

Abstract
The heat-shock, or HSF1-mediated proteotoxic stress, response (HSR/HPSR) is characterized by induction of heat-shock proteins (HSPs). As molecular chaperones, HSPs facilitate the folding, assembly, transportation and degradation of other proteins. In mammals, heat shock factor 1 (HSF1) is the master regulator of this ancient transcriptional programme. Upon proteotoxic insults, the HSR/HPSR is essential to proteome homeostasis, or proteostasis, thereby resisting stress and antagonizing protein misfolding diseases and ageing. Contrasting with these benefits, an unexpected pro-oncogenic role of the HSR/HPSR is unfolding. Whereas HSF1 remains latent in primary cells without stress, it becomes constitutively activated within malignant cells, rendering them addicted to HSF1 for their growth and survival. Highlighting the HSR/HPSR as an integral component of the oncogenic network, several key pathways governing HSF1 activation by environmental stressors are causally implicated in malignancy. Importantly, HSF1 impacts the cancer proteome systemically. By suppressing tumour-suppressive amyloidogenesis, HSF1 preserves cancer proteostasis to support the malignant state, both providing insight into how HSF1 enables tumorigenesis and suggesting disruption of cancer proteostasis as a therapeutic strategy. This review provides an overview of the role of HSF1 in oncogenesis, mechanisms underlying its constitutive activation within cancer cells and its pro-oncogenic action, as well as potential HSF1-targeting strategies.
This article is part of the theme issue ‘Heat shock proteins as modulators and therapeutic targets of chronic disease: an integrated perspective’.
Keywords: amyloids, the heat-shock response, HSF1, proteostasis, proteotoxic stress, tumorigenesis
1. Introduction
Every biological process is dynamic and needs to stay homeostatic [1], a state essential to cellular and organismal fitness and survival. Disruption of this equilibrium inevitably provokes stress and elicits stress responses, through which cells and organisms can counter stresses and reinstate the homeostatic state.
Among the various cellular responses to stress is the heat-shock, or HSF1-mediated proteotoxic stress, response (HSR/HPSR) [2,3], an evolutionarily conserved defensive mechanism. Upon challenge by proteotoxic stressors, such as heat shock, cells mobilize the HSR/HPSR to produce a large amount of heat-shock proteins (HSPs), or molecular chaperones [2,3]. HSPs are a group of proteins specializing in facilitating the folding, trafficking, complex assembly, and ubiquitination and degradation of other proteins [2,3]. Therefore, HSPs ensure the quality of the cellular proteome and preserve proteome homeostasis, or proteostasis [4,5]. Of note, two categories of HSPs, constitutively expressed and stress-inducible, exist inside cells. Whereas the constitutively expressed HSPs, such as HSC70 and HSP90β, supply the basal chaperoning activity, the stress-inducible ones, such as HSP27 and HSP72, are necessary for meeting the extra demand for chaperoning activity, owing to elevated protein misfolding and aggregation, under proteotoxic conditions. Accordingly, the HSR/HPSR is dispensable under basal growth conditions but becomes indispensable under stressful conditions.
2. Transcriptional governance of the HSR/HPSR
The HSR/HPSR is primarily a transcriptional programme, characterized by induced HSP mRNAs [2,3]. A small family of transcription factors, named heat shock factors (HSFs), have been implicated in controlling the HSR/HPSR in response to proteotoxic stress [6,7]. While only a single HSF gene exists in yeasts and invertebrates, at least nine HSF paralogues have been identified in vertebrates to date, among which HSF1 is the most conserved and regarded as the prototype [6–8]. In mammals, HSF1 has proved to be the master regulator of the HSR/HPSR, as genetic deletion of Hsf1 abolishes the induction of HSPs by heat shock in mice [9,10]. By contrast, the HSR/HPSR is still mounted in mice deficient for Hsf2 or Hsf4 [11,12]. Of note, although HSF2 can form heterotrimers with HSF1 to modulate the HSR/HPSR, this interplay requires the presence of HSF1 [13]. While HSF3 is pivotal to the HSR/HPSR in avian cells, it appears to regulate the stress-inducible expression of non-Hsp genes in mice [14]. Despite being dispensable for the HSR/HPSR, HSF4 is required for normal lens development in mice [15]. Moreover, the functions of HSF5, HSFX1/2 and HSFY1/2 remain unknown [8].
Highly conserved among the HSF family of proteins are several functional domains, including the N-terminal helix–turn-helix DNA-binding domain, hydrophobic heptad repeats (HR)-enriched trimerization domain and C-terminal transactivation domain [7]. HSFs trimerize through their HR domains, a configuration crucial to their DNA binding [7,16]. Following nuclear translocation, trimeric HSFs bind to the consensus heat-shock elements (HSEs), typically consisting of contiguous inverted arrays of 5′-nGAAn-3′ motif [7,16], in gene promoters. Following DNA binding and subsequent stress-inducible phosphorylation, HSF1 recruits positive transcription elongation factor b (P-TEFb) to phosphorylate paused RNA polymerase II (RNAP II) at the proximal promoters of HSP genes, resulting in RNAP II pause release and transcription elongation [16].
3. Complex regulations of HSF1
In yeast HSF is constitutively active [17]; however, in vertebrates HSF1 becomes mobilized upon challenge by stressors [7,16]. As the principal regulator of the HSR/HPSR, HSF1 is subject to multilayer regulations.
In mammals, HSF1 remains latent under normal non-stressful conditions. By forming a protein complex with HSP90 and co-chaperones, HSF1 is repressed in the monomeric state [18]. Following proteotoxic stress, however, this repressive complex is disrupted, partially owing to the titration of HSPs away from the complexes by the accumulation of misfolded proteins. Subsequently, monomeric HSF1 is released and undergoes trimerization [18]. In agreement with this model, inhibition of HSP90 alone is sufficient to activate HSF1 in the absence of environmental stressors [18]. However, in yeast Hsf1 seems to predominantly interact with cytosolic Hsp70 and this repressive association is transiently disrupted by heat shock [19].
Following trimerization, HSF1 enters the nucleus and becomes competent for DNA binding. In addition, post-translational modifications (PTMs) are important for complete HSF1 activation. Among the various types of modifications reported, the best studied is phosphorylation. Of note, HSF1 undergoes both stimulatory and inhibitory phosphorylation. Interestingly, recent studies suggest that phosphorylation, although not required, is a fine-tuning mechanism for HSF1 activation [19]. Beyond phosphorylation, HSF1 is subject to acetylation and sumoylation [7,16]. These diverse PTMs likely impact different steps of HSF1 activation, including nuclear translocation, DNA binding and transactivation.
It is widely believed that HSF1 is regulated at the point of activation primarily; nonetheless, emerging evidence has started unveiling additional mechanisms for regulating HSF1 by altering its expression levels. For instance, polyubiquitination destabilizes HSF1 proteins and, therefore, impaired ubiquitination may contribute to elevated HSF1 proteins in human cancers [20–22]. Furthermore, in many human cancers HSF1 mRNA levels are increased too [23–25]. HSF1 gene amplification may be one of the underlying mechanisms [25]. It was also reported that the splicing factor SF3B1, genetic mutations of which occur in chronic lymphocytic leukaemia recurrently [26], can regulate HSF1 mRNA levels [27], suggesting a role of RNA splicing in regulating HSF1 expression. Together, these findings indicate that HSF1 can be regulated at the levels of both expression and activation.
4. HSF1 acts as a potent pro-oncogenic factor
Beyond enhancing cellular and organismal survival of stress, HSF1 prolongs the lifespan in nematodes [28,29]. In addition, HSF1 can protect neurons against protein aggregation and degeneration. In transgenic R6/2 mice, a Huntington's disease model, Hsf1 deficiency causes increased aggregates and inclusions of huntingtin proteins in the brain, shortening animal survival [30]. Conversely, activation of HSF1 by a small molecule HSF1A not only suppresses the aggregation of polyQ-huntingtin proteins and toxicity in rat neuronal precursor PC-12 cells, but also ameliorates the neurotoxicity in a fly model of spinocerebellar ataxia type 3 [31]. Moreover, pharmacological inhibition of HSP90 activates HSF1, thereby rescuing synaptic dysfunction and memory loss in a mouse model of Alzheimer's disease [32]. Clearly, all these effects of HSF1 are beneficial.
By stark contrast, recent studies have revealed a surprising role of HSF1 in enabling oncogenesis. In 2007, two independent studies demonstrated this unexpected deleterious action of HSF1. In one study, Hsf1 knockout selectively impaired lymphomagenesis in Trp53-deficient mice [33]. In the other study, Hsf1-deficient mice were markedly refractory to the DMBA-induced skin carcinogenesis and the tumorigenesis initiated by Trp53R172H mutation [34]. Subsequent studies in diverse mouse cancer models supported these initial findings. For example, diethylnitrosamine-induced hepatocellular carcinogenesis was suppressed in Hsf1-deficient mice [35]. Furthermore, Hsf1 deficiency impaired the development of malignant peripheral nerve sheath tumours (MPNSTs) in mice due to loss of the tumour suppressor neurofibromatosis type I (Nf1) [20]. Moreover, two independent groups demonstrated that deletion of Hsf1 significantly delayed mammary tumorigenesis in MMTV-Her2/Neu transgenic mice [36,37].
In addition to these spontaneous tumorigenesis models, various xenograft models have been employed to interrogate the role of HSF1 in oncogenesis. For example, RNA interference (RNAi)-mediated HSF1 depletion impeded the in vivo growth of transplanted human mammary epithelial cells overexpressing HER2/NEU [38]. Similarly, HSF1 knockdown resulted in impaired growth, invasion, and metastasis of xenografted human hepatocellular carcinoma (HCC) and melanoma cells in immunocompromised mice [39–42]. These studies clearly support an important role for HSF1 in the maintenance and progression of established malignancies. Moreover, HSF1 overexpression suffices to enhance the malignant phenotypes of xenografted human melanoma cells in vivo [43–45]. Thus, these findings collectively highlight the potent pro-oncogenic effects of HSF1.
In line with these in vivo findings, in vitro studies confirmed that HSF1 becomes indispensable for the growth and survival of established cancer cells. HSF1 depletion, via either lentiviral small hairpin RNAs (shRNAs) or small interfering RNAs (siRNAs), markedly impaired the growth and survival of a wide array of human cancer cell lines, including breast cancer cells, MPNST cells, melanoma cells, multiple myeloma cells, HCC cells and pancreatobiliary cancer cells [20,34,41,46–48]. In sharp contrast to the HSF1 addiction of malignant cells, HSF1 depletion exerted little or no impact on non-transformed cells [20,34]. The vast differences in HSF1 dependency are congruent with their distinct intrinsic levels of proteotoxic stress and states of HSF1 activation.
Unlike HSF1, HSF2 appears to suppress malignant progression [49]; moreover, HSF4 slightly promotes tumorigenesis in Trp53- or Arf-deficient mice [50]. In addition, the roles of other members of the HSF family in malignancy remain unknown.
5. Constitutive, autonomous activation of HSF1 within cancer cells
Canonically, HSF1 is activated by various environmental stressors, an acute and transient process. By contrast, within cancer cells HSF1 appears to remain constitutively activated [20,34,51], suggesting a state of chronic stress.
On the one hand, this chronic stress could be induced by acidic and hypoxic tumour microenvironments, which inflict protein damage inside cancer cells. On the other hand, proteotoxic stress could arise from within the tumour cell in the absence of environmental insults. A number of mechanisms may account for this intrinsic stress within cancer cells, including heightened global protein synthesis driven by mTORC1 hyper-activation, exacerbated proteomic imbalance due to aneuploidy, destabilized protein conformations resulting from numerous genetic mutations and oxidative protein damage caused by oxidative stress [8]. As a result, HSF1 activation is widespread in human cancers [23,51].
Although it is well known that proteotoxic stressors, such as heat shock, potently activate HSF1, our understanding of the underlying molecular mechanisms remains incomplete. For example, HSF1 hyper-phosphorylation is important to its full transcriptional activation and a number of phosphorylating events have been defined to date (figure 1) [52]. The stimulatory events include Ser230, Ser419, Thr142, Ser320 and Ser326 phosphorylation. By contrast, the inhibitory events include Ser303, Ser307, Ser363 and Ser121 phosphorylation. Nonetheless, it remains obscure how these events regulate HSF1 activity.
Figure 1.
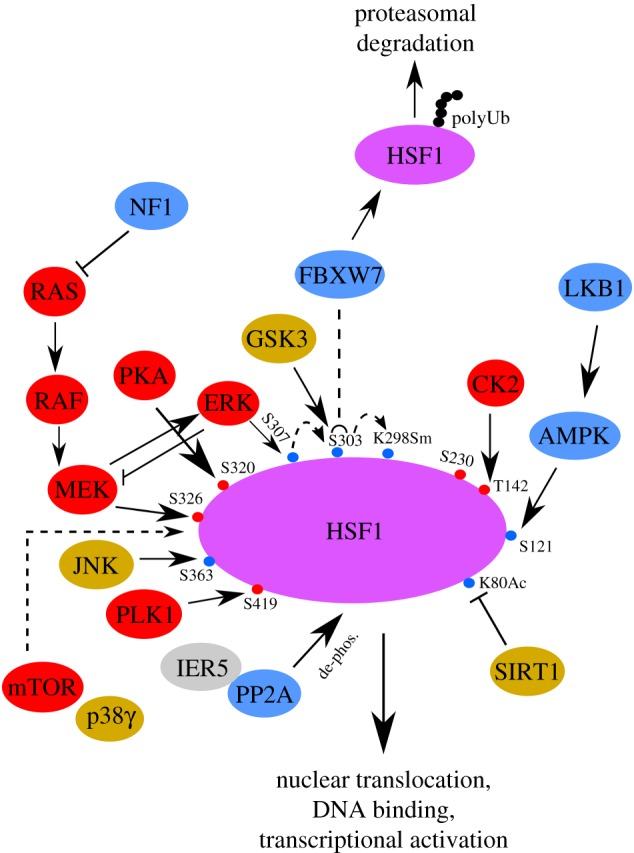
Summary of various post-translational modifications potentially implicated in constitutive activation of HSF1 within cancer cells. Refer to the main text for detailed regulations. Modifications stimulating HSF1 activation are marked in red and modifications inactivating HSF1 are marked in blue. Accordingly, oncoproteins and tumour suppressors are labelled in red and blue, respectively. Proteins displaying both oncogenic and tumour-suppressive roles are labelled in brown. Ac, acetylation; de-phos., de-phosphorylation; K, lysine; polyUb, polyubiquitination; S, serine; T, threonine; Sm, sumoylation.
Although these phosphorylating events occur either under physiological conditions or in the face of environmental stressors, it remains elusive whether cancer cells exploit the very same mechanisms to render HSF1 constitutively active. Of interest, some of the signalling pathways implicated in HSF1 regulation are frequently altered in human malignancy, such as the RAS/MAPK and LKB1/AMPK signalling cascades.
(a). Oncogenic RAS signalling directly activates HSF1 through phosphorylation
RAS/MAPK signalling plays a prominent role in oncogenesis, highlighted by the fact that RAS mutations occur in up to 30% of all human cancers [53]. These mutations all lead to hyper-activation of RAS/MAPK signalling, which, in turn, mobilizes numerous downstream effectors that control a plethora of cellular processes to drive malignant transformation collectively [54,55]. In addition to RAS mutations, frequent aberrations in upstream receptor tyrosine kinases (RTKs) and loss of the tumour suppressor NF1 can also activate RAS/MAPK signalling [20].
Does the oncogenic RAS/MAPK signalling pathway control HSF1 activation? It has been reported that ERK1/2, the widely believed ultimate effector of this signalling cascade, phosphorylates HSF1 at Ser307, a constitutively repressive modification under basal growth conditions [56]. Thus, HSF1 would be repressed by the most important oncogenic signalling pathway. Nonetheless, our and others' studies demonstrated that oncogenic RAS/MAPK signalling directly activates HSF1 and its mediated HSR/HPSR [20,21,57]. Surprisingly, our study uncovered that it is MEK, rather than ERK, that physically interacts with and activates HSF1 [21]. Thus, in parallel with ERK, HSF1 is a second physiological substrate for MEK. Our study indicated that heat shock activates RAS/MAPK signalling and induces physical MEK–HSF1 interactions, which leads to HSF1 phosphorylation at Ser326 [21], a modification known to be critical to its activation by heat shock [52]. Moreover, our study revealed that ERK suppresses HSF1 activation indirectly via a negative feedback mechanism. That is, ERK phosphorylates MEK at Thr292/386 to repress the MEK-mediated HSF1 Ser326 phosphorylation [21].
Interestingly, another report proposed that mTOR also phosphorylates HSF1 at Ser326 [58]. mTOR, a serine/threonine kinase, assembles into two distinct large protein complexes, mTORC1 and mTORC2, controlling protein translation, lipogenesis, autophagy and survival [59]. Unsurprisingly, mTOR signalling is pro-oncogenic [60]. In line with a role of mTOR signalling in HSF1 activation, the dual PI3 K/mTOR inhibitor BEZ235 blocks HSF1 activation due to HSP90 inhibition [61]. Moreover, p38γ was recently reported to phosphorylate Ser326 under heat shock [62]. It is possible that multiple kinases can phosphorylate Ser326. Despite phosphorylation demonstrated in vitro, the key evidence showing physical interactions between HSF1 and mTOR or p38γ inside cells is still missing. Thus, it remains unclear whether mTOR and p38γ regulate HSF1 directly or indirectly in vivo. Further detailed studies are necessary to clarify these issues.
(b). Tumour-suppressive LKB1/AMPK signalling represses HSF1 through phosphorylation
In response to elevated cellular AMP/ATP or ADP/ATP ratio, signals indicating cellular energy stress, AMP-activated protein kinase (AMPK) becomes activated [63]. In turn, AMPK phosphorylates numerous downstream effectors, eliciting a systemic cellular response that aims to conserve ATP consumption and enhance ATP production simultaneously [64]. Thereby, AMPK initiates the metabolic stress response and preserves energy homeostasis. Following binding of AMP or ADP to its γ subunits, full activation of AMPK requires phosphorylation of its α subunits at Thr172 by its upstream kinase liver kinase B1 (LKB1), a tumour suppressor causally implicated in human Peutz–Jeghers syndrome [65]. Notable downstream effectors of AMPK include acetyl CoA carboxylase 1 (ACC1), regulatory associated protein of mTOR (RAPTOR), sterol regulatory element binding protein 1c (SREBP1c) and Unc-51-like autophagy activating kinase 1 (ULK1) [65–67], which are involved in lipogenesis, protein translation and autophagy. In contrast with its well-known activation by proteotoxic stressors, it remains unknown how the HSR/HPSR responds to metabolic stressors.
Our recent study revealed that glucose and amino acid deprivation both suppress the HSR/HPSR [43]. Moreover, the widely prescribed anti-diabetic drug metformin exerted a similar effect [43]. Metformin, a mitochondrial toxin that disrupts cellular energy homeostasis by depleting ATP, has been recently shown to display promising anti-neoplastic effects [68]. Mechanistically, our study indicated that both nutrient deprivation and metformin activate AMPK, which subsequently phosphorylates HSF1 at Ser121 [43]. As a negative modification, Ser121 phosphorylation impairs the nuclear translocation and stability of HSF1 [43]. Accordingly, metabolic stressors suppress the HSR/HPSR triggered by heat shock, exacerbating proteomic perturbation and impairing survival [43]. Through the same mechanism, metformin at a clinically relevant dose suppresses the constitutive HSF1 activation in diverse human cancer cell lines and in xenografted human melanomas, provoking global protein ubiquitination [43]. Conversely, HSF1 overexpression renders human melanoma cells refractory to the inhibition of anchorage-independent growth by metformin in vitro and to the tumour-suppressive effect of metformin in vivo [43]. In line with its role in activating AMPK, LKB1 deficiency not only enhances the HSR/HPSR triggered by heat shock but also heightens the constitutive HSF1 activation in malignant cells (KH V, S Dai, Z Tang, C Dai 2017, unpublished manuscript).
Taken together, these findings uncover a previously unrecognized metabolic control of the HSR/HPSR via the AMPK-HSF1 interplay. In addition to its activation by proteotoxic stressors, metabolic stressors suppress HSF1. Although most of the anti-neoplastic effects of metformin have been ascribed to its metabolic impacts, our study pinpoints a new mechanism of action of metformin—disruption of cancer proteostasis. This action is probably applicable to metabolic stressors in general.
(c). GSK3 signalling suppresses HSF1 through phosphorylation
Glycogen synthase kinase 3 (GSK3), a key serine/threonine kinase involved in glycogen synthesis, regulates a wide variety of cellular functions and has been implicated in many human pathological conditions including neurodegenerative disorders and diabetes [69]. However, its roles in cancer remain controversial; and GSK3 seems to function as both a tumour suppressor and a tumour promoter. On the one hand, GSK3 can activate tumour suppressors, including TP53, TSC2 and RBL2 [70], and inactivate oncoproteins, including c-MYC, cyclin D1 and HIF-1α [70]. On the other hand, GSK3 activates some oncoproteins, including p70S6 K and MDM2 [70], and inactivates certain tumour suppressors, including PTEN and p27KIP1 [70].
It was reported that GSK3β phosphorylates HSF1 at Ser303, a constitutively negative modification promoting the nuclear exit of HSF1 via recruiting 14-3-3 proteins [56]. Interestingly, this GSK3-mediated HSF1 phosphorylation suppresses the expression of RNF126, an E3 ubiquitin ligase, to stabilize IGF-IIR proteins, thereby supporting hypertension-induced cardiomyocyte hypertrophy [71]. Moreover, another study showed that Ser303 phosphorylation is required for subsequent sumoylation of HSF1 at Lys298, which is inhibitory to its transactivation [72]. This negative regulation of HSF1 suggests a new role of GSK3 in regulating proteostasis. Therefore, in some human cancers inactivated GSK3 signalling may contribute to malignant transformation at least in part via HSF1 activation.
(d). JNK signalling suppresses HSF1 through phosphorylation
c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK), a multifaceted serine/threonine kinase, responds to numerous extracellular and intracellular cues, including growth factors, inflammatory cytokines, UV radiation and cellular stresses, including oxidative, osmotic, endoplasmic reticulum and proteotoxic stress [73,74]. Following activation, JNK phosphorylates a list of downstream effectors, including c-JUN, ELK1, ATF2 and TP53, to regulate differentiation, growth and apoptosis [75]. The role of JNK signalling in human cancer is context-dependent, exerting both tumour-suppressive and tumour-promoting effects [75]. In support of its tumour-suppressive role, inactivating mutations in MKK4 and MKK7, the genes encoding two key upstream kinases activating JNK, have been found in human cancer [76,77].
Interestingly, one of the JNK targets is HSF1. It was reported that JNK phosphorylates HSF1 at Ser363, leading to its inhibition [78]. Congruent with this negative regulation, JNK deficiency activates HSF1 [79]. Furthermore, our recent study showed that HSF1 prevents JNK activation reciprocally [79], revealing a mutual suppression between JNK and HSF1. Thus, these findings suggest that in some human cancers with inactivated JNK signalling HSF1 is mobilized to support malignancy.
(e). PKA signalling activates HSF1 through phosphorylation
Cyclic adenosine 3′,5′ monophosphate (cAMP), the first identified intracellular second messenger, plays a pivotal role in the signal transductions triggered by hormones and neurotransmitters [80]. In mammalian cells, the primary effector of cAMP is protein kinase A (PKA), a ubiquitous tetrameric cAMP-binding kinase [80]. Among the key PKA substrates are GSK3 and cAMP-response element binding protein [80]. Importantly, activation of PKA signalling has been implicated in tumour initiation and progression [81]. In human Carney complex syndrome, germline mutations in PRKAR1A, which encodes the type 1A regulatory subunit of PKA, result in hyper-activation of PKA and, ultimately, development of endocrine tumours in the testicle, thyroid and pancreas [82].
Of note, a recent study reported that PKA activates HSF1 through Ser320 phosphorylation, a modification promoting its nuclear translocation and DNA binding [83]. In addition, PKA may activate HSF1 indirectly, via suppressing GSK3 [84]. These findings suggest that PKA signalling can promote tumorigenesis, in part, by activating HSF1.
(f). PLK1 regulates HSF1 through phosphorylation
Polo-like kinase 1 (PLK1) is essential to cell cycle progression and mitosis by regulating maturation of mitotic centrosomes, assembly of mitotic spindle, as well as cytokinesis [85,86]. Unsurprisingly, PLK1 plays a key role in maintaining genomic stability. Prominent PLK1 substrates include CDC25, cyclin B1, MYT1/WEE1, NLP, APC/C and NUDC [85,86]. Whereas its expression remains low in most primary adult tissues, PLK1 is frequently overexpressed in human cancer tissues, which is associated with tumour progression and poor prognosis [86].
Congruent with its tumour-promoting effects, it was reported that PLK1 phosphorylates HSF1 at Ser419, a modification enhancing HSF1 nuclear translocation induced by heat shock [87]. Furthermore, another study showed that during mitosis PLK1 phosphorylates HSF1 at Ser216, which blocks the SCFβ-TrCP-mediated ubiquitination and subsequent degradation of HSF1 by inducing physical HSF1–CDC20 interactions [88]. This interaction sequesters CDC20 away from the anaphase promoting complex/cyclosome (APC/C), blocking mitotic exit and inducing aneuploidy [88]. These findings suggest that HSF1 acts as a mitotic regulator independently of its transcriptional regulation of the HSR/HPSR. Further studies are necessary to fully delineate the PLK1-mediated HSF1 regulations; nonetheless, current evidence suggests that HSF1 may contribute to the tumour-promoting effects of PLK1.
(g). CK2 signalling activates HSF1 through phosphorylation
Casein kinase II (CK2) is a constitutively active serine/threonine kinase closely associated with enhanced cell proliferation and survival [89]. CK2 normally exists as a heterotetrameric complex comprising two catalytic subunits, α and α′, and two regulatory β subunits [89]. CK2 can phosphorylate a myriad of substrates, and accumulated evidence has demonstrated the oncogenic potential of CK2. For example, CK2α overexpression accelerated the development of acute lymphoblastic leukaemia in TAL-1 transgenic mice [90]. Furthermore, MMTV-CK2α transgenic mice developed mammary gland hyperplasia and adenocarcinomas [91]. Consistent with its pro-oncogenic potential, CK2 expression is elevated in a large diversity of human cancers [89].
It was reported that heat shock triggers the nuclear translocation and activation of CK2 [92]. Importantly, CK2 phosphorylates HSF1 at Thr142, a modification necessary for its DNA binding and HSP gene transcription under heat shock [92]. Thus, it is conceivable that HSF1 activation may contribute to the oncogenic property of CK2.
(h). IER5 activates HSF1 through de-phosphorylation
In contrast to the well-recognized HSF1 activation via phosphorylation, a recent study uncovered that HSF1 can also be activated via de-phosphorylation. Immediate early response 5 (IER5), a transcriptional target of TP53, acts as an activator of HSF1 by forming a ternary complex with HSF1 and the phosphatase PP2A [93]. In consequence, HSF1 becomes hypo-phosphorylated but active [93]. Although the underlying mechanisms remain unclear, it is possible that the IER5-PP2A complex alleviates some of the inhibitory phosphorylation events on HSF1. Importantly, IER5 is often transcriptionally upregulated in various human cancers [93], which may also contribute to the widespread constitutive activation of HSF1 in cancer via this de-phosphorylating mechanism.
(i). SIRT1 promotes HSF1 activation via deacetylation
Sirtuin 1 (SIRT1), the mammalian orthologue of Sir2p in yeast, is an NAD+-dependent protein deacetylase controlling DNA repair, cellular metabolism, longevity and stress responses [94]. TP53, FOXO, KU70, PGC1α and LXR are among the notable SIRT1 substrates [94]. By preventing MDM2 binding to TP53-responsive promoters, acetylation of the tumour suppressor TP53 is essential to its transcriptional activation [95]. In support of its oncogenic potential, SIRT1 deacetylates Lys382 to impair the transcriptional activity of TP53 [96]. Moreover, SIRT1 can promote epithelial–mesenchymal transition (EMT) [97]. Paradoxically, SIRT1 can also act as a tumour suppressor. Sirt1-deficiency led to increased genomic instability and accelerated tumorigenesis in Trp53+/− mice [98]. Conversely, Sirt1 overexpression impaired intestinal tumorigenesis in APCmin/+ mice [99]. Thus, the roles of SIRT1 in cancer are complex, likely tissue- and context-dependent.
It has been shown that heat stress induces Lys80 acetylation of HSF1 by p300 or cyclic AMP response element-binding protein, a modification negatively regulating its DNA binding [100]. Of interest, SIRT1 deacetylates Lys80, thereby maintaining HSF1 in a DNA-binding competent state [100]. While this mechanism likely underlies the beneficial effects of SIRT1 on stress-resistance and longevity [100], it may also serve to promote malignancy. Importantly, it remains elusive whether SIRT1 plays a role in protecting cancer proteostasis via HSF1.
(j). HDAC6 senses protein aggregation to de-repress HSF1
In addition to SIRT1, histone deacetylase 6 (HDAC6) can regulate HSF1 activation as well; however, this action is independent of its deacetylase activity. Under non-stress conditions, HDAC6 and its interacting partner p97/VCP, an AAA+-ATPase, associate with the repressive HSP90–HSF1 protein complex [101]. Upon accumulation of ubiquitinated protein aggregates inside cells, HDAC6 senses protein aggregation via its ubiquitin-binding domain and dissociates itself from p97/VCP, thereby enabling p97/VCP to disrupt HSP90–HSF1 interactions via its ATPase activity [101]. Thus, HDAC6 is required for disassembly of the repressive HSP90-HSF1 complexes to unleash HSF1 for activation in the face of protein aggregation. In the light of its upregulated expression in various human cancers [102], it is conceivable that HDAC6 may contribute to constitutive HSF1 activation in malignancies.
(k). EEF1A1 enhances the HSR/HPSR both transcriptionally and translationally
A recent study revealed that, beyond its well-defined function in protein translation, eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) is also actively involved in regulating the HSF1-mediated HSR/HPSR. During proteotoxic stress, EEF1A1 helps to recruit HSF1 to the HSP72 gene promoter to initiate the transcription; subsequently, it stabilizes and transports HSP72 mRNAs to translating ribosomes by binding to their 3′ untranslated regions (UTRs) [103]. Thereby, EEF1A1 assists the HSR/HPSR to enhance thermotolerance. Although it remains unclear whether this mechanism operates in the context of cancer, it is tempting to speculate that EEF1A1 may support oncogenesis in part by heightening the HSF1-mediated HSR/HPSR.
(l). Stabilization of HSF1 proteins in cancer
Canonically, HSF1 regulation is thought to occur at the activation step primarily; however, elevated mRNAs and proteins of HSF1 have been noticed in human cancers [21–23]. Whereas the mechanisms underlying upregulated HSF1 mRNAs remain elusive, emerging evidence has highlighted a role of the ubiquitin–proteasome system (UPS) in regulating HSF1 protein stability.
Our study indicated that the MEK-mediated Ser326 phosphorylation stabilizes HSF1 by blocking its polyubiquitination and subsequent proteasomal degradation [21]. Furthermore, another study reported that filamin A-interacting protein 1-like (FILIP1 L) interacts with HSF1 to promotes its ubiquitination and proteasomal degradation [104]. FILIP1 L, whose expression is downregulated in several human cancers, inhibits the migration, invasion and metastasis of various human cancer cell lines [105]. Thus, FILIP1 L acts like a tumour suppressor to destabilize HSF1. Moreover, a recent study identified F-box and tryptophan/aspartic acid (WD) repeat domain-containing 7 (FBXW7) as an E3 ligase responsible for HSF1 ubiquitination [22]. FBXW7 is a tumour suppressor targeting several key proto-oncoproteins, including c-MYC, cyclin E and SREBP1, for proteasomal degradation [106]. This study demonstrated that FBXW7 physically binds to HSF1 via a conserved degron motif (aa 303–307), which is phosphorylated by both GSK3β and ERK1 [56]. Another new study indicated that CK2α’ can also phosphorylate Ser303/307 to recruit FBXW7 for HSF1 ubiquitination [107]. In a xenograft model, FBXW7 deficiency led to nuclear accumulation of HSF1 and enhanced lung metastasis of human melanoma cells [22]. Interestingly, our study indicated that the MEK-mediated Ser326 phosphorylation diminishes HSF1 Ser307 phosphorylation [21]. Thus, it is possible that MEK stabilizes HSF1, in part, by impeding the FBXW7-mediated ubiquitination.
In aggregate, a growing body of evidence indicates that both HSF1 activity and expression are upregulated in human cancers via diverse mechanisms.
6. How does HSF1 empower tumorigenesis?
Given that HSPs chaperone a vast number of cellular proteins, unsurprisingly, the impacts of HSF1 on tumorigenesis are very broad and diverse.
(a). Transcription-dependent, cell-autonomous pro-oncogenic effects
Naturally, it has been believed that HSF1 promotes oncogenesis primarily through its transcriptional action (figure 2). Congruent with its role in regulating HSP transcription, αB-crystallin/Hspb5 expression is diminished in Hsf1−/− mouse embryonic fibroblasts [108]. It is known that αB-crystallin complexes with FBX4, an E3 ubiquitin ligase, to promote protein ubiquitination [109]. Of note, one of the FBX4 targets is the tumour suppressor TP53 [108]. Thus, decreased αB-crystallin expression impairs TP53 protein ubiquitination, leading to TP53 accumulation in Hsf1−/− cells [108]. This result suggests that HSF1 promotes malignancy, in part, by enhancing TP53 degradation.
Figure 2.

Diverse transcription-dependent mechanisms through which HSF1 potently promotes oncogenesis. Through induction of both HSPs and non-HSPs, within cancer cells HSF1 maintains oncogenic signalling, enhances EMT and angiogenesis, promotes genomic instability and preserves proteomic stability. In addition, HSF1 activation within tumour-associated stromal cells can support tumour progression in a non-cell-autonomous fashion.
Furthermore, HSF1 is required for malignant transformation of immortalized mammary epithelial MCF-10A cells driven by the HER2/NEU oncogene. Mechanistically, HSF1 antagonizes HER2/NEU-induced cellular senescence [38]. This can be, in part, ascribed to elevated HSP expression owing to HSF1 activation, as depletion of either HSP27 or HSP72 by shRNAs sensitizes MCF-10A cells to senescence induced by HER2/NEU [38].
Moreover, through induction of HSP90α expression, HSF1 can promote oncogenic RAS signalling indirectly via stabilizing kinase suppressor of RAS 1 (KSR1) [20], a client protein of HSP90. KSR1 is a scaffolding protein providing docking sites for RAF, MEK and ERK oncoproteins to undergo serial activating phosphorylation events [110]. Congruently, HSF1 deficiency diminishes KSR1 protein and impairs ERK phosphorylation, impeding the tumorigenesis driven by hyper-activation of RAS signalling due to Nf1 deficiency [20]. In addition, mitigation of RAS signalling may underlie the migration defect of Hsf1-deficient cells stimulated with epidermal growth factor (EGF) [111]. Similarly, HSF1 deficiency can destabilize other client proteins of HSP90, including AKT, EGFR and MIF [112–114], all of which are important players in oncogenesis.
Given its regulation of several classes of chaperones and co-chaperones, HSF1 deficiency is expected to influence numerous chaperone client proteins and their mediated biological pathways, therefore bringing forth systemic impacts. Indeed, our recent study revealed that HSF1 compromise, via either shRNA, AMPK activation or MEK blockade, induces global protein ubiquitination and aggregation in malignant cells, accompanied by diminished cellular chaperoning capacity [21,43]. Moreover, beyond protein destabilization and aggregation, HSF1 deficiency provokes amyloidogenesis [21], marking the state of utmost proteomic chaos. Whereas amyloidogenesis frequently occurs in neural cells and has been closely associated with human neurodegenerative disorders [115], it appears that cancer cells are also susceptible to amyloidogenesis. Our study revealed that both HSF1 and proteasome operate in concert to contain amyloidogenesis at a low level not obviously detrimental to cancer cells. Nonetheless, amyloids are still elevated in malignant cells compared with their non-transformed counterparts [21]. Of note, this fragile cancer proteostasis is highly vulnerable to proteomic perturbations. Either HSF1 compromise, proteasome inhibition or both combined induces amyloid formation, leading to toxicity in both cancer cells in vitro and melanomas in vivo [21]. Moreover, our study indicated that amyloidogenesis is tumour-suppressive, impeding melanoma growth and metastasis in vivo [21].
Conceptually, our study suggests that proteostasis enables oncogenesis. Furthermore, our study not only establishes that HSF1 acts as a generic pro-oncogenic factor, by safeguarding proteostasis in cancer, but also suggests that disrupting proteostasis and provoking amyloidogenesis may be a novel therapeutic strategy to combat malignancies.
In addition to their prominent cytosolic localization and roles as molecular chaperones, a fraction of HSPs are associated with membranes in tumour cells. For example, HSP72 proteins have been found both in lipid rafts of the plasma membrane and on the lysosomal membrane. While the plasma membrane-bound HSP72 can both protect tumour cells against radiation and function as a target structure recognized by natural killer cells [116], the lysosomal membrane-bound HSP72 inhibits lysosomal membrane permeabilization and subsequent cathepsin release [117]. Thus, through regulation of membrane-bound HSPs, HSF1 may modulate the anti-tumour immune response and promote tumour cell survival.
Accumulating evidence also indicates that HSF1 can regulate non-HSP genes, particularly in the context of cancer [53]. In addition to suppressing the initiation of mammary carcinomas in MMTV-Her2/Neu transgenic mice, Hsf1 deficiency impeded tumour progression by impairing angiogenesis [36]. Mechanistically, HSF1 controls the transcription of human antigen R (HuR), an RNA-binding protein specifically recognizing the AU-rich elements located at the 3′ UTR of mRNAs [118]. HuR is known to regulate the stability and/or translation of many mRNAs, including HIF-1α and VEGF [119,120]. Thus, Hsf1 deficiency diminishes cellular HuR expression, leading to reduced HIF-1α protein translation and impaired tumour angiogenesis [36]. Similarly, HSF1 can control β-catenin mRNA translation via HuR in mammary cancer cells [121].
HSF1 also promotes EMT. It was reported that Hsf1 deficiency impedes the EMT and migration of mammary epithelial cells derived from MMTV-Her2/Neu transgenic mice, contributing to impaired mammary tumorigenesis and metastasis [37]. Similarly, it was reported that HSF1 knockdown mitigates the transcription of several master inducers of EMT, including SLUG, SNAIL, TWIST1 and ZEB1, thereby blocking the EMT and migration induced by transforming growth factor beta (TGFβ) in ovarian cancer cell lines [22]. HSF1 can also promote HCC cell migration and invasion through transcriptional induction of miR-135b, a microRNA targeting RECK and EVI5 [122].
Interestingly, HSF1 can also promote the transcription of telomeric repeat containing RNA and telomere protection under heat stress [123]. Given the suppression of tumorigenesis by telomere shortening [124], it is plausible to postulate that the HSF1-mediated telomere protection may contribute to malignant transformation.
(b). Transcription-dependent, non-cell-autonomous pro-oncogenic effects
Undoubtedly, HSF1 is capable of promoting malignant phenotypes in a cell-autonomous fashion; however, emerging evidence also points to a non-cell-autonomous action of HSF1 in oncogenesis (figure 2). It was reported that HSF1 is activated in cancer-associated stromal fibroblasts and, importantly, deletion of HSF1 in fibroblasts impedes the in vivo growth of xenografted MCF-7 breast cancer cells [125]. Mechanistically, HSF1 appears to drive a transcriptional programme in stromal fibroblasts that induces the expression of TGFβ and SDF1 to support the malignant growth of adjacent cancer cells [125]. Interestingly, our recent study also uncovered that Jnk1 deficiency causes HSF1 activation in non-parenchymal cells, a group of diverse cell populations that critically support hepatocytes, in mouse livers, leading to increased transcription of hepatocyte growth factor (Hgf) [79]. In a paracrine manner, enhanced HGF production in non-parenchymal cells, in turn, stimulates c-MET signalling in adjacent hepatocytes to drive their proliferation [79]. Thus, it is conceivable that this non-cell-autonomous mechanism could be operating in the context of liver carcinogenesis, given that c-MET is a potent proto-oncogene [126]. Furthermore, HGF secreted by microenvironments may render tumour cells resistant to therapeutic agents [127].
In addition, accumulating evidence indicates that HSPs can be secreted into the extracellular space via exosomes [128]. Extracellular HSPs can not only induce proinflammatory cytokines but also suppress protein misfolding and aggregation in recipient cells through exosome-mediated transmission [129,130]. Thus, through the non-cell-autonomous actions of HSPs, HSF1 may promote tumour progression by creating an inflammatory microenvironment and maintaining global proteomic stability in tumours.
(c). Transcription-independent pro-oncogenic effects
Previously, it has been shown that a dominant negative HSF1 mutant is able to impair cyclin B1 degradation and suppress aneuploidy in prostate cancer cell lines in vitro [131]. Another study reported that PLK1 phosphorylates HSF1 at Ser216, sequestering CDC20 away from the APC/C [88]. Thus, phosphorylated HSF1 heightens mitotic checkpoint activation to promote aneuploidy, independently of its transcriptional action (figure 3a).
Figure 3.
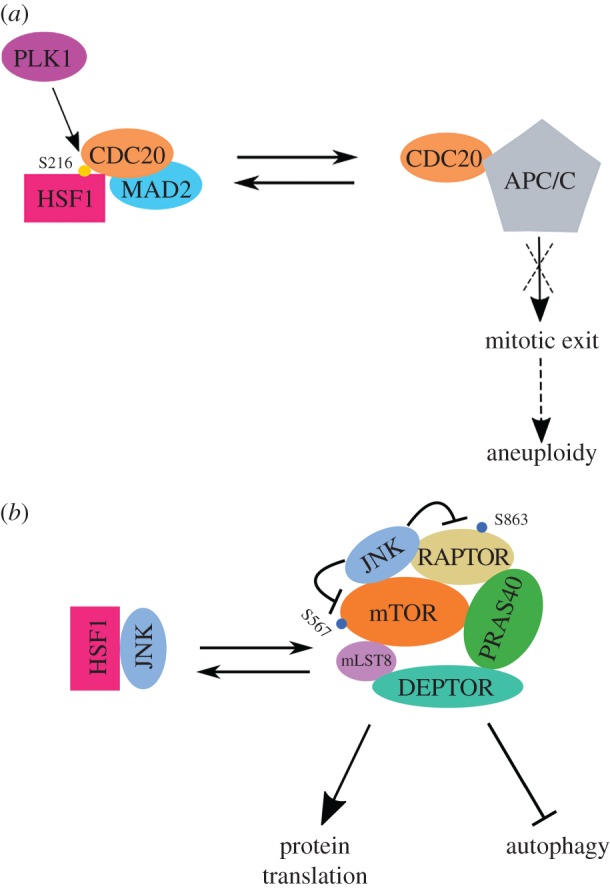
HSF1 can exert oncogenic effects via transcription-independent mechanisms. (a) Phosphorylated HSF1 sequesters CDC20 away from APC/C, blocking mitotic exit to promote aneuploidy. (b) Through physical sequestration of JNK apart from mTORC1, HSF1 promotes robust protein synthesis and suppresses autophagy, thereby supporting malignant growth.
Our recent study also indicates that HSF1 supports robust protein translation mediated by mTORC1 in a transcription-independent fashion (figure 3b). Mechanistically, the multifaceted stress-responsive kinase JNK constitutively associates with mTORC1 [79]. Upon activation by proteotoxic stress, JNK directly phosphorylates both RAPTOR at Ser863 and mTOR at Ser567, resulting in selective exclusion of mTOR from the complex and subsequent mTORC1 suppression [81]. Although not part of mTORC1, HSF1, through physical interactions, sequestrates JNK apart from mTORC1, thereby de-repressing mTORC1 [79]. This mechanism not only averts deep repression of mTORC1 under proteotoxic stress, enhancing stress-resistance by ensuring efficient translation of induced HSP mRNAs, but also supports cellular and organismal growth under normal conditions, controlling cell and body size [79]. Given the important role of mTORC1 in malignancy [59], it is conceivable that this very same mechanism could contribute to oncogenesis. Indeed, our results show that in diverse human cancer cell lines, HSF1 depletion by lentiviral shRNAs leads to JNK activation and suppressed global protein translation (KH Su, J Cao, C Dai 2017, unpublished manuscript).
Collectively, these recent findings pinpoint a key transcription-independent mode of action of HSF1, in addition to its well-appreciated transcriptional action.
7. Therapeutic targeting of HSF1 in cancer
Compelling evidence has indicated the potent pro-oncogenic role of HSF1 [8,132]. It is natural to consider HSF1 as a potential anti-cancer therapeutic target. Furthermore, targeting HSF1 is being considered for use in combinatorial therapies to mitigate the counterproductive HSF1 activation triggered by proteasome and HSP90 inhibitors [61]. In fact, in recent years increasing efforts have been invested in developing various strategies to target HSF1 for cancer therapies.
(a). Targeting HSF1 mRNAs
One means to block the HSF1 pathway is to deplete HSF1 mRNAs via RNAi. This strategy has been successfully applied in many studies to demonstrate the pro-oncogenic effects of HSF1. Owing to its relatively greater target specificity compared with small-molecule drugs, there is a considerable amount of interest in developing RNAi therapeutics for various human diseases including cancer [133]. Importantly, in the light of the emerging transcription-independent action of HSF1, one evident advantage of the RNAi-based therapies is depletion of HSF1 proteins, which is typically difficult to achieve with small molecules.
(b). Targeting the HSF1-mediated transcription or translation of HSP mRNAs
Still, small-molecule drugs are the mainstream pharmaceutical approach. To date, a number of compounds displaying inhibitory effects on the HSF1-mediated transcription or translation of HSP mRNAs have been reported (table 1), although most of them suffer from lack of target specificity or poorly defined mechanisms of action.
Table 1.
Small molecules inhibiting the HSF1-mediated HSR/HPSR. n.d., not determined.
| drug-like compounds | chemical class | mechanisms of action | molecular targets | references |
|---|---|---|---|---|
| quercetin | flavonoid | reduction of HSF1 expression | many | [134,135] |
| KNK437 | benzylidene lactam | blockade of HSF1-mediated HSP transcription | n.d. | [136] |
| triptolide | diterpenoid epoxide | global transcriptional arrest | XPB/ERCC3 | [137,138] |
| KRIBB11 | pyridinediamine | blockade of HSF1-dependent recruitment of P-TEFb to the HSP72 promoter | HSF1 in cell lysates | [139] |
| fisetin | dietary flavonoid | blockade of HSF1 binding to the HSP72 promoter | n.d. | [140] |
| NZ28 and emunin | emetine | inhibition of HSP mRNA translation | n.d. | [141] |
| rohinitib | rocaglate | blockade of genome-wide HSF1 DNA binding | EIF4A | [142] |
Recently, a new strategy based on RNA aptamer technology has emerged. It was reported that RNA aptamers bind to HSF1 proteins avidly in vitro and block the binding of HSF1 to HSP genomic loci in vivo [143]. Like HSF1-targeting RNAi, RNA aptamers effectively impede the malignant phenotypes of human cancer cell lines [143]. Thus, RNA aptamers may represent a promising class of HSF1 inhibitors with improved target specificity.
8. Concluding remarks and perspectives
Unequivocally, cancer is a genetic disease. Whereas genomic instability has been causally associated with tumorigenesis, little is known of the role of proteomic stability or proteostasis in cancer. Now, emerging evidence suggests that proteostasis enables malignancy, sharply contrasting with its beneficial roles in antagonizing neurodegeneration and ageing. The cellular proteostasis network consists of translation, chaperoning and proteolytic machineries, among which HSF1 governs the stress-inducible, but not the basal, chaperoning capacity. Owing to the chronic proteotoxic stress endured by malignant cells, HSF1 is obligated to remain constitutively active to supply the additional chaperoning capacity, which is necessary to accomplish and sustain malignant transformation. Thus, the proteostasis in cancer is constrained and fragile, distinct from that in primary non-transformed cells, which is robust and capable of buffering a considerable degree of proteomic perturbation. This key distinction may offer a promising opportunity for proteostasis-targeted anti-cancer therapies.
Acknowledgement
The author sincerely apologizes to those whose work could not be cited in this review because of space limitations.
Data accessibility
This article has no additional data.
Competing interests
I declare I have no competing interests.
Funding
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute and Center for Cancer Research.
Disclaimer
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
References
- 1.Cannon WB. 1932. The wisdom of the body. New York, NY: WW Norton & Co. [Google Scholar]
- 2.Morimoto RI. 2011. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 91–99. ( 10.1101/sqb.2012.76.010637) [DOI] [PubMed] [Google Scholar]
- 3.Lindquist S. 1986. The heat-shock response. Annu. Rev. Biochem. 55, 1151–1191. ( 10.1146/annurev.bi.55.070186.005443) [DOI] [PubMed] [Google Scholar]
- 4.Balch WE, Morimoto RI, Dillin A, Kelly JW. 2008. Adapting proteostasis for disease intervention. Science 319, 916–919. ( 10.1126/science.1141448) [DOI] [PubMed] [Google Scholar]
- 5.Labbadia J, Morimoto RI. 2015. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. ( 10.1146/annurev-biochem-060614-033955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujimoto M, Nakai A. 2010. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 277, 4112–4125. ( 10.1111/j.1742-4658.2010.07827.x) [DOI] [PubMed] [Google Scholar]
- 7.Akerfelt M, Morimoto RI, Sistonen L. 2010. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555. ( 10.1038/nrm2938) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai C, Sampson SB. 2016. HSF1: Guardian of proteostasis in cancer. Trends Cell Biol. 26, 17–28. ( 10.1016/j.tcb.2015.10.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. 1999. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 18, 5943–5952. ( 10.1093/emboj/18.21.5943) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Huang L, Zhang J, Moskophidis D, Mivechi NF. 2002. Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J. Cell. Biochem. 86, 376–393. ( 10.1002/jcb.10232) [DOI] [PubMed] [Google Scholar]
- 11.Paslaru L, Morange M, Mezger V. 2003. Phenotypic characterization of mouse embryonic fibroblasts lacking heat shock factor 2. J. Cell. Mol. Med. 7, 425–435. ( 10.1111/j.1582-4934.2003.tb00245.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujimoto M, Oshima K, Shinkawa T, Wang BB, Inouye S, Hayashida N, Takii R, Nakai A. 2008. Analysis of HSF4 binding regions reveals its necessity for gene regulation during development and heat shock response in mouse lenses. J. Biol. Chem. 283, 29 961–29 970. ( 10.1074/jbc.M804629200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostling P, Bjork JK, Roos-Mattjus P, Mezger V, Sistonen L. 2007. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 282, 7077–7086. ( 10.1074/jbc.M607556200) [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto M, et al. 2010. A novel mouse HSF3 has the potential to activate nonclassical heat-shock genes during heat shock. Mol. Biol. Cell 21, 106–116. ( 10.1091/mbc.E09-07-0639) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimoto M, et al. 2004. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 23, 4297–4306. ( 10.1038/sj.emboj.7600435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anckar J, Sistonen L. 2011. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 80, 1089–1115. ( 10.1146/annurev-biochem-060809-095203) [DOI] [PubMed] [Google Scholar]
- 17.Sorger PK, Pelham HR. 1988. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54, 855–864. ( 10.1016/S0092-8674(88)91219-6) [DOI] [PubMed] [Google Scholar]
- 18.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. 1998. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480. ( 10.1016/S0092-8674(00)81588-3) [DOI] [PubMed] [Google Scholar]
- 19.Zheng X, Krakowiak J, Patel N, Beyzavi A, Ezike J, Khalil AS, Pincus D. 2016. Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. Elife 5, e18638 ( 10.7554/eLife.18638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L, Lindquist S. 2012. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Invest. 122, 3742–3754. ( 10.1172/JCI62727) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z, Dai S, He Y, Doty RA, Shultz LD, Sampson SB, Dai C. 2015. MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160, 729–744. ( 10.1016/j.cell.2015.01.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kourtis N, et al. 2015. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat. Cell Biol. 17, 322–332. ( 10.1038/ncb3121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santagata S, et al. 2011. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl Acad. Sci. USA 108, 18 378–18 383. ( 10.1073/pnas.1115031108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engerud H, et al. 2014. High level of HSF1 associates with aggressive endometrial carcinoma and suggests potential for HSP90 inhibitors. Br. J. Cancer 111, 78–84. ( 10.1038/bjc.2014.262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powell CD, Paullin TR, Aoisa C, Menzie CJ, Ubaldini A, Westerheide SD. 2016. The heat shock transcription factor HSF1 induces ovarian cancer epithelial-mesenchymal transition in a 3D spheroid growth model. PLoS ONE 11, e0168389 ( 10.1371/journal.pone.0168389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, et al. 2016. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell 30, 750–763. ( 10.1016/j.ccell.2016.10.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Guisbert KS, Guisbert E. 2017. SF3B1 is a stress-sensitive splicing factor that regulates both HSF1 concentration and activity. PLoS ONE 12, e0176382 ( 10.1371/journal.pone.0176382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu AL, Murphy CT, Kenyon C. 2003. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145. ( 10.1126/science.1083701) [DOI] [PubMed] [Google Scholar]
- 29.Morley JF, Morimoto RI. 2004. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 15, 657–664. ( 10.1091/mbc.E03-07-0532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimoto M, Takaki E, Hayashi T, Kitaura Y, Tanaka Y, Inouye S, Nakai A. 2005. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 280, 34 908–34 916. ( 10.1074/jbc.M506288200) [DOI] [PubMed] [Google Scholar]
- 31.Neef DW, Turski ML, Thiele DJ. 2010. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 8, e1000291 ( 10.1371/journal.pbio.1000291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang B, et al. 2016. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer's mouse model via an HSF1-mediated mechanism. Mol. Psychiatry 22, 990–1001. ( 10.1038/mp.2016.104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF. 2007. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 26, 5086–5097. ( 10.1038/sj.onc.1210317) [DOI] [PubMed] [Google Scholar]
- 34.Dai C, Whitesell L, Rogers AB, Lindquist S. 2007. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130, 1005–1018. ( 10.1016/j.cell.2007.07.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin X, Moskophidis D, Mivechi NF. 2011. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 14, 91–103. ( 10.1016/j.cmet.2011.03.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabai VL, Meng L, Kim G, Mills TA, Benjamin IJ, Sherman MY. 2012. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol. Cell. Biol. 32, 929–940. ( 10.1128/MCB.05921-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xi C, Hu Y, Buckhaults P, Moskophidis D, Mivechi NF. 2012. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J. Biol. Chem. 287, 35 646–35 657. ( 10.1074/jbc.M112.377481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng L, Gabai VL, Sherman MY. 2010. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 29, 5204–5213. ( 10.1038/onc.2010.277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang F, Chang R, Yang L. 2012. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 118, 1782–1794. ( 10.1002/cncr.26482) [DOI] [PubMed] [Google Scholar]
- 40.Chen Y, et al. 2013. Targeting HSF1 sensitizes cancer cells to HSP90 inhibition. Oncotarget 4, 816–829. ( 10.18632/oncotarget.991) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujimoto M, Takaki E, Takii R, Tan K, Prakasam R, Hayashida N, Iemura S, Natsume T, Nakai A. 2012. RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol. Cell 48, 182–194. ( 10.1016/j.molcel.2012.07.026) [DOI] [PubMed] [Google Scholar]
- 42.Nakamura Y, Fujimoto M, Fukushima S, Nakamura A, Hayashida N, Takii R, Takaki E, Nakai A, Muto M. 2014. Heat shock factor 1 is required for migration and invasion of human melanoma in vitro and in vivo. Cancer Lett. 354, 329–335. ( 10.1016/j.canlet.2014.08.029) [DOI] [PubMed] [Google Scholar]
- 43.Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, Dai C. 2015. Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J. 34, 275–293. ( 10.15252/embj.201489062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott KL. et al. 2011. Proinvasion metastasis drivers in early-stage melanoma are oncogenes. Cancer Cell 20, 92–103. ( 10.1016/j.ccr.2011.05.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toma-Jonik A, Widlak W, Korfanty J, Cichon T, Smolarczyk R, Gogler-Piglowska A, Widlak P, Vydra N. 2015. Active heat shock transcription factor 1 supports migration of the melanoma cells via vinculin down-regulation. Cell. Signal. 27, 394–401. ( 10.1016/j.cellsig.2014.11.029) [DOI] [PubMed] [Google Scholar]
- 46.Heimberger T, et al. 2013. The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br. J. Haematol. 160, 465–476. ( 10.1111/bjh.12164) [DOI] [PubMed] [Google Scholar]
- 47.Chuma M, et al. 2014. Heat shock factor 1 accelerates hepatocellular carcinoma development by activating nuclear factor-κB/mitogen-activated protein kinase. Carcinogenesis 35, 272–281. ( 10.1093/carcin/bgt343) [DOI] [PubMed] [Google Scholar]
- 48.Dudeja V, Chugh RK, Sangwan V, Skube SJ, Mujumdar NR, Antonoff MB, Dawra RK, Vickers SM, Saluja AK. 2011. Prosurvival role of heat shock factor 1 in the pathogenesis of pancreatobiliary tumors. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G948–G955. ( 10.1152/ajpgi.00346.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjork JK, Akerfelt M, Joutsen J, Puustinen MC, Cheng F, Sistonen L, Nees M. 2016. Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene 35, 1770–1784. ( 10.1038/onc.2015.241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin X, Eroglu B, Cho W, Yamaguchi Y, Moskophidis D, Mivechi NF. 2012. Inactivation of heat shock factor Hsf4 induces cellular senescence and suppresses tumorigenesis in vivo. Mol. Cancer Res. 10, 523–534. ( 10.1158/1541-7786.MCR-11-0530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendillo ML, et al. 2012. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562. ( 10.1016/j.cell.2012.06.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guettouche T, Boellmann F, Lane WS, Voellmy R. 2005. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 6, 4 ( 10.1186/1471-2091-6-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. 2014. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851. ( 10.1038/nrd4389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhillon AS, Hagan S, Rath O, Kolch W. 2007. MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. ( 10.1038/sj.onc.1210421) [DOI] [PubMed] [Google Scholar]
- 55.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. 2011. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. ( 10.1038/nrc3106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Grammatikakis N, Siganou A, Calderwood SK. 2003. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3ɛ binding, and cytoplasmic sequestration of heat shock factor 1. Mol. Cell. Biol. 23, 6013–6026. ( 10.1128/MCB.23.17.6013-6026.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li D, Yallowitz A, Ozog L, Marchenko N. 2014. A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis. 5, e1194 ( 10.1038/cddis.2014.158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chou SD, Prince T, Gong J, Calderwood SK. 2012. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS ONE 7, e39679 ( 10.1371/journal.pone.0039679) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zoncu R, Efeyan A, Sabatini DM. 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35. ( 10.1038/nrm3025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wander SA, Hennessy BT, Slingerland JM. 2011. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J. Clin. Invest. 121, 1231–1241. ( 10.1172/JCI44145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Acquaviva J, He S, Sang J, Smith DL, Sequeira M, Zhang C, Bates RC, Proia DA. 2014. mTOR inhibition potentiates HSP90 inhibitor activity via cessation of HSP synthesis. Mol. Cancer Res. 12, 703–713. ( 10.1158/1541-7786.MCR-13-0605) [DOI] [PubMed] [Google Scholar]
- 62.Dayalan Naidu S, et al. 2016. Heat shock factor 1 is a substrate for p38 mitogen-activated protein kinases. Mol. Cell. Biol. 36, 2403–2417. ( 10.1128/MCB.00292-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hardie DG. 2015. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 33, 1–7. ( 10.1016/j.ceb.2014.09.004) [DOI] [PubMed] [Google Scholar]
- 64.Hardie DG. 2013. The LKB1-AMPK pathway—friend or foe in cancer? Cancer Cell 23, 131–132. ( 10.1016/j.ccr.2013.01.009) [DOI] [PubMed] [Google Scholar]
- 65.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. ( 10.1016/j.molcel.2008.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Y, et al. 2011. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388. ( 10.1016/j.cmet.2011.03.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. ( 10.1038/ncb2152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pernicova I, Korbonits M. 2014. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 10, 143–156. ( 10.1038/nrendo.2013.256) [DOI] [PubMed] [Google Scholar]
- 69.Jope RS, Yuskaitis CJ, Beurel E. 2007. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 32, 577–595. ( 10.1007/s11064-006-9128-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCubrey JA, et al. 2014. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 5, 2881–2911. ( 10.18632/oncotarget.2037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang CY. 2017. HSF1 phosphorylation by ERK/GSK3 suppresses RNF126 to sustain IGF-IIR expression for hypertension-induced cardiomyocyte hypertrophy. J. Cell. Physiol. ( 10.1002/jcp.25945) [DOI] [PubMed] [Google Scholar]
- 72.Hietakangas V, et al. 2003. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol. Cell. Biol. 23, 2953–2968. ( 10.1128/MCB.23.8.2953-2968.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dhanasekaran DN, Reddy EP. 2008. JNK signaling in apoptosis. Oncogene 27, 6245–6251. ( 10.1038/onc.2008.301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su KH, Dai C. 2017. mTORC1 senses stresses: coupling stress to proteostasis. Bioessays 39, 1600268 ( 10.1002/bies.201600268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bubici C, Papa S. 2014. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br. J. Pharmacol. 171, 24–37. ( 10.1111/bph.12432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parsons DW, et al. 2005. Colorectal cancer: mutations in a signalling pathway. Nature 436, 792 ( 10.1038/436792a) [DOI] [PubMed] [Google Scholar]
- 77.Greenman C, et al. 2007. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. ( 10.1038/nature05610) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dai R, Frejtag W, He B, Zhang Y, Mivechi NF. 2000. c-Jun NH2-terminal kinase targeting and phosphorylation of heat shock factor-1 suppress its transcriptional activity. J. Biol. Chem. 275, 18 210–18 218. ( 10.1074/jbc.M000958200) [DOI] [PubMed] [Google Scholar]
- 79.Su KH, Cao J, Tang Z, Dai S, He Y, Sampson SB, Benjamin IJ, Dai C. 2016. HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat. Cell Biol. 18, 527–539. ( 10.1038/ncb3335) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu B, Ragazzon B, Rizk-Rabin M, Bertherat J. 2012. Protein kinase A alterations in endocrine tumors. Horm. Metab. Res. 44, 741–748. ( 10.1055/s-0032-1316292) [DOI] [PubMed] [Google Scholar]
- 81.Naviglio S, et al. 2009. Protein kinase A as a biological target in cancer therapy. Expert Opin. Ther. Targets 13, 83–92. ( 10.1517/14728220802602349) [DOI] [PubMed] [Google Scholar]
- 82.Correa R, Salpea P, Stratakis CA. 2015. Carney complex: an update. Eur. J. Endocrinol. 173, M85–M97. ( 10.1530/EJE-15-0209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murshid A, Chou SD, Prince T, Zhang Y, Bharti A, Calderwood SK. 2010. Protein kinase A binds and activates heat shock factor 1. PLoS ONE 5, e13830 ( 10.1371/journal.pone.0013830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. 2000. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3β. Mol. Cell. Biol. 20, 9356–9363. ( 10.1128/MCB.20.24.9356-9363.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Vugt MA, Medema RH. 2005. Getting in and out of mitosis with polo-like kinase-1. Oncogene 24, 2844–2859. ( 10.1038/sj.onc.1208617) [DOI] [PubMed] [Google Scholar]
- 86.Strebhardt K, Ullrich A. 2006. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 6, 321–330. ( 10.1038/nrc1841) [DOI] [PubMed] [Google Scholar]
- 87.Kim SA, Yoon JH, Lee SH, Ahn SG. 2005. Polo-like kinase 1 phosphorylates heat shock transcription factor 1 and mediates its nuclear translocation during heat stress. J. Biol. Chem. 280, 12 653–12 657. ( 10.1074/jbc.M411908200) [DOI] [PubMed] [Google Scholar]
- 88.Kim EH, Lee YJ, Bae S, Lee JS, Kim J, Lee YS. 2009. Heat shock factor 1-mediated aneuploidy requires a defective function of p53. Cancer Res. 69, 9404–9412. ( 10.1158/0008-5472.CAN-09-1411) [DOI] [PubMed] [Google Scholar]
- 89.Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. 2009. Protein kinase CK2 in health and disease. CK2: a key player in cancer biology. Cell. Mol. Life Sci. 66, 1858–1867. ( 10.1007/s00018-009-9154-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kelliher MA, Seldin DC, Leder P. 1996. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIα. EMBO J. 15, 5160–5166. [PMC free article] [PubMed] [Google Scholar]
- 91.Landesman-Bollag E, Romieu-Mourez R, Song DH, Sonenshein GE, Cardiff RD, Seldin DC. 2001. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 20, 3247–3257. ( 10.1038/sj.onc.1204411) [DOI] [PubMed] [Google Scholar]
- 92.Soncin F, Zhang X, Chu B, Wang X, Asea A, Stevenson MA, Sacks DB, Calderwood SK. 2003. Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem. Biophys. Res. Commun. 303, 700–706. ( 10.1016/S0006-291X(03)00398-X) [DOI] [PubMed] [Google Scholar]
- 93.Asano Y, et al. 2016. IER5 generates a novel hypo-phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci. Rep. 6, 19174 ( 10.1038/srep19174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brooks CL, Gu W. 2009. How does SIRT1 affect metabolism, senescence and cancer? Nat. Rev. Cancer 9, 123–128. ( 10.1038/nrc2562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. 2008. Acetylation is indispensable for p53 activation. Cell 133, 612–626. ( 10.1016/j.cell.2008.03.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. 2001. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159. ( 10.1016/S0092-8674(01)00527-X) [DOI] [PubMed] [Google Scholar]
- 97.Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, Dai Y. 2012. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 31, 4619–4629. ( 10.1038/onc.2011.612) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang RH, et al. 2008. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312–323. ( 10.1016/j.ccr.2008.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Firestein R, et al. 2008. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3, e2020 ( 10.1371/journal.pone.0002020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. 2009. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323, 1063–1066. ( 10.1126/science.1165946) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Boyault C. et al. 2007. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 21, 2172–2181. ( 10.1101/gad.436407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aldana-Masangkay GI, Sakamoto KM. 2011. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 875824 ( 10.1155/2011/875824) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vera M, Pani B, Griffiths LA, Muchardt C, Abbott CM, Singer RH, Nudler E. 2014. The translation elongation factor eEF1A1 couples transcription to translation during heat shock response. Elife 3, e03164 ( 10.7554/eLife.03164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu Y, Mivechi NF. 2011. Promotion of heat shock factor Hsf1 degradation via adaptor protein filamin A-interacting protein 1-like (FILIP-1 L). J. Biol. Chem. 286, 31 397–31 408. ( 10.1074/jbc.M111.255851) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kwon M, Libutti SK. 2014. Filamin A interacting protein 1-like as a therapeutic target in cancer. Expert Opin Ther. Targets 18, 1435–1447. [DOI] [PubMed] [Google Scholar]
- 106.Welcker M, Clurman BE. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93. ( 10.1038/nrc2290) [DOI] [PubMed] [Google Scholar]
- 107.Gomez-Pastor R, et al. 2017. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington's disease. Nat. Commun. 8, 14405 ( 10.1038/ncomms14405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin X, Moskophidis D, Hu Y, Phillips A, Mivechi NF. 2009. Heat shock factor 1 deficiency via its downstream target gene αB-crystallin (Hspb5) impairs p53 degradation. J. Cell. Biochem. 107, 504–515. ( 10.1002/jcb.22151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.den Engelsman J, Keijsers V, de Jong WW, Boelens WC. 2003. The small heat-shock protein αB-crystallin promotes FBX4-dependent ubiquitination. J. Biol. Chem. 278, 4699–4704. ( 10.1074/jbc.M211403200) [DOI] [PubMed] [Google Scholar]
- 110.Zhang H, Koo CY, Stebbing J, Giamas G. 2013. The dual function of KSR1: a pseudokinase and beyond. Biochem. Soc. Trans. 41, 1078–1082. ( 10.1042/BST20130042) [DOI] [PubMed] [Google Scholar]
- 111.O'Callaghan-Sunol C, Sherman MY. 2006. Heat shock transcription factor (HSF1) plays a critical role in cell migration via maintaining MAP kinase signaling. Cell Cycle 5, 1431–1437. ( 10.4161/cc.5.13.2915) [DOI] [PubMed] [Google Scholar]
- 112.Sato S, Fujita N, Tsuruo T. 2000. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl Acad. Sci. USA 97, 10 832–10 837. ( 10.1073/pnas.170276797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ahsan A, et al. 2012. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 14, 670–677. ( 10.1593/neo.12986) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schulz R, Streller F, Scheel AH, Ruschoff J, Reinert MC, Dobbelstein M, Marchenko ND, Moll UM. 2014. HER2/ErbB2 activates HSF1 and thereby controls HSP90 clients including MIF in HER2-overexpressing breast cancer. Cell Death Dis. 5, e980 ( 10.1038/cddis.2013.508) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Knowles TP, Vendruscolo M, Dobson CM. 2014. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. ( 10.1038/nrm3810) [DOI] [PubMed] [Google Scholar]
- 116.Gehrmann M, Marienhagen J, Eichholtz-Wirth H, Fritz E, Ellwart J, Jaattela M, Zilch T, Multhoff G. 2005. Dual function of membrane-bound heat shock protein 70 (Hsp70), Bag-4, and Hsp40: protection against radiation-induced effects and target structure for natural killer cells. Cell Death Differ. 12, 38–51. ( 10.1038/sj.cdd.4401510) [DOI] [PubMed] [Google Scholar]
- 117.Nylandsted J, et al. 2004. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 200, 425–435. ( 10.1084/jem.20040531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barreau C, Paillard L, Osborne HB. 2005. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33, 7138–7150. ( 10.1093/nar/gki1012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Galban S, et al. 2008. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1α. Mol. Cell. Biol. 28, 93–107. ( 10.1128/MCB.00973-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Levy NS, Chung S, Furneaux H, Levy AP. 1998. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J. Biol. Chem. 273, 6417–6423. ( 10.1074/jbc.273.11.6417) [DOI] [PubMed] [Google Scholar]
- 121.Chou SD, Murshid A, Eguchi T, Gong J, Calderwood SK. 2015. HSF1 regulation of β-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 34, 2178–2188. ( 10.1038/onc.2014.177) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Y, et al. 2015. MicroRNA-135b, a HSF1 target, promotes tumor invasion and metastasis by regulating RECK and EVI5 in hepatocellular carcinoma. Oncotarget 6, 2421–2433. ( 10.18632/oncotarget.2965) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Koskas S, Decottignies A, Dufour S, Pezet M, Verdel A, Vourc'h C, Faure V. 2017. Heat shock factor 1 promotes TERRA transcription and telomere protection upon heat stress. Nucleic Acids Res. 45, 6321–6333. ( 10.1093/nar/gkx208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Artandi SE, DePinho RA. 2010. Telomeres and telomerase in cancer. Carcinogenesis 31, 9–18. ( 10.1093/carcin/bgp268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Scherz-Shouval R, et al. 2014. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158, 564–578. ( 10.1016/j.cell.2014.05.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. 2012. Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer 12, 89–103. ( 10.1038/nrc3205) [DOI] [PubMed] [Google Scholar]
- 127.Straussman R, et al. 2012. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504. ( 10.1038/nature11183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lancaster GI, Febbraio MA. 2005. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J. Biol. Chem. 280, 23 349–23 355. ( 10.1074/jbc.M502017200) [DOI] [PubMed] [Google Scholar]
- 129.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. 2000. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6, 435–442. ( 10.1038/74697) [DOI] [PubMed] [Google Scholar]
- 130.Takeuchi T, Suzuki M, Fujikake N, Popiel HA, Kikuchi H, Futaki S, Wada K, Nagai Y. 2015. Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl Acad. Sci. USA 112, E2497–E2506. ( 10.1073/pnas.1412651112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Y, Theriault JR, He H, Gong J, Calderwood SK. 2004. Expression of a dominant negative heat shock factor-1 construct inhibits aneuploidy in prostate carcinoma cells. J. Biol. Chem. 279, 32 651–32 659. ( 10.1074/jbc.M401475200) [DOI] [PubMed] [Google Scholar]
- 132.Dai C, Dai S, Cao J. 2012. Proteotoxic stress of cancer: implication of the heat-shock response in oncogenesis. J. Cell. Physiol. 227, 2982–2987. ( 10.1002/jcp.24017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wittrup A, Lieberman J. 2015. Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet. 16, 543–552. ( 10.1038/nrg3978) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nagai N, Nakai A, Nagata K. 1995. Quercetin suppresses heat shock response by down regulation of HSF1. Biochem. Biophys. Res. Commun. 208, 1099–1105. ( 10.1006/bbrc.1995.1447) [DOI] [PubMed] [Google Scholar]
- 135.Wang RE, Hunt CR, Chen J, Taylor JS. 2011. Biotinylated quercetin as an intrinsic photoaffinity proteomics probe for the identification of quercetin target proteins. Bioorg. Med. Chem. 19, 4710–4720. ( 10.1016/j.bmc.2011.07.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yokota S, Kitahara M, Nagata K. 2000. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 60, 2942–2948. [PubMed] [Google Scholar]
- 137.Westerheide SD, Kawahara TL, Orton K, Morimoto RI. 2006. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 281, 9616–9622. ( 10.1074/jbc.M512044200) [DOI] [PubMed] [Google Scholar]
- 138.Titov DV, et al. 2011. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 7, 182–188. ( 10.1038/nchembio.522) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, Han DC. 2011. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem. 286, 1737–1747. ( 10.1074/jbc.M110.179440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim JA, Lee S, Kim DE, Kim M, Kwon BM, Han DC. 2015. Fisetin, a dietary flavonoid, induces apoptosis of cancer cells by inhibiting HSF1 activity through blocking its binding to the hsp70 promoter. Carcinogenesis 36, 696–706. ( 10.1093/carcin/bgv045) [DOI] [PubMed] [Google Scholar]
- 141.Zaarur N, Gabai VL, Porco JA Jr, Calderwood S, Sherman MY. 2006. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 66, 1783–1791. ( 10.1158/0008-5472.CAN-05-3692) [DOI] [PubMed] [Google Scholar]
- 142.Santagata S, et al. 2013. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341, 1238303 ( 10.1126/science.1238303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Salamanca HH, Antonyak MA, Cerione RA, Shi H, Lis JT. 2014. Inhibiting heat shock factor 1 in human cancer cells with a potent RNA aptamer. PLoS ONE 9, e96330 ( 10.1371/journal.pone.0096330) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.