

Abstract
Influenza is a serious hazard to human health that causes hundreds of thousands of deaths annually. Though vaccines and current therapeutics can blunt some of the perilous impact of this viral infection, new treatments are needed due to the constantly evolving nature of this virus. Recently, our growing understanding of an essential influenza viral protein, PA, has led to the development of focused libraries of new small molecules that specifically target the active site of the PA influenza endonuclease, which we report here. Our overarching approach has been to proactively develop lead inhibitors that are less likely to rapidly develop clinical resistance by optimizing inhibitors that retain activity against induced resistant mutants. Here, we report details behind the discovery of new potent inhibitors of wild type and resistant mutant endonucleases along with their high-resolution co-crystal structure-activity relationships. These results add to our understanding of nuclease protein targets and potentially serve as starting points for a new therapeutic approach to the treatment of influenza.
Introduction
Influenza is an infectious disease associated with 500,000 deaths and 3–4 million severe illnesses annually worldwide, despite the availability of vaccines and antiviral drugs. During the 1918 ‘Spanish flu’ pandemic, it is estimated that between 20 and 40 million people died in just eight months1. Other major influenza pandemics in the 20th century include the 1957 ‘Asian flu,’ and the 1968 ‘Hong Kong flu’, with each resulting in approximately 1 million deaths2–4. In each case, 20–30% of the global population was infected within a year of the outbreak. One approach to rapidly addressing pandemics is to develop small molecule antivirals that have broad activity against all strains of influenza.
Most small molecule anti-influenza drugs currently on the market act as neuraminidase inhibitors (zanamivir, oseltamivir, peramivir) or target the M2-ion channel (amantadine, rimantadine)5. However, these targets, particularly the latter, are prone to rapid mutations that can confer antiviral resistance, due to the inability of the viral RNA dependent RNA polymerase (RdRp) to proofread during RNA replication. In fact, the World Health Organization’s Global Influenza Program reported that >99% of seasonal influenza A strains are now resistant to amantadine and rimantadine6. This has led to the search for new antiviral compounds that target other essential viral processes7. The influenza RdRp is itself an attractive drug target, because it is relatively slow to develop drug resistance, is conserved across genotypes, and is essential for viral replication. The influenza virus RdRp is a heterotrimer that includes the polymerase catalytic subunit (PB1), the ‘cap-binding’ subunit (PB2), and the endonuclease-containing (PA) subunit. The ‘cap-binding’ and endonuclease functionalities of RdRp work in concert to perform the essential ‘cap snatching’ of host mRNAs to generate primers for viral transcription8,9. In the last decade, our understanding of influenza viral RdRp has dramatically expanded through the elucidation of the high-resolution architecture of influenza endonuclease8,10 and, most recently, the unveiling of the complete RdRp heterotrimer structure by Cusack and coworkers11.
The importance of the RdRp to influenza virus viability has spawned a number of recent drug discovery efforts. Favipiravir, a broad-spectrum drug that targets numerous viral RdRps including influenza RdRp, was approved in Japan in 2014 for emergency use in the event of influenza pandemics, despite some significant concerns regarding this drug’s toxicity12. Also, a promising drug (VX-787)13 that targets the ‘cap-binding’ site of influenza RdRp is currently in advanced clinical trials14. However, no new influenza drug is currently available to the general population7.
The essential endonuclease domain within the PA subunit is a particularly attractive drug target. It has no eukaryotic homolog, so the potential for toxicity due to off-target effects is reduced for small molecules that target its active site9. Using the structure of the domain determined in isolation10,15 and in the context of the trimeric complex8,11 a number of groups, including ours, have successfully used structure-assisted approaches to develop potent inhibitors16–23. Our efforts build off of the foundational work of Tomassini and coworkers at Merck and their early report of 2,4-dioxobutanoic acid inhibitors containing a two-metal binding pharmacophore24–26. We have structurally characterized the binding mode of L-742,001 (1, Fig. 1), the most potent inhibitor in this class,17 and shown that it engages highly conserved active site residues. Our recent demonstration that L-742,001 does not readily generate resistance mutations in the RdRp27 has provided fresh impetus to the development of endonuclease inhibitors and this effort has recently been exhaustively reviewed28. Figure 1 shows five endonuclease inhibitors (1, 2, 3, 4, and 5) that have been reported to show antiviral activity in vitro.
Figure 1.

Compounds targeting the two-metal nuclease active site, including the FDA approved HIV integrase inhibitor raltegravir (Isentress)35, along with the first influenza lead compound in this class, L-742,001 (1)46, and several recently developed experimental influenza endonuclease inhibitors (2 17, 3 21, 4 47, and 5 48). The general structure 6 shows the structure of the active inhibitors reported in this publication.
Our overarching approach has been to apply structure-based design, while optimizing inhibitors that retain activity against induced resistant mutants, in order to proactively develop lead inhibitors that are less likely to rapidly develop clinical resistance27. An important aspect of this work is the discovery of inhibitors that are able to potently engage previously identified sites that are conserved in resistant mutations. Here, we describe the further optimization of such a series of new endonuclease inhibitors that are an extension of our earlier published studies17,29. The chemical scaffold for our studies is 4,5-dihydroxypyrimidine-6-carboxamide (DHPC), which was originally developed by Summa and coworkers (Merck, Italy) into the first-in-class HIV two-metal integrase inhibitor raltegravir (Isentriss)30 (Fig. 1). We have previously reported active influenza endonuclease inhibitory compounds based on this DHPC scaffold17,31, such as 2, and report here the identification of more potent inhibitors through iterative cycles of library synthesis, FP binding assays, FRET nuclease assays, cell-based plaque assays and inspection of inhibitor/endonuclease co-crystal structures. An important feature in the discovery of new antivirals is the ability to show potency in the face of resistance mutations. It has previously been reported that T20A mutations in influenza virus endonuclease can result from viral selection32 using the Merck endonuclease inhibitor33 (L-742,001, compound 1, Fig. 1) and we have recently reported the mutagenesis and selection of novel compound 1 resistant mutations that also contain the T20A background27. For our approach, we utilized the sequence background of a wild type strain (A/California/04/2009 H1N1) that has the T20A sequence. This construct was used in our biochemical assays (FP and FRET) and in our crystal structures, so in the following discussion we refer to this as the ‘wild type’ strain. Thus, our approach starts with endonucleases that incorporate single point mutation or two point mutations so that we can identify inhibitors that can retain significant activity even when these two known resistant mutations are combined in the influenza viral endonuclease construct. We focused our attention on the E119D resistance mutation, since this mutation showed the greatest resistance in our previous studies27. In the following sections, we discuss our exploration and optimization chemistry in the context of a wide range of inhibitor/protein co-crystal structures which provides insight into the structure-activity relationships.
Results
First Generation Analogs
Subsequent to our previous reports17,34 we prepared and screened focused libraries that included a diverse set of optically pure carboxylate analogs. Some of these carboxylates (see Supplementary Scheme 6) for example 6a: R1 = 2-chlorophenoxymethyl; R = OH) showed potent PA N-terminal construct (PAN) binding (Ki < 10 nM) in our fluorescent polarization (FP) assay17 (which is near the lower limit of this assay) and good activity in the FRET nuclease assay (IC50 = 45 nM) For comparison, L-742,001 shows a Ki of 346 nM in the FP assay under the same assay conditions. For carboxylate analogs (e.g. general structure 6R = OH: 6b: R1 = naphthalen-1-oxymethyl, 6c: R1 = 4-chorobenzyl, and 6d: R1 = phenoxymethyl), we generally observed the DHPC warhead engaging the two-metal center, as expected, and the R1 group stacking onto Tyr24 in the crystal structures of the inhibitors in the PAN binding site (Supplementary Results, Supplementary Fig. S1). This mimics the binding of a nucleotide base in the native RNA substrate, which may account for the potency of these inhibitors20,27. As anticipated for a carboxylate, no significant activity was observed in the viral plaque inhibition assay at 50 μM. These observations led us to initiate the synthesis of a series of diamide derivatives using previously published chemistry29,35–37 as shown in Supplementary Scheme 1–5. Such derivatives allowed us to introduce a second functional group at the R2 position to survey other binding opportunities within the endonuclease active site cavity. The structures and fluorescent binding data of a selection of the 1st generation diamide analogs are shown in Fig. 2: Table A. In addition to the FP data in Table A, the biochemical inhibition of nuclease activity was confirmed for the most important compounds using the previously reported FRET assay27 (see Supplementary Table S1).
Figure 2.

Tables A–D. Fluorescent polarization structure-activity relationships of inhibitors for wild type PA organized by small molecule 2D structural type. Table A. Initial diamides. Table B. 3,5-Dichloroisonicotinyl and N-methyl derivatives. Table C. Linker heteroatom exploration. Table D. R2 optimization. The fluorescent polarization assay conditions have previously been reported17,27.
Initial diamide compounds such as 6e (Fig. 1, general structure 6: R1 = phenoxyacetyl, R2 = 2-methoxy ethylamine) showed relatively weak activity in the FP assay (Ki = 1.6 μM), but compounds such as 6f (Fig. 1, general structure 6: R1 = 2,6-dichlorobenzoyl, R2 = 2-phenethylamine) showed improved binding (Ki = 0.36 μM) for the amide series, although the potency was generally not as high as the corresponding carboxylate analogs. In order to determine the optimal R1 functionality for the pyrrolidine amide, a variety of compounds with different side chains were synthesized, while the R2 phenethyl side chain was held constant. Based on the potency of 6f, a number of similar R1 aryl groups, 7b-7e, were tested (see Fig. 2: Table A).
In this series, the identity and locations of the halogens were changed and a heteroatom was added to help improve solubility. Overall, the presence of a nitrogen heterocycle improved potency in the FP assay and the position and number of halogens was found to be extremely important for potency (as seen in 7d vs. 7e). The difluoro compound (7b) was observed to be significantly more potent than the initial dichloro compound (7a), so the crystal structures of these compounds in the PAN active site were obtained to investigate their modes of binding (see Fig. 3A, Fig. S1b). Although the protein structures, including the two metal binding sites, are nearly identical, the R1 group of 7b adopts both cis (44%) and trans (56%) amide bond conformations; while 7a only adopts the trans pyrrolidine amide conformation. For simplicity we will refer to these conformations in the following discussion simply as cis and trans, respectively. In the cis conformation of 7b the difluorophenyl group stacks against the metal-chelating heterocylic core, while in the trans conformations of 7a and 7b, the R1 aryl group extends into a pocket created by Tyr24, Glu26, and Lys34. Regardless of the cis or trans conformation, the R2 2-phenethyl stacks against Tyr24. While this interaction is clearly important to the potency of these inhibitors, we wanted to see if we could improve the tightness of this interaction or pick up any new interactions in this binding pocket, by modification of the R2 group.
Figure 3.

The wild type PAN high affinity bound conformations of inhibitors from high-resolution co-crystal structures. (A) The PAN inhibitor co-crystal structures of 7b (left panel, which shows the cis (44%) and trans (56%) bound conformations) and 7a (right panel, which shows the trans bound conformation). (B) The bound conformation of 8f (left panel) in the PAN active site and the bound conformation of 8e (right panel) in the wild type PAN construct showing potential hydrogen-bond interactions with crystallographic water molecules, active-site side chains, and a main chain interaction. Both structures feature H-bond donation from Lys34 to the pyrrolidine amide carbonyl (3.4 Å and 3.3 Å, respectively), and numerous interactions near the two-metal site, which are common to all DHPC inhibitors. In 8e, note the main chain (Leu106) H-bond (3.1 Å) to one sulfone oxygen and the H-bond (3.0 Å) donation from Tyr24 the other sulfone oxygen. Mn2++ ions are shown as violet spheres and Mg2++ as green spheres. See Supporting Figure S1b for electron density maps for examples of these and other relevant structures.
Second Generation Diamide Analogs
We next chose to explore alternatives to the R2 phenethyl group by preparing the analogs shown in Fig. 2: Table B, while holding the R1 group constant as 3,5-dichloroisonicotinyl to give compounds of the general structure 8. The goal of this stage of exploration was to identify higher affinity compounds by the introduction of conformational constraints and groups that could enhance aqueous solubility. First, we explored conformational constraints of the phenethyl group, which led to the synthesis of indanyl derivatives 8f and 8h, which were found to be potent endonuclease binders. 8f was one of our most potent inhibitors (Ki = 9 nM) and so we obtained a crystal structure of it in complex with the wild type PAN complex (Fig. 3B). 8f adopts the cis conformation observed in 7b, with a few key differences in the R1 positioning: the pyrrolidine is now positioned in a way that places the R1 carbonyl close enough to Lys34 (3.4 Å) to pick up a hydrogen-bond. Additionally, due to the conformational constraint, the R2 phenyl ring is now in a position to stack directly underneath Tyr24, creating a tight binding interaction.
We also explored the alkylation of the 5,6-dihydroxypyrimidine amide nitrogen, since another consideration in the development of orally active compounds is the reduction of the number of hydrogen-bond donors (as part of the rule of five)38. As shown by compounds 8b and 8g, the presence of the methyl group severely impacts the potency of these compounds, resulting in a dramatic loss of binding affinity as measured by FP when compared to the corresponding unmethylated analogs 7c and 8f. This is consistent with our observation of the co-crystal structures in which the amide NH group interacts with the OH group of Tyr130.
Notably, the R2 sulfone derivative 8e (Fig. 3B and Fig. S1b), which was introduced to improve solubility, showed very potent binding with Ki = 19 nM. The co-crystal structure of 8e revealed a remarkable stacked ‘sandwich’ structure in which the R1 and R2 groups stack on opposite surfaces of the warhead. This bound conformation may be driven, in part, by the energetic benefit of the two sulfone oxygens, which accept hydrogen bonds from the OH group of Tyr24 (3.0 Å) and the backbone amide of Leu106 (3.1 Å). In order to make the hydrogen bond with the sulfone oxygen, Tyr24 moves closer to the warhead than in similar structures such as 7a or 8f. Additionally, the cis conformation of this inhibitor places the pyrrolidine amide in a position to pick up a hydrogen bond between the carbonyl and Lys34 (3.3 Å).
In order to evaluate the antiviral activity relationships of these inhibitors on viral replication in living cells, we also performed plaque inhibition assays on selected FP active compounds from Fig. 2: Tables A and B (also see Supplementary Fig. S2A) against wild type A/Puerto Rico/8/34 (H1N1)27. Inhibition of wild type virus growth in the micromolar range was seen for almost all of these active inhibitors apart from 8e, which showed no effect. With an FP Ki value in the low nanomolar range but no inhibition in plaque assay, it is likely that 8e has particularly poor transport into cells. However, we were not discouraged from using the sulfone group as we hoped to improve the cell permeability through the use of more polar R1 groups. The results up to this point, particularly the success of the indanyl and sulfonyl R2 groups, were used to guide the design of the next iteration of endonuclease inhibitors.
Aryl Linker Exploration
In order to expand the diversity of options for lead optimization, we next sought to explore R1 groups with similarities to our initial carboxylate hits, such as 6d. For this series we chose to alter the aryl linker atom, X1, and the halogen substituent X2 (Fig. 2: Table C). We also hoped that the addition of an electron donor to the R2 phenyl ring would increase the affinity of this group for Tyr24, based on the interactions that we observed with compounds such as 7a, so we tested both R4 = Ph and R4 = OPh. The relevant compound library synthesized and their FP binding affinities are summarized in Fig. 2: Table C. The following general trends were observed: the optimal X1 linker atom is sulfur, chlorine is preferred over fluorine as an X2 substituent, and a phenoxy group is preferred over a phenyl group in the R2 substituent. Thus compound 9k, which combines all of these features, shows the most potent binding for this series with a Ki = 151 nM.
In regard to the binding conformations of similar analogs, we found that the co-crystal structures of inhibitors that differ only by a single atom type (Cl vs F, or CH2 vs O vs S) show very similar bound conformations to each other. For example, the phenoxy analogs with X2 = Cl and different X1 groups, 9b (X1 = CH2), 9f (X1 = O), and 9k (X1 = S) all have a cis pyrrolidine amide bond, which orients the R1 group beneath the heterocyclic core, and also have a similar tight stacking interaction of the phenoxy group with Tyr24 (see Supplementary Fig. S3). Similarly, the change from X2 = Cl to X2 = F does not seem to have any impact on the binding of these compounds, as seen in the similar binding conformations of 9f (X1 = O, X2 = Cl) and 9h (X1 = O, X2 = F). In contrast, the inhibitor-protein crystal structure of the less potent phenethyl analog 9e (X1 = O, X2 = Cl) shows a very different protein-bound conformation when compared to the phenoxy analogs (see Supplementary Fig. S3). In this case, the trans conformation is adopted and the phenyl groups from R1 and R2 both interact with Tyr24.
Aryl Linker Optimization
The ultimate goal in the development of an endonuclease inhibitor is the identification of a potent, selective and orally active agent; we therefore focused on the introduction of functionality that should improve the aqueous solubility of our compounds while maintaining potent target (PAN) binding. As shown in Fig. 2: Table C, we prepared a set of compounds that combine the features found in our most potent compounds from Fig. 2: Table C, while incorporating strategic modifications to the R2 group designed to enhance solubility. Due to the potency of 9f and 9k, we first looked at making compounds with groups that might have similar tight stacking interactions with Tyr24. This led us to replace the oxygen with sulfur, which decreased the potency. However, a single ‘oxidation’ of 10a yielded the sulfoxide 10c, which is significantly more potent in FP binding and should improve solubility. Further ‘oxidation’ produced the corresponding sulfone derivative, 10d, which showed additional improvement in binding potency (see Fig. 2: Table C). When a methoxy group was added to the phenyl ring of the sulfone to give 10f, the potency continued to improve. A similar trend was also observed for the X1 = S derivatives (i.e. 9k, 10b, and 10e) which led to the discovery of 10g, which reached the lower limit of our FP assay (~5 nM). This ‘oxidation’ also had a significant impact on the bound conformation of the inhibitor (see Fig. 3A and Fig. S1c). Instead of stacking with Tyr24 as in 9k, the phenyl ring in 10e stacks above the warhead, in a similar position to our previous sulfone compound 8e. The two bound conformations of 10e, which are present in equal amounts, have nearly identical placement of the R2 group and the warhead with the same new hydrogen bonds from Tyr24 and the backbone of Leu106 to the sulfone oxygens. However, in the cis conformation, the R1 aryl group stacks beneath the warhead, while in the trans conformation, it stacks with Tyr24. This allows the cis conformation to pick up a hydrogen bond with Lys34 and the R1 carbonyl, and the trans conformation to obtain a favorable interaction between the aryl chloride and Glu26.
We next tested compounds with conformational constraints, due to our success with these groups when R1 = 3,5-dichloroisonicotinyl (e.g. 8f). In this case, the 2-indanyl analogs 10h and 10i were not as potent as the conformationally unconstrained phenethyl analogs. However, when two methoxy groups were added to the indane (10j) the binding activity improved significantly. Because of the dramatic improvement in potency, we obtained the crystal structures to see if there was any change in the binding modes of the inhibitors. We found that 10j (Fig. 4B) binds the endonuclease in a very similar fashion to 9e, with a trans configuration and the R1 and R2 groups sharing the interaction with Tyr24. In this structure, the R1 carbonyl has a close interaction with Lys34 (2.7 Å) and the R1 chlorine has an interaction with Glu26. In contrast, the less potent inhibitors with dihydrobenzofuran (10k) or indane (10i) substituents in R2, adopt a cis conformation with the R1 aryl chloride stacking underneath the warhead and the R2 group stacking against Tyr24, in a similar position to the R1 aryl chloride in 10j. When compared to 8f, these two inhibitors have identical positioning of the warhead and the R2 indanyl groups, but they differ in the positioning of the pyrrolidine ring, causing the loss of the carbonyl-Lys34 interaction that was found in 8f. As a part of our effort to improve solubility, we also tested pyridine analogs 10m and 10n, which showed improved potency over the corresponding phenyl analog 9g.
Figure 4.

Examples of some aryl linker bound conformations with wild type PA. (A) A depiction of the co-crystal structure showing two binding modes of 10e with comparable binding energies in the wild type PAN; showing active-site side-chains Tyr111 on the left and Tyr24 on the right. (B) Top Panel: Overlay of the bound conformation of conformationally constrained phenethylamine analogs 10i (tan) and 10j (cyan). Bottom Panel: Overlay of 10j (cyan) with 10k (purple).
Structure-activity Relationships with the Point Mutant E119D and Comparison of Wild Type and E119D Mutant Inhibitor Co-crystal Structures
We also chose to examine the crystal structures of various inhibitors in complex with the E119D mutant. Overall, the impact of this point mutation on the bound conformations of our inhibitors is highly variable. For example, two of our sulfone inhibitors (8e and 10e) respond in different ways to the mutation. In the case of 8e (Supplementary Fig. S4), which has a cis “sandwich” structure, only a small change in the pyrrolidine ring was observed in the complex with the E119D mutant, while the R1 aryl ring and the R2 phenyl sulfone remained in the same position. For 10e (Fig. 5 and Fig S1d), which has a more flexible R1 group, the mutant has two conformations that both differ from the wild type and are present in nearly equal amounts (cis = 49%, inverted = 51%). In the cis wild type and mutant conformations, the R2 group maintains its position and the “sandwich” structure is adopted, but the 2-chloro aryl group in the mutant is flipped so that the chlorine points towards the R1 carbonyl, picking up a favorable intramolecular interaction. This cis conformation with the chlorine-carbonyl interaction is not found in any of the wild type structures we obtained. The other conformation adopted by the 10e mutant complex is very unusual, with the R1 and R2 groups in inverted positions: the R2 sulfone stacks underneath the warhead and the R1 aryl chloride stacks above the warhead.
Figure 5.
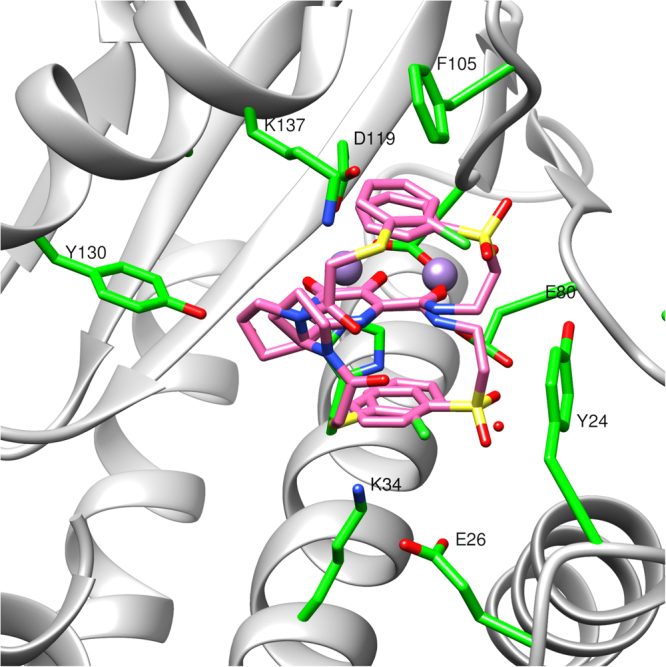
The bound conformations (cis = 49%, inverted = 51%) of 10e in complex with the E119D mutant of PAN construct. The cis mutant conformation includes a chloro carbonyl intramolecular interaction that is not present in the wild type structure with the same inhibitor.
Another set of compounds that undergo significant changes in the mutant structures are 9k and 9b (Fig. 6 and Fig. S1e). In the wild type and the mutant structures for these compounds, the R2 phenoxy group maintains the same position, stacking with Tyr24, and the R1 2-chloro aryl group adopts the cis conformation and stacks underneath the warhead. However, for the wild type structures, the 2-chloro group is pointed towards the R2 substituent, while in the mutant structures the 2-chloro group is directed away from R2. For one of our most potent inhibitors, 10j (Fig. 2: Table D), the mutant and the wild type structures both have the R2 dimethoxy indane group and the R1 2-chloro aryl group in similar positions, interacting with Tyr24 (see Supplementary Fig. 5). The most significant difference between these structures lies in the position of Lys34: only the wild type inhibitor is in a position to pick up a key interaction between the R1 carbonyl and Lys34.
Figure 6.

A comparison of bound high affinity wild type versus E119D inhibitor conformations. Left Panel: The co-crystal structure of 9k (tan) overlaid with the conformation 9b (cyan) in the wild type PAN construct. Right Panel: The conformation of 9b (cyan) overlaid with the conformation of 9k (tan) in the E119D mutant. Note that the amide bond with the 2-Cl phenyl group is cis in both the wild type and mutant structures.
Comparison of Wild Type Versus E119D Mutant Endonuclease FP Binding and Cell-based Activity of Inhibitors
We also obtained FP binding data for many of the inhibitors against the E119D mutant (see Supplementary Table S2). As seen by inspection of this table, the affinity of these inhibitors is somewhat reduced, as expected27, but the activity ranking of FP potency is very similar for both wild type and mutant. In the mutant FP binding assay, our most potent compounds from the wild type assay including sulfone compounds 8e, 10e, and 9k and indane 8f retain their potency. In contrast, indanyl derivatives 10i and 10k and phenyl derivative 9e see the largest drops in potency. We also tested many of the compounds from Fig. 2: Table C and Fig. 2: Table D in the plaque inhibition assay against both wild type A/Puerto Rico/8/34 (H1N1) and the endonuclease mutant bearing an E119D PA substitution27 (Supplementary Fig. S2B). In the wild type assay, most of the compounds showed IC50’s in the low micromolar range (IC50 = 6.6–38.0 µM). Notably, some compounds such as 9k and 9b with relatively high FP Ki values (0.151 µM and 0.376 µM respectively) show similar plaque IC50 values (7.2 µM and 6.6 µM respectively) to compounds with lower FP Ki values, such as 10g (FP Ki = < 0.005 µM, plaque IC50 = 9.1 µM). This may indicate that 9k and 9b have better cell permeability than 10g. These results emphasize the importance of the early integration of cell-based assays into lead optimization, rather than the sole reliance on cell-free biochemical assays for this purpose39. Overall linker optimization targeting increased solubility generally led to improvements in inhibition of viral growth, especially for compounds like 9k and 10j, in line with the results of the FP data and crystal structures. Generally, activity against the E119D mutant was reduced in comparison to the wild type, but at comparable levels to L-742,001.
Discussion
First and Second Generation Endonuclease inhibitors
The initial diamide inhibitor FP binding data (Fig. 2: Table A) showed the importance of the aryl ortho halogens and, independently, the benefit of having heteroatoms in the aryl ring for tight binding. Aryl heterocycles may also improve aqueous solubility, which is one of our key design considerations since water solubility is an important property that is associated with good oral activity in drug leads40.
For this type of inhibitor, with R1 = aryl and R2 = 2-phenethyl, we observed that the two bound conformations of 7a and 7b (Fig. 3A) appear to be representative of the most common binding modes for the series. However, it is not clear what causes an inhibitor to adopt the cis conformation versus the trans conformation, and in general, there does not appear to be a significant advantage to binding in one conformation over the other. These results starkly demonstrate that a modest change (e.g. from chlorine to fluorine) can have a dramatic impact on the small molecule bound conformation and that there is a need for caution when assuming that an analog series share a similar and common binding mode, which is the usual assumption in medicinal chemistry when structural data are not available.
When we moved to other R2 groups, we observed that the length of the linker to the phenyl ring from the amide is very important; since shorter (e.g. 8a) chains generally showed lower potency than their counterparts with 2–3 atom linkers. This suggests that a 2–3 atom linker is likely the ideal range to orient the phenyl ring so that it can interact favorably with Tyr24. The sulfone R2 group was an interesting discovery due to its atypical ‘sandwich’ structure and its high potency. This increase in potency is likely due to the new hydrogen bonding interactions the sulfone oxygens are able to achieve in the binding site with the backbone amide of Leu106 and with the OH of Tyr24. It should be noted that the sulfone is analogous to a phosphate group and its mode of binding could therefore reveal how the phosphate of the upstream nucleotide of the mRNA substrate binds adjacent to the active site.
Aryl Linker Optimization
Examining a selection of crystal structures from Fig. 2: Tables C and D, we found that most of the inhibitors with this type of R1 group, with the exception of the sulfones, adopt binding modes similar to either 9b or 9e (Supplementary Fig. S2). For compounds in which the R2 phenyl group interacts weakly with Tyr24, such as 9e and 10j, the R1 amide bond adopts a trans conformation, which allows the R1 phenyl group to interact with Tyr24 as well. In contrast to this observation we noted that if the stacking interaction with R2 is tight, as for 9b or 10i, the R1 group will adopt a cis conformation and the R1 phenyl ring will stack beneath the warhead. The exception to this is the sulfone in this series, 10e, which adopts two significantly different binding modes (Fig. 5A) that are more similar to 8e, with the sulfone stacked above the warhead. For the trans conformation, the R1 group is still in an identical position to 9e; but for the cis conformation, the R1 group is much farther underneath the warhead than in 9b, almost directly underneath the R2 phenyl sulfone.
As previously discussed, we discovered that the most potent R1 linker was one with X1 = S and X2 = Cl, even though there is often no significant difference between crystal structures with this R1 group and other similar R1 groups (see 9b vs 9f vs 9h vs 9k, Supplementary Fig. S2). The most potent binding inhibitors in this compound series, 10e, 10 g and 10j (Fig. 2: Table D), all contain sulfur and chlorine atoms at the X1 and X2 positions of the R1 group, respectively. Also, the sulfone group once again seems to be extremely favorable in terms of binding affinity.
Structure-activity Relationships with the Point Mutant E119D and Comparison of Wild Type and E119D Mutant Inhibitor Co-crystal Structures
Previous studies identified an active-site residue mutation (T20A) in PA that was induced under pressure of an inhibitor32 and is also observed in some wild type influenza strains27. We reported that L-742,001 pressure combined with random mutagenesis was required to generate the novel resistant mutation E119D, which displayed significant resistance to L-742,00127. Though we found it possible to induce this mutation in the active site of the RdRp, less than 1% of sequenced influenza viruses contain the resistant residue naturally27. The E119D mutation is directly adjacent to the two-metal site of the endonuclease and we expected a large effect on the binding modes of inhibitors since this mutation has been shown to affect the binding of mononucleotides27. In order to design inhibitors that can regain activity against influenza strains with this mutation, we focused on exploiting the pocket created by the highly conserved Tyr24, Glu26, and Lys34 residues, which form key interactions with the bound mononucleotide and are therefore may be less likely to mutate under clinical drug pressure.
In summary, we have synthesized and tested a number of focused 2-(pyrrolidin-2-yl)-5,6-dihydroxypyrimidine-4-carboxamide libraries in a structure-guided fashion and obtained compounds with potent binding activity that also show strong biochemical nuclease inhibition and antiviral activity in cells in vitro. Several inhibitors have been identified that show >40 fold increase in the binding Ki when compared to the well-studied Merck inhibitor, L-742,00124. The approach we followed has been to proactively develop lead inhibitors that are less likely to quickly develop clinical resistance by selecting compounds that retain good activity against artificially induced resistant mutant proteins and mutant virus in vitro 27. We have also developed an improved understanding of the molecular determinants of inhibition for both the wild type and E119D mutant inhibitor resistant influenza endonucleases while we explored several lead chemotypes and pharmacophores that interact with conserved residues in the active site of PA. In the course of this work we have also observed unique binding conformations of small molecule inhibitors bound to the endonuclease constructs and fortuitously discovered several new types of tight-binding interactions. We believe that the results presented in these studies will be useful for other members of the scientific community involved in the discovery of improved inhibitors of endonuclease and other homologous targets. Additionally, these new inhibitors together with the associated atomic resolution 3D structure-activity data, may dramatically facilitate the development of an important new therapeutic approach to the treatment of influenza.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Fluorescence Polarization Binding Assay
Wild type PAN protein and E119D-PAN ΔLoop were used to obtain the dissociation constants of the compounds. The reagents, experimental protocol and data analysis were performed as previously reported17,27.
PAN nuclease activity assay
The nuclease activity assay was slightly modified from previously reported methods17,27. The fluorescently-labeled synthetic oligonucleotide 5′-Cy5-GAATACTCAAGCTATGCATC-3IAbRQSp was obtained from Integrated DNA Technologies (Coralville, IA) and dissolved in 10 mM Tris-HCl, pH 7.0. 2 μl of 4 µM substrates (10x with 400 nM final concentration) were added to a 384-well, black, low volume microplate (Greiner #784076). Subsequently, 2 µl compounds in DMSO at 10x final concentrations were added to the plate. The reactions were started by adding 31.25 nM wild type PAN ΔLoop protein (1.25x with 25 nM final concentration) in 16 μl per well master mix. The master mix contained 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10 mM β-mercaptoethanol, 2.5 mM MnCl2, and 0.25 mg/mL bovine serum albumin. Plates were covered with clear adhesive film and the fluorescence was monitored using an EnVision Multilabel reader (Perkin-Elmer, Waltham, MA) at 37 °C by exciting Fluorescein at 485 ± 7 nm (Perkin-Elmer filter#102), and monitoring fluorescence intensity at 535 ± 12.5 nm (Perkin Elmer filter#206). The plate was read every minute for 60 minutes and signal gain was determined by scanning the plate for the strongest signal. All assays were carried out in triplicate. Relative fluorescence units (RFU) between 10 to 60 minutes were fitted with linear regression to obtain the reaction rate. Reaction rates were averaged and plotted against the corresponding compound concentration and fitted with four parameter logistic curve to obtain IC50.
Cloning, expression and purification of PAN endonuclease domain
PAN endonuclease domain from A/California/04/2009 H1N1 (wild type PAN), PAN domain with a loop deletion (PAN ΔLoop, residues 51–72 replaced with a GGS linker), and E119D-PAN ΔLoop were cloned in pET52b vector with a cleavable C-Terminal His-tag and transformed into BL21(DE3). The protein was expressed in LB medium overnight at 18 °C after induction at an OD600 ~0.8 with 0.2 mM isopropyl-β-thiogalactopyranoside (IPTG). The protein was purified from cell lysates by HisTrap affinity chromatography and the His-tag was removed by digestion with thrombin. The protein was further purified by gel filtration using a Superdex 75 size-exclusion chromatography column in 20 mM Tris pH 8.0, 150 mM NaCl and 1 mM TCEP. The same domain was also cloned in pET28a vector with a N-Terminal His-tag, purified similarly in a buffer containing 20 mM HEPES pH 7.8, 200 mM NaCl, 2 mM TCEP and 1 mM EDTA, but without cleaving the His-tag and used for co-crystallization of the protein in complex with various inhibitors.
Crystallization of PAN endonuclease domain with inhibitors
The proteins from either construct were concentrated to ~10 mg/ml for crystallization. For crystal structure determination, the proteins were either pre-incubated with 1.2 mM inhibitor or crystals of the holo-enzyme were soaked in the crystallization solution containing 3 mM inhibitor overnight. Crystals were obtained by using hanging drop vapor diffusion method. 2 μl drops were set up by mixing protein and crystallization solution in 1:1 or 2:1 ratio on a cover slide that hangs over 500 μl well solution. Crystals appeared in 1–3 days and grew to maximum in 4–6 days. For the X-ray diffraction experiment, crystals were cryo-protected in crystallization solution containing 25% ethylene glycol (PEG conditions) and 30% glycerol (Ammonium sulfate condition), or a mixture of crystallization solution, 3.4 M Sodium malonate (pH 7.0) and glycerol in 1:1:1 ratio (Succinic acid condition), and flash frozen in liquid nitrogen. Detailed information on crystallization solution for each protein-inhibitor complex is provided in Supplementary Table S3.
X-Ray data collection and refinement
The X-ray diffraction data were collected at a wavelength of 1.000, 0.97903 or 0.9789 Å at a temperature of 100 Kelvin at the 22-ID and 22-BM beam lines maintained by SERCAT (Southeast Regional Collaborative Access Team) at the Advanced Photon Source, Argonne National Laboratory, USA. The data were indexed, integrated and scaled using the HKL2000 suite of programs41. After molecular replacement with Phaser42, refinement and model building was completed using Refmac43 or Phenix44 and Coot45. The details of the data collection, refinement statistics and the PDB IDs of the crystal structures deposited at the wwPDB are given in Supplementary Tables S4–S12.
Compound Library Synthesis
Compounds were prepared as previously reported29. Detailed experimental procedures and characterization data can be found in the Supplementary Materials file.
Plaque reduction assay
Egg propagated stocks of influenza virus A/Puerto Rico/8/34 (H1N1), or a reverse genetics version containing the E119D PA mutant, were diluted to 100 pfu/ml in infection medium (minimum essential medium (MEM; Invitrogen), 4% BSA, 100 units/ml penicillin, 100 units/ml streptomycin, 0.25 µg/ml Amphotericin B, 1x MEM vitamins, 2 mM l-glutamine) and 1.0 ml inoculated onto confluent MDCK (ATCC; CCL-34) cells in 6-well plates. Inoculations were performed for 1 hour at 37 °C/5% CO2, then wells washed with PBS twice before overlaying with infection medium containing: drug, 1% low melting point agarose (Lonza), and 1 µg/ml TPCK-trypsin. Agarose overlays were set at room temperature for 15 min and incubated at 37 °C/5% CO2 for 72 hours. Plaques were visualized by removing overlay and staining with a 1% crystal violet/2% formalin solution in PBS for 1 hour. Plaques were then gently washed in RO water and allowed to dry. Plaques were counted manually and percent reduction calculated compared to infected only control. Statistical analyses were performed using GraphPad Prism using a two-way ANOVA (α = 0.05) with Sidak post-hoc tests to compare wild type and E119D for each drug concentration.
Cytotoxicity assay
Toxicity of drugs was assessed using a MTT based in vitro toxicology assay kit (Sigma-Aldrich) using MDCK cells incubated with the drug at the indicated concentrations for 72 hours at 37 °C/5% CO2 with infection medium to mimic infection conditions. After 72 hours MTT was added and incubated for a further 3 hours. Formazan crystals were dissolved and absorbance read at 570 nm (background 690 nm) on a Biotek Synergy 2 plate reader. Cytotoxicity was assumed when a statistical difference was seen between a drug concentration and untreated controls using a two-way ANOVA (α = 0.05) with Sidak post-hoc tests. Drug concentrations that showed cytotoxicity were excluded from further analysis.
Data accessibility statement
All data generated or analyzed during this study are included in this published article, the Protein Data Bank (PDB) (and the associated Supplementary Information files).
Electronic supplementary material
Acknowledgements
We thank Dr. Jamie Jaruziewicz and Dr. Brandon Young for the synthesis of the initial carboxylate endonuclease inhibitors discussed in this work. This work was supported by NIH Grant AI098757 (to T.R.W.).
Author Contributions
T.R.W., R.J.W. and S.W.W. designed experiments in this study. T.R.W. led the chemistry and biochemistry along the writing of the manuscript. G.K. performed the protein structural work, which was directed by S.W.W. D.B. was responsible for final edits of the manuscript and the bulk of the synthetic work. J.P. and C.L. performed some of the organic synthesis of inhibitors. W.Z. performed the FP experiments and the FRET nuclease assays. R.H. and T.J. performed the plaque assays. All authors discussed results and contributed to the writing and editing of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Diane Beylkin, Gyanendra Kumar and Wei Zhou contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17419-6.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Reid AH, Taubenberger JK, Fanning TG. The 1918 Spanish influenza: integrating history and biology. Microbes Infect. 2001;3:81–7. doi: 10.1016/S1286-4579(00)01351-4. [DOI] [PubMed] [Google Scholar]
- 2.Viboud C, et al. Global Mortality Impact of the 1957-1959 Influenza Pandemic. J Infect Dis. 2016;213:738–45. doi: 10.1093/infdis/jiv534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viboud C, et al. Multinational impact of the 1968 Hong Kong influenza pandemic: evidence for a smoldering pandemic. J Infect Dis. 2005;192:233–48. doi: 10.1086/431150. [DOI] [PubMed] [Google Scholar]
- 4.Taubenberger JK, Reid AH, Janczewski TA, Fanning TG. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci. 2001;356:1829–39. doi: 10.1098/rstb.2001.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden FG. Antivirals for influenza: historical perspectives and lessons learned. Antiviral Res. 2006;71:372–8. doi: 10.1016/j.antiviral.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Barr IG, et al. WHO recommendations for the viruses used in the 2013–2014 Northern Hemisphere influenza vaccine: Epidemiology, antigenic and genetic characteristics of influenza A(H1N1)pdm09, A(H3N2) and B influenza viruses collected from October 2012 to January 2013. Vaccine. 2014;32:4713–25. doi: 10.1016/j.vaccine.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 7.Yen HL. Current and novel antiviral strategies for influenza infection. Curr Opin Virol. 2016;18:126–134. doi: 10.1016/j.coviro.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Pflug A, Guilligay D, Reich S, Cusack S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature. 2014;516:355–60. doi: 10.1038/nature14008. [DOI] [PubMed] [Google Scholar]
- 9.Ju, H. et al. Inhibitors of Influenza Virus Polymerase Acidic (PA) Endonuclease: Contemporary Developments and Perspectives. J Med Chem (2017). [DOI] [PubMed]
- 10.Dias A, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–8. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 11.Hengrung N, et al. Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature. 2015;527:114–7. doi: 10.1038/nature15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagata T, Lefor AK, Hasegawa M, Ishii M. Favipiravir: a new medication for the Ebola virus disease pandemic. Disaster Med Public Health Prep. 2015;9:79–81. doi: 10.1017/dmp.2014.151. [DOI] [PubMed] [Google Scholar]
- 13.Clark MP, et al. Discovery of a novel, first-in-class, orally bioavailable azaindole inhibitor (VX-787) of influenza PB2. J Med Chem. 2014;57:6668–78. doi: 10.1021/jm5007275. [DOI] [PubMed] [Google Scholar]
- 14.Byrn RA, et al. Preclinical activity of VX-787, a first-in-class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit. Antimicrob Agents Chemother. 2015;59:1569–82. doi: 10.1128/AAC.04623-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan P, et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–13. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 16.Carcelli M, et al. N-acylhydrazone inhibitors of influenza virus PA endonuclease with versatile metal binding modes. Sci Rep. 2016;6:31500. doi: 10.1038/srep31500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baughman BM, et al. Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS Chem Biol. 2012;7:526–34. doi: 10.1021/cb200439z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fudo S, et al. Two Distinctive Binding Modes of Endonuclease Inhibitors to the N-Terminal Region of Influenza Virus Polymerase Acidic Subunit. Biochemistry. 2016;55:2646–60. doi: 10.1021/acs.biochem.5b01087. [DOI] [PubMed] [Google Scholar]
- 19.Fudo S, et al. Structural and computational study on inhibitory compounds for endonuclease activity of influenza virus polymerase. Bioorg Med Chem. 2015;23:5466–75. doi: 10.1016/j.bmc.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 20.Kowalinski E, et al. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS Pathog. 2012;8:e1002831. doi: 10.1371/journal.ppat.1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauman JD, et al. Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem Biol. 2013;8:2501–8. doi: 10.1021/cb400400j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sagong HY, et al. Phenyl substituted 4-hydroxypyridazin-3(2H)-ones and 5-hydroxypyrimidin-4(3H)-ones: inhibitors of influenza A endonuclease. J Med Chem. 2014;57:8086–98. doi: 10.1021/jm500958x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sagong HY, et al. 3-Hydroxyquinolin-2(1H)-ones As Inhibitors of Influenza A Endonuclease. ACS Med Chem Lett. 2013;4:547–50. doi: 10.1021/ml4001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hastings JC, Selnick H, Wolanski B, Tomassini JE. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob Agents Chemother. 1996;40:1304–7. doi: 10.1128/aac.40.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkes KE, et al. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J Med Chem. 2003;46:1153–64. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- 26.Singh SB, Tomassini JE. Synthesis of natural flutimide and analogous fully substituted pyrazine-2,6-diones, endonuclease inhibitors of influenza virus. J Org Chem. 2001;66:5504–16. doi: 10.1021/jo015665d. [DOI] [PubMed] [Google Scholar]
- 27.Song MS, et al. Identification and characterization of influenza variants resistant to a viral endonuclease inhibitor. Proc Natl Acad Sci USA. 2016;113:3669–74. doi: 10.1073/pnas.1519772113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ju H, et al. Inhibitors of Influenza Virus Polymerase Acidic (PA) Endonuclease: Contemporary Developments and Perspectives. J Med Chem. 2017;60:3533–3551. doi: 10.1021/acs.jmedchem.6b01227. [DOI] [PubMed] [Google Scholar]
- 29.Boyd VA, et al. 2-Substituted-4,5-dihydroxypyrimidine-6-carboxamide antiviral targeted libraries. J Comb Chem. 2009;11:1100–4. doi: 10.1021/cc900111u. [DOI] [PubMed] [Google Scholar]
- 30.Hsieh HP, Hsu JT. Strategies of development of antiviral agents directed against influenza virus replication. Curr Pharm Des. 2007;13:3531–42. doi: 10.2174/138161207782794248. [DOI] [PubMed] [Google Scholar]
- 31.Mallipeddi PL, Kumar G, White SW, Webb TR. Recent advances in computer-aided drug design as applied to anti-influenza drug discovery. Curr Top Med Chem. 2014;14:1875–89. doi: 10.2174/1568026614666140929153812. [DOI] [PubMed] [Google Scholar]
- 32.Nakazawa M, et al. PA subunit of RNA polymerase as a promising target for anti-influenza virus agents. Antiviral Res. 2008;78:194–201. doi: 10.1016/j.antiviral.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Tomassini JE, et al. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob Agents Chemother. 1996;40:1189–93. doi: 10.1128/aac.40.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DuBois RM, et al. Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog. 2012;8:e1002830. doi: 10.1371/journal.ppat.1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Summa V, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51:5843–55. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 36.Pace P, et al. Dihydroxypyrimidine-4-carboxamides as novel potent and selective HIV integrase inhibitors. J Med Chem. 2007;50:2225–39. doi: 10.1021/jm070027u. [DOI] [PubMed] [Google Scholar]
- 37.Gardelli C, et al. Discovery and synthesis of HIV integrase inhibitors: development of potent and orally bioavailable N-methyl pyrimidones. J Med Chem. 2007;50:4953–75. doi: 10.1021/jm0704705. [DOI] [PubMed] [Google Scholar]
- 38.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 39.Moore K, Rees S. Cell-based versus isolated target screening: how lucky do you feel? J Biomol Screen. 2001;6:69–74. doi: 10.1177/108705710100600202. [DOI] [PubMed] [Google Scholar]
- 40.Stegemann S, Leveiller F, Franchi D, de Jong H, Linden H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31:249–61. doi: 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, et al. Acta Crystallogr D Biol Crystallogr. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution; pp. 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomassini J, et al. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob Agents Chemother. 1994;38:2827–37. doi: 10.1128/AAC.38.12.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Credille CV, Chen Y, Cohen SM. Fragment-Based Identification of Influenza Endonuclease Inhibitors. J Med Chem. 2016;59:6444–54. doi: 10.1021/acs.jmedchem.6b00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones, J.C. et al. A novel endonuclease inhibitor exhibits broad-spectrum anti-influenza activity in vitro. Antimicrob Agents Chemother (2016). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article, the Protein Data Bank (PDB) (and the associated Supplementary Information files).