess a LytF homolog associated with competence (21, 27). Furthermore, S. oralis encodes a CbpD homolog. All of these bacteria are oral commensal organisms, and the fact that each has a competence-controlled murein hydrolase indicates that LytF and CbpD play an important biological role in oral biofilm formation. Under oral biofilm-forming conditions, LytF appears to contribute to overall maintenance and structure. When competence activation is altered due to environmental stimuli, the amount of LytF secreted from cells would change, possibly changing population dynamics within the biofilm, since numerous oral bacteria contain similarly controlled proteins. Since LytF can act on multiple oral bacteria, due to the structural similarity of the peptidoglycans, it is likely that eDNA is released from a variety of oral streptococci due to the promiscuous activity of LytF. In addition, oral streptococci share many regions of high chromosomal homology, which would make this released eDNA suitable for horizontal gene transfer, in addition to its structural function in biofilms. The exchange of genes from eDNA release after H2O2 induction was shown previously by Itzek et al. (18). In an increasingly anaerobic biofilm lacking H2O2 stimulation, presumably LytF activity would mediate comparable genetic exchange.
The hydrolytic activity of LytF, its association with competence, and its ability to induce the release of eDNA might lead one to describe LytF as a fratricin, i.e., a hydrolytic enzyme responsible for the lysis of genetically identical cells (22). Although we did not examine the metabolic activity of cells from our mutants or cells after LytF treatment, from the SEM images and survival data it is apparent that not all cells are morphologically affected and not all treated cells die. It is known that, in S. pneumoniae, the activity of the murein hydrolase CbpD is blocked by an immunity protein termed ComM (33, 34). We were unable to identify a ComM homolog through a search of the competence regulon in S. sanguinis,. Therefore, we do not consider LytF of S. sanguinis to function as a CbpD-like fratricin, as proposed previously for LytF of S. gordonii (27). Since competence is not expressed in all cells within a biofilm at once, LytF must be subject to alternative regulation that would control its activities during competence and biofilm development. Although it apparently lacks an immunity protein and we predict that LytF does not fully lyse its targets, our hypothesis remains that the basic function of LytF is as a fratricin, acting on genetically similar cells and in turn releasing eDNA.
In all, we have determined that LytF of S. sanguinis is capable of inducing eDNA release, potentially contributing to horizontal gene transfer, and is involved in biofilm formation. Based on its muralytic activities and its association with competence, LytF is also involved in transformation. We conclude that LytF is involved in the release of eDNA; however, improper regulation of the competence pathway, and therefore lytF expression, leads to inappropriate cell division, uncontrolled eDNA release, and effects on overall biofilm formation with S. sanguinis.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Streptococcus sanguinis SK36 and its derivatives, Streptococcus gordonii DL1, Streptococcus mutans UA159, and Streptococcus oralis J22 (Table 1) were routinely grown in Bacto brain heart infusion (BHI) medium or Todd-Hewitt (TH) medium, where indicated, at 37°C with 5% CO2. For antibiotic selection, cultures were supplemented with the following antibiotics: erythromycin at 3 μg ml−1, spectinomycin at 500 μg ml−1, and kanamycin at 300 μg ml−1. For expression experiments, strains were grown to mid-log phase (OD600 of 0.4) in TH medium and then synthetic CSP was added to a final concentration of 0.5 μg ml−1. Cultures were incubated for an additional 2 h prior to RNA extraction. CDM supplemented with 50 mM sucrose was used for biofilm growth (35, 36). Exact components and concentrations can be found elsewhere. Biofilms were grown in CDM from a 1:60 dilution of overnight cultures.
TABLE 1.
Bacterial strains used
| Strain | Relevant characteristicsa | Source |
|---|---|---|
| Streptococcus sanguinis SK36 | 42 | |
| Streptococcus gordonii DL1 | 43 | |
| Streptococcus mutans UA159 | 44 | |
| Streptococcus oralis J22 | 45 | |
| S. sanguinis ΔlytF | ssa_0036 deleted by replacement with ermAM; Ermr | This study |
| S. sanguinis lytF-c | ΔlytF with pDL278/ssa_0036, transformed via concatenation; Ermr, Spcr | This study |
Erm, erythromycin; Spc, spectinomycin.
Construction of mutant strains.
All oligonucleotides were designed based on sequence data obtained from the NCBI and were synthesized by IDT DNA (Table 2). PCR was performed using Accuprime Pfx Supermix (Invitrogen), unless otherwise stated. For the lytF mutant, regions of approximately 500 bp directly upstream and downstream of lytF were amplified. Primers for these regions were designed with overlapping regions corresponding to an erythromycin resistance cassette. The antibiotic resistance cassette, ermAM, was also amplified. Using overlap extension PCR, the three PCR fragments were combined 1:1:1 for ligation of the construct. The resulting PCR product was transformed into wild-type S. sanguinis SK36 cells. Transformants were selected on BHI agar containing erythromycin and were confirmed, via PCR, for the lack of lytF and the presence of ermAM. To create the lytF-complemented strain, the entirety of lytF and its native promoter were amplified using Phusion polymerase (New England BioLabs). Simultaneously, the linearized shuttle vector pDL278 was amplified. The amplified lytF fragment and vector were combined in a 2:1 ratio and were PCR amplified using an extended Phusion polymerase reaction for concatenation, as described previously (37). Successful concatenation appears as high-molecular-weight smears on agarose gels. This product was transformed directly into the S. sanguinis lytF mutant strain to complement lytF. Transformants were confirmed via PCR. All constructs were additionally confirmed via genetic sequencing. Flanking genes and intergenic regions were unaltered, and the complemented sequence was identical to the wild-type sequence.
TABLE 2.
Primers used
| Primer name | Oligonucleotide sequence (5′ to 3′)a | Purposeb | Reference |
|---|---|---|---|
| Strep. 16S rRNA-RT-F | AAGCAACGCGAAGAACCTTA | qRT-PCR | 4 |
| Strep. 16S rRNA-RT-R | GTCTCGCTAGAGTGCCCAAC | qRT-PCR | 4 |
| Ss lytF F | CTCCTGCGTCATACCACTGA | qRT-PCR | |
| Ss lytF R | CAGGAGGTACAGGACGCATT | qRT-PCR | |
| Ss comC RTF | TGAAAATCTATTCTTTTCAAATTGC | qRT-PCR | |
| Ss comC RTR | CCATGGATTTGGAACACCTC | qRT-PCR | |
| Ss comD RTF | GGAGATTCAGCTTTAAGGAGTGTC | qRT-PCR | 31 |
| Ss comD RTR | ACAACTTGATTGGAAGGCGTTC | qRT-PCR | 31 |
| Ss comE RTF | TTGGAATTGACATCCAGGTTC | qRT-PCR | |
| Ss comE RTR | TGTTTCCTCCCCCTTAATGTC | qRT-PCR | |
| Ss comX RTF | CAAGAAAGCCAAAAGCGAAA | qRT-PCR | |
| Ss comX RTR | TCGCTTCTCTGAAGGCAACT | qRT-PCR | |
| Ss comYA RTF | TGATTAGGCAGGCTCGGCAAGAAG | qRT-PCR | 31 |
| Ss comYA RTR | AAGCGGCGTTCATCACCAATTCTC | qRT-PCR | 31 |
| Ss purD RTF | GCGTGGCTTCTGTCAAGATT | qRT-PCR | |
| Ss purD RTR | CCAGCTTCTGCGATTTCTTC | qRT-PCR | |
| Ss purH RTF | GAGCTGGAGTGGTCCAAAGA | qRT-PCR | |
| Ss purH RTR | TCCCTGCTTTTCGATGTAGG | qRT-PCR | |
| Ss lytF US-F | TGGAATGTTCGGAAATTCCA | lytF KO | |
| Ss lytF US-R | ATCAAACAAATTTTGGGCCCGGGATATTTCTTCATAAAACTC | lytF KO | |
| Ss lytF DS-F | ATTCTATGAGTCGCTGCCGACTCCTAAATAAGTCGCTTTCGC | lytF KO | |
| Ss lytF DS-R | CGAGTGGCTTTGGAATTTGC | lytF KO | |
| Erm-L | CCGGGCCCAAAATTTGTTTGAT | Erythromycin cassette | |
| Erm-R | AGTCGGCAGCGACTCATAGAAT | Erythromycin cassette | |
| SS lytF-c_L | AGTGAATTCGAGCTCGGTACCCGGACCAAGCTAACACATCACGTA | lytF complementation | |
| SS lytF-c_R | AACAGCTATGACCATGATTACGCCGAGCAGTCTGCTCAAAGTAG | lytF complementation | |
| pDL278_1L | TCCCCGGGTACCGAGCTCGAATTCACT | Concatenation | |
| pDL278_1R | CTTGGCGTAATCATGGTCATAGCTGTT | Concatenation | |
| 5′ SS lytF gs-F | ATTATCCGGAGCTGTCCTAGGT | 5′ RACE | |
| 5′ SS lytF gs inner-R | AAGCTTGATAAGGTTCTTGAGCTTGAGAACG | 5′ RACE | |
| 5′ SS lytF gs outer-R | AGCGCTCTCCTATCCGTATGTAAG | 5′ RACE |
Oligonucleotides were created for this study unless otherwise specified.
KO, knockout.
RNA isolation and qRT-PCR.
RNA was extracted using a phenol-chloroform method published previously (38, 39). Briefly, cells were harvested via centrifugation at 4,000 rpm for 15 min at 4°C. Pellets were resuspended in TRI reagent (Sigma) and stored at −80°C. Bacteria were lysed in a Precellys Evolution homogenizer in the presence of 0.1 mm zirconia beads (BioSpec Products). Chloroform was added, and the samples were centrifuged at 13,000 rpm for 10 min at 4°C. RNA was precipitated from the aqueous layer by the addition of isopropanol and washing with ice-cold 75% ethanol. RNA was resuspended in nuclease-free, molecular-grade water (Corning). RNA was treated with Turbo DNase (Ambion) and cleaned with the Qiagen RNeasy kit. cDNA was synthesized using the qScript cDNA synthesis kit (Quanta). Quantitative real-time PCR (qRT-PCR) was conducted using the PerfeCTa SYBR green supermix for iQ (Quanta), in a Bio-Rad CFX Connect real-time PCR system; 16S rRNA was used as the reference gene. The primers used can be found in Table 2.
5′-RACE PCR.
RACE PCR was conducted using the FirstChoice RLM-RACE kit (Ambion), according to the manufacturer's instructions. Products were sequenced by EuroFins Genomics.
Transformation efficiencies.
Transformation efficiencies were calculated as described previously, with slight modifications (31). Briefly, overnight cultures were inoculated 1:40 in fresh TH medium supplemented with 2.5% heat-inactivated horse serum (Sigma-Aldrich) and were grown to an OD600 of 0.08. Chromosomal DNA containing a kanamycin resistance cassette (aphAIII) and 0.5 μg ml−1 CSP were added to 1-ml samples, and cells were further incubated for 2 h. Cells were plated on selective and nonselective TH agar plates. Transformation efficiency was calculated as the number of kanamycin-resistant CFU relative to the CFU on nonselective agar.
Fluorescent staining and microscopy.
From overnight cultures, cells were inoculated 1:40 in fresh BHI medium and grown to mid-log phase (OD600 of 0.4). For cell wall visualization, cells were pelleted by centrifugation and resuspended in 1× phosphate-buffered saline (PBS) (pH 7.4), 1 μg ml−1 vancomycin-BODIPY-FL conjugate (Molecular Probes) was added, and the cells were incubated at room temperature for 30 min, with rocking and protection from light. Cells were imaged using a 100× oil immersion lens with an Olympus IX73 microscope, Olympus digital camera, and cellSens standard software.
Scanning electron microscopy.
For SEM, 1 ml of bacterial culture in CDM was added to sterile, cell-culture-treated, 13-mm Thermanox discs (ThermoFisher) housed in a 24-well plate. Where indicated, strain-specific precipitated proteins were added to a final concentration of 100 μg ml−1 or purified protein to 12.1 μg ml−1. After incubation at 37°C in 5% CO2 for 18 h, unattached cells and medium were removed and then biofilms were washed twice with Sorensen's buffer (pH 7.2). Biofilms were fixed for 24 h at 4°C with 2% (vol/vol) glutaraldehyde in Sorensen's buffer. Biofilms were washed once with 0.1 M sodium acetate (pH 7.2). The Oregon Health and Science University Multiscale Microscopy Core dehydrated biofilms with ethanol gradients. Samples were further processed using a critical point dryer (Leica CPD300) prior to sputter coating with 10-nm-thick carbon (ACE600 coater). Imaging was completed using a Helios Nanolab 660 dual-beam scanning electron microscope (FEI).
Biofilm quantification.
Eighteen-hour biofilms in a 96-well plate were washed once with 1× PBS (pH 7.4) and then stained for 15 min with 0.1% safranin. Biofilms were washed and then air dried. Biofilm formation was quantified by measuring the absorbance at 490 nm in a GloMax Discover microplate reader.
Zymograms.
Zymogram assays were used to determine the muralytic activity of potential murein hydrolases in S. sanguinis. The zymogram protocol was adapted from the methods described by Berg et al. and Leclerc and Asselin (27, 40). Whole-cell extracts were prepared from 50-ml mid-log-phase cultures (OD600 of 0.4) grown in TH medium (pH 6.8) with 0.5 μg ml−1 CSP. Cells were harvested by centrifugation at 4,000 rpm for 15 min at 4°C, and the pellet was resuspended in 50 μl of 6× Laemmli sample buffer plus 50 μl of distilled water. Whole-cell extracts were heated at 95°C for 10 min. Substrate cells for polyacrylamide gel incorporation were prepared from wild-type strains. Cultures (100 ml) were grown to an OD600 of 0.4, and then cells were harvested as described above. The pellets were washed in 1.5 ml of ice-cold 20 mM Tris-HCl (pH 7.4) with 100 mM NaCl. Cells were centrifuged and then resuspended in 500 μl of 1.5 M Tris-HCl (pH 8.8). Substrate cells were heat inactivated at 95°C for 10 min.
Substrate cells were incorporated into a 10% resolving gel prior to polymerization. Whole-cell extracts or protein samples were separated by SDS-PAGE, using the substrate-cell-incorporated gel with a 4% stacking layer, at a constant current of 35 mA. The polyacrylamide gel was washed briefly in distilled water prior to incubation in refolding buffer (50 mM NaCl, 20 mM MgCl2, 0.5% Triton X-100, 20 mM Tris-HCl [pH 7.4]) overnight at 37°C, with gentle rocking. Hydrolytic activity was observed as clearances in the opaque gel, and zymograms were photographed on a black background with a Nikon D80 camera.
Precipitation and purification of LytF.
Since LytF is predicted to be a secreted protein, supernatants from actively growing cultures were used for protein precipitation. A protein precipitation protocol was adapted from the method described by Duong-Ly and Gabelli (41). S. sanguinis SK36 wild-type, lytF mutant, and lytF-complemented overnight cultures were inoculated 1:40 in 50 ml fresh BHI medium and incubated to mid-log phase (OD600 of 0.4). CSP was added to a final concentration of 0.5 μg ml−1, and cultures were further incubated for 30 min. Cultures were centrifuged at 4,000 rpm for 15 min 4°C to remove bacteria, and the supernatants were collected for protein precipitation. Ammonium sulfate (EMD Millipore) was added to the supernatants to 30% saturation, and the solution was constantly stirred for 30 min at 4°C. To collect precipitated proteins, supernatants were centrifuged at 10,000 rpm for 30 min at 4°C. Supernatants were collected, and the resulting protein pellets were resuspended in 1 ml of 1× PBS (pH 7.4). The collected supernatants were used for further precipitation by adding ammonium sulfate to 60% saturation and treating the solution as described above. Precipitated proteins were evaluated via SDS-PAGE and stored at −20°C for further usage.
To further purify LytF, precipitated proteins from the lytF-complemented strain were dialyzed against an MES buffer (20 mM MES [pH 5.35], 1 mM dithiothreitol [DTT]) overnight at 4°C. FPLC was conducted using an SP Sepharose cation-exchange column with a 0 to 400 mM NaCl gradient in MES buffer. Fractions were collected and analyzed using SDS-PAGE. Protein activity was examined using zymograms.
Mass spectrometry.
The Oregon Health and Science University Proteomics Shared Resource analyzed excised SDS-PAGE gel fragments for protein identification. Proteins were identified using a database of Swiss-Prot/TrEMBL entries for S. sanguinis SK36, with an appendix to exclude known contaminants.
eDNA quantification.
One milliliter of precipitated or purified LytF protein in buffer was used as a treatment (at final concentrations of 100 μg ml−1 and 12.1 μg ml−1, respectively). Specifically, overnight cultures were inoculated into fresh BHI medium and grown to an OD600 of 1.0. Bacteria were centrifuged and resuspended in fresh BHI medium with or without protein treatments. One-milliliter samples were collected at the indicated times, and eDNA was precipitated as discussed by Kreth et al. (17), with an added filtration step for the bacterial supernatant, using 0.2-μm syringe filters (VWR), prior to eDNA precipitation. qRT-PCR was performed using 1 μl of precipitated eDNA, with 1 cycle of 95°C for 3 min followed by 40 cycles of 95°C for 15 s and 55°C for 1 min. The quantity of eDNA was calculated relative to the cycle threshold values of chromosomal DNA dilutions using universal 16S rRNA primers. eDNA samples (1 μl) were examined via electrophoresis on 1% agarose gels.
Survival assay.
S. sanguinis SK36 was inoculated into fresh BHI medium and grown with or without 100 μg ml−1 precipitated protein samples. After an 18-hour incubation, cell chains and clumps were dispersed via sonication (successful cell separation was monitored microscopically), and cells were serially diluted and plated on BHI agar. CFU per milliliter were calculated.
Statistical analysis.
Statistical analysis of data was performed using the QuickCalcs online calculator (http://www.graphpad.com/quickcalcs/index.cfm); t tests were performed to compare the means of two groups, and data were considered significantly different if the two-tailed P value was ≤0.05.
Supplementary Material
ACKNOWLEDGMENTS
Electron microscopy was performed at the Multiscale Microscopy Core, with technical support from the Oregon Health and Science University-FEI Living Lab and the Oregon Health and Science University Center for Spatial Systems Biomedicine.
This work was supported by NIH NIDCR grant DE021726 to J.K. and NIH NIDCR grants DE018893 and DE022083 to J.M.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01709-17.
REFERENCES
- 1.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J Bacteriol 192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jakubovics NS, Yassin SA, Rickard AH. 2014. Community interactions of oral streptococci. Adv Appl Microbiol 87:43–110. doi: 10.1016/B978-0-12-800261-2.00002-5. [DOI] [PubMed] [Google Scholar]
- 3.Sbordone L, Bortolaia C. 2003. Oral microbial biofilms and plaque-related diseases: microbial communities and their role in the shift from oral health to disease. Clin Oral Invest 7:181–188. doi: 10.1007/s00784-003-0236-1. [DOI] [PubMed] [Google Scholar]
- 4.Kreth J, Zhang Y, Herzberg MC. 2008. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J Bacteriol 190:4632–4640. doi: 10.1128/JB.00276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrero ER, Slomka V, Bernaerts K, Boon N, Hernandez-Sanabria E, Passoni BB, Quirynen M, Teughels W. 2016. Antimicrobial effects of commensal oral species are regulated by environmental factors. J Dent 47:23–33. doi: 10.1016/j.jdent.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Claverys JP, Prudhomme M, Martin B. 2006. Induction of competence regulons as a general response to stress in Gram-positive bacteria. Annu Rev Microbiol 60:451–475. doi: 10.1146/annurev.micro.60.080805.142139. [DOI] [PubMed] [Google Scholar]
- 7.Johnsborg O, Havarstein LS. 2009. Regulation of natural genetic transformation and acquisition of transforming DNA in Streptococcus pneumoniae. FEMS Microbiol Rev 33:627–642. doi: 10.1111/j.1574-6976.2009.00167.x. [DOI] [PubMed] [Google Scholar]
- 8.Gaustad P. 1979. Genetic transformation in Streptococcus sanguis: distribution of competence and competence factors in a collection of strains. Acta Pathol Microbiol Scand B 87B:123–128. [PubMed] [Google Scholar]
- 9.Havarstein LS, Hakenbeck R, Gaustad P. 1997. Natural competence in the genus Streptococcus: evidence that streptococci can change pherotype by interspecies recombinatorial exchanges. J Bacteriol 179:6589–6594. doi: 10.1128/jb.179.21.6589-6594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Havarstein LS, Gaustad P, Nes IF, Morrison DA. 1996. Identification of the streptococcal competence-pheromone receptor. Mol Microbiol 21:863–869. doi: 10.1046/j.1365-2958.1996.521416.x. [DOI] [PubMed] [Google Scholar]
- 11.Pestova EV, Havarstein LS, Morrison DA. 1996. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol Microbiol 21:853–862. doi: 10.1046/j.1365-2958.1996.501417.x. [DOI] [PubMed] [Google Scholar]
- 12.Lee MS, Morrison DA. 1999. Identification of a new regulator in Streptococcus pneumoniae linking quorum sensing to competence for genetic transformation. J Bacteriol 181:5004–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das T, Sehar S, Manefield M. 2013. The roles of extracellular DNA in the structural integrity of extracellular polymeric substance and bacterial biofilm development. Environ Microbiol Rep 5:778–786. doi: 10.1111/1758-2229.12085. [DOI] [PubMed] [Google Scholar]
- 14.Dominiak DM, Nielsen JL, Nielsen PH. 2011. Extracellular DNA is abundant and important for microcolony strength in mixed microbial biofilms. Environ Microbiol 13:710–721. doi: 10.1111/j.1462-2920.2010.02375.x. [DOI] [PubMed] [Google Scholar]
- 15.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 16.Boles BR, Horswill AR. 2011. Staphylococcal biofilm disassembly. Trends Microbiol 19:449–455. doi: 10.1016/j.tim.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreth J, Vu H, Zhang Y, Herzberg MC. 2009. Characterization of hydrogen peroxide-induced DNA release by Streptococcus sanguinis and Streptococcus gordonii. J Bacteriol 191:6281–6291. doi: 10.1128/JB.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itzek A, Zheng L, Chen Z, Merritt J, Kreth J. 2011. Hydrogen peroxide-dependent DNA release and transfer of antibiotic resistance genes in Streptococcus gordonii. J Bacteriol 193:6912–6922. doi: 10.1128/JB.05791-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnsborg O, Eldholm V, Havarstein LS. 2007. Natural genetic transformation: prevalence, mechanisms and function. Res Microbiol 158:767–778. doi: 10.1016/j.resmic.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Berg KH, Biornstad TJ, Johnsborg O, Havarstein LS. 2012. Properties and biological role of streptococcal fratricins. Appl Environ Microbiol 78:3515–3522. doi: 10.1128/AEM.00098-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cullin N, Merritt J, Kreth J. 2017. Beyond cell division: the ecological roles of autolysins in oral biofilm communities. Curr Oral Health Rep 4:14–21. doi: 10.1007/s40496-017-0118-2. [DOI] [Google Scholar]
- 22.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 23.Moscoso M, Claverys JP. 2004. Release of DNA into the medium by competent Streptococcus pneumoniae: kinetics, mechanism and stability of the liberated DNA. Mol Microbiol 54:783–794. doi: 10.1111/j.1365-2958.2004.04305.x. [DOI] [PubMed] [Google Scholar]
- 24.Eldholm V, Johnsborg O, Haugen K, Ohnstad HS, Havarstein LS. 2009. Fratricide in Streptococcus pneumoniae: contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology 155:2223–2234. doi: 10.1099/mic.0.026328-0. [DOI] [PubMed] [Google Scholar]
- 25.Wei H, Havarstein LS. 2012. Fratricide is essential for efficient gene transfer between pneumococci in biofilms. Appl Environ Microbiol 78:5897–5905. doi: 10.1128/AEM.01343-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marquis RE. 1995. Oxygen metabolism, oxidative stress and acid-base physiology of dental plaque biofilms. J Ind Microbiol 15:198–207. doi: 10.1007/BF01569826. [DOI] [PubMed] [Google Scholar]
- 27.Berg KH, Ohnstad HS, Havarstein LS. 2012. LytF, a novel competence-regulated murein hydrolase in the genus Streptococcus. J Bacteriol 194:627–635. doi: 10.1128/JB.06273-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Layec S, Decaris B, Leblond-Bourget N. 2008. Diversity of Firmicutes peptidoglycan hydrolases and specificities of those involved in daughter cell separation. Res Microbiol 159:507–515. doi: 10.1016/j.resmic.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez AM, Callahan JE, Fawcett P, Ge X, Xu P, Kitten T. 2011. Physiological and molecular characterization of genetic competence in Streptococcus sanguinis. Mol Oral Microbiol 26:99–116. doi: 10.1111/j.2041-1014.2011.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vickerman MM, Iobst S, Jesionowski AM, Gill SR. 2007. Genome-wide transcriptional changes in Streptococcus gordonii in response to competence signaling peptide. J Bacteriol 189:7799–7807. doi: 10.1128/JB.01023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu L, Zhang Y, Fan J, Herzberg MC, Kreth J. 2011. Characterization of competence and biofilm development of a Streptococcus sanguinis endocarditis isolate. Mol Oral Microbiol 26:117–126. doi: 10.1111/j.2041-1014.2010.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeDent AC, McAdow M, Schneewind O. 2007. Distribution of protein A on the surface of Staphylococcus aureus. J Bacteriol 189:4473–4484. doi: 10.1128/JB.00227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Straume D, Stamsas GA, Salehian Z, Havarstein LS. 2017. Overexpression of the fratricide immunity protein ComM leads to growth inhibition and morphological abnormalities in Streptococcus pneumoniae. Microbiology 163:9–21. doi: 10.1099/mic.0.000402. [DOI] [PubMed] [Google Scholar]
- 34.Havarstein LS, Martin B, Johnsborg O, Granadel C, Claverys JP. 2006. New insights into the pneumococcal fratricide: relationship to clumping and identification of a novel immunity factor. Mol Microbiol 59:1297–1307. doi: 10.1111/j.1365-2958.2005.05021.x. [DOI] [PubMed] [Google Scholar]
- 35.Standar K, Kreikemeyer B, Redanz S, Munter WL, Laue M, Podbielski A. 2010. Setup of an in vitro test system for basic studies on biofilm behavior of mixed-species cultures with dental and periodontal pathogens. PLoS One 5:e13135. doi: 10.1371/journal.pone.0013135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Rijn I, Kessler RE. 1980. Growth characteristics of group A streptococci in a new chemically defined medium. Infect Immun 27:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Z, Qi F, Merritt J. 2013. Cloning-independent plasmid construction for genetic studies in streptococci. J Microbiol Methods 94:77–82. doi: 10.1016/j.mimet.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z, Itzek A, Malke H, Ferretti JJ, Kreth J. 2012. Dynamics of speB mRNA transcripts in Streptococcus pyogenes. J Bacteriol 194:1417–1426. doi: 10.1128/JB.06612-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Itzek A, Malke H, Ferretti JJ, Kreth J. 2013. Multiple roles of RNase Y in Streptococcus pyogenes mRNA processing and degradation. J Bacteriol 195:2585–2594. doi: 10.1128/JB.00097-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leclerc D, Asselin A. 1989. Detection of bacterial cell wall hydrolases after denaturing polyacrylamide gel electrophoresis. Can J Microbiol 35:749–753. doi: 10.1139/m89-125. [DOI] [PubMed] [Google Scholar]
- 41.Duong-Ly KC, Gabelli SB. 2014. Salting out of proteins using ammonium sulfate precipitation. Methods Enzymol 541:85–94. doi: 10.1016/B978-0-12-420119-4.00007-0. [DOI] [PubMed] [Google Scholar]
- 42.Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, Hendricks S, Wang Y, Chaplin MD, Akan D, Paik S, Peterson DL, Macrina FL, Buck GA. 2007. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol 189:3166–3175. doi: 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pakula R, Walczak W. 1963. On the nature of competence of transformable streptococci. J Gen Microbiol 31:125–133. doi: 10.1099/00221287-31-1-125. [DOI] [PubMed] [Google Scholar]
- 44.Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A 99:14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cisar JO, Kolenbrander PE, McIntire FC. 1979. Specificity of coaggregation reactions between human oral streptococci and strains of Actinomyces viscosus or Actinomyces naeslundii. Infect Immun 24:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.