

Abstract
We demonstrate rapid quantitative measurements of site-resolved paramagnetic relaxation enhancements (PREs), which are a source of valuable structural restraints corresponding to electron-nucleus distances in the ~10–20 Å regime, in solid-state nuclear magnetic resonance (NMR) spectra of proteins containing covalent Cu2+-binding tags. Specifically, using protein GB1 K28C-EDTA-Cu2+ mutant as a model, we show the determination of backbone amide 15N longitudinal and 1H transverse PREs within a few hours of experiment time based on proton-detected 2D or 3D correlation spectra recorded with magic-angle spinning frequencies ≥ ~60 kHz for samples containing ~10–50 nanomoles of 2H,13C,15N-labeled protein back-exchanged in H2O. Additionally, we show that the electron relaxation time for the Cu2+ center, needed to convert PREs into distances, can be estimated directly from the experimental data. Altogether, these results are important for establishing solid-state NMR based on paramagnetic-tagging as a routine tool for structure determination of natively diamagnetic proteins.
Keywords: Magic-angle spinning solid-state NMR, Protein structure, Metalloprotein, Metal binding tag, Spin label
Graphical Abstract
Recent advances in magic-angle spinning (MAS) solid-state nuclear magnetic resonance (NMR) spectroscopy have enabled structural and dynamic studies to be performed with atomic resolution for a broad range of biological macromolecules and supramolecular assemblies,1–6 many of which present considerable challenges for other techniques. Among these advances are solid-state NMR methods which permit site-specific structural information on length scales on the order of ~20 Å to be accessed by utilizing paramagnetic moieties, such as metal centers in metalloproteins or covalent spin label or metal-binding tags incorporated into otherwise diamagnetic proteins, and measurements of nuclear pseudocontact shifts (PCSs) and paramagnetic relaxation enhancements (PREs).7–11 Indeed, the initial successful applications of paramagnetic MAS solid-state NMR to the determination of protein three-dimensional structures12–17 and intermolecular interactions18–19 have already been reported, clearly illustrating the promise of this methodology.
In the context of structure determination of natively diamagnetic proteins based on measurements of site-resolved PREs,13,16–17 the need to extract quantitative relaxation data from series of multidimensional chemical shift correlation spectra for multiple paramagnetic analogues of the protein of interest generally presents a major bottleneck. For example, the initial measurements of residue-specific longitudinal backbone 15N PREs for Cys-EDTA-Cu2+ mutants of the B1 immunoglobulin binding domain of protein G (GB1) in microcrystalline phase,20 based on conventional 2D 15N-13C correlation spectra with 13C detection at relatively slow (~10 kHz) MAS rates, were prohibitively time-consuming from the practical standpoint requiring on the order of a week of measurement time per mutant as well as samples containing ~1 μmol of 13C,15N-labeled protein. The measurement times and sample quantities needed for such experiments could be considerably reduced (typically to ~24–48 h and ~0.1–0.2 μmol of 13C,15N-labeled protein per sample)13,21–22 by employing probes equipped with smaller diameter rotors and recording 13C-detected spectra at ~40 kHz MAS rates using low-power radiofrequency pulse sequences23 and condensed data collection schemes24–25 with recycle delays of a few hundred milliseconds, facilitated by the enhanced amide proton longitudinal relaxation caused by the presence of the covalent paramagnetic EDTA-Cu2+ tags and efficient 1H-1H spin diffusion.
The latest developments in fast MAS (~60–110 kHz) probe technology combined with proton detection26–28 have been shown to permit the rapid acquisition of solid-state NMR spectra with high sensitivity, and such studies have been previously applied to quantitative measurements of PCSs29 and 15N and 13C PREs14 in a perdeuterated metalloenzyme, superoxide dismutase, back-exchanged in H2O. Here we explore the utility of proton-detected solid-state NMR schemes of this type for the rapid determination of residue-specific PREs in Cu2+-tagged proteins with high resolution and sensitivity, and report several advances in this regard that are important for the routine high-throughput application of this paramagnetic solid-state NMR methodology to natively diamagnetic proteins. Specifically, we demonstrate the quantitative measurements of intramolecular amide 15N longitudinal and 1H transverse PREs for a model Cys-EDTA-Cu2+ mutant of 2H,13C,15N-GB1 back-exchanged in H2O and diluted in a fully protonated diamagnetic protein matrix by using pulse schemes based on series of 2D 15N-1H or 3D 13C-15N-1H spectra (the latter offering enhanced resolution due to the additional chemical shift dimension), samples containing tens of nanomoles of isotope labeled paramagnetic protein and measurement times as short as ~1–2 hours. In addition, we show that the 15N and 1H PREs, which yield valuable long-distance structural restraints, can be used to estimate the electron relaxation time constant for the Cu2+ center directly from experimental data.
Uniformly 2H,13C,15N-labeled K28C mutant of GB1 back-exchanged in 100% H2O to replace the labile amide deuterons with protons was used as the model protein in our studies, with the cysteine residue further modified with a covalent EDTA-Cu2+ tag via thiol-disulfide chemistry as described previously.20–21,30 For brevity, this protein is referred to as 28EDTA-Cu2+. To attenuate the influence of intermolecular electron-nucleus interactions on the PRE measurements,31 the protein microcrystals used for the solid-state NMR analysis were generated by co-precipitating ~0.35 mg (~50 nmol) of the H2O back-exchanged 2H,13C,15N-28EDTA-Cu2+ with natural abundance GB1 in a ~1:3 molar ratio. An analogous 28EDTA-Zn2+ diamagnetic control protein, with EDTA loaded with Zn2+ instead of Cu2+, containing ~0.1 mg (~15 nmol) of the H2O back-exchanged 2H,13C,15N-28EDTA-Zn2+ diluted in natural abundance GB1 was also prepared. The 1H longitudinal relaxation time constants (T1) for the 28EDTA-Cu2+ and 28EDTA-Zn2+ proteins were found to be ~240 ms and ~770 ms, respectively, at 60 kHz MAS by using the usual inversion-recovery measurements, and for optimum signal-to-noise (S/N) ratio per unit time the recycle delays were set to ~1.3 × 1H T1 for all experiments.
In Figures 1A and 1B we show 2D 15N-1H spectra of 28EDTA-Zn2+ and 28EDTA-Cu2+, respectively, recorded at 800 MHz 1H frequency and 60 kHz MAS using the CP-HSQC pulse scheme32 (see Supporting Information (SI) Figure S1A). Owing to the use of proton detection and short recycle delays, the 28EDTA-Cu2+ spectrum displaying high resolution in both 15N and 1H dimensions could be recorded in less than 3 minutes, with 50 resolved 15N-1H cross-peaks (out of the 55 possible in GB1) having S/N ratios ranging from 37 to 142 for correlations corresponding to residues Q32 and E56, respectively, and an average S/N ratio of ~90 ± 30. Although further sensitivity gains, on the order of ~10% on average (see SI Figure S2), are realized by increasing the MAS rate to 65 kHz, the fact that comparable data are obtained at 60 and 65 kHz MAS is useful from the practical standpoint given that spinning instabilities can occur for some samples near the ~65 kHz MAS rate limit associated with 1.3 mm diameter rotors; on the other hand, the average S/N ratio was found to decrease by ~25–30% when spinning the sample at 55 kHz vs. 60 kHz (SI Figure S2), indicating that the use of MAS rates in the ~60–65 kHz regime is ideal for these experiments.
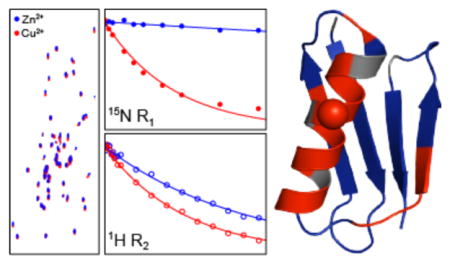
Figure 1.

Two-dimensional 15N-1H spectra of 2H,13C,15N-labeled 28EDTA-Zn2+ (A) and 28EDTA-Cu2+ (B) back-exchanged in H2O. The spectra were recorded at 800 MHz 1H frequency and 60 kHz MAS rate using the pulse scheme shown in SI Figure S1A with t1,max (15N) = 25 ms and t2,max (1H) = 30 ms, and 2 scans per row, 325 ms recycle delay and total measurement time of ~2.5 min for 28EDTA-Cu2+ and 16 scans per row, 1 s recycle delay and total measurement time of ~45 min for 28EDTA-Zn2+. The spectra were processed with cosine-bell window functions in both dimensions and are shown with the first contour drawn at ~35 times the rms noise level. (C) Representative strips from a 3D 13CO-15N-1H spectrum of 28EDTA-Cu2+ recorded at 800 MHz 1H frequency and 60 kHz MAS rate using the pulse scheme in SI Figure S1B with t1,max (13CO) = 4 ms, t2,max (15N) = 5 ms and t3, max (1H) = 30 ms, 2 scans per row, 325 ms recycle delay and total measurement time of ~10 min. The spectrum was processed with 81º-shifted sine-bell window functions in all dimensions and is shown with the first contour drawn at ~15 times the rms noise level. (D, E) Measurements of backbone amide 15N longitudinal (D) and 1H transverse (E) relaxation trajectories for representative residues in 28EDTA-Cu2+ (red open and filled circles) and 28EDTA-Zn2+ (blue open and filled circles) using the pulse schemes shown in SI Figure S3A and S3B. The total measurement times used to collect the complete longitudinal 15N and transverse 1H relaxation trajectories for the paramagnetic 28EDTACu2+ sample were approximately 3 h and 1.5 h, respectively (c.f., SI Figures S4 and S5). Solid lines of corresponding color indicate decaying single exponential fits of the experimental data.
The high resolution and sensitivity afforded by the 2D 15N-1H spectra collected with minimal acquisition times enables rapid measurements of spin relaxation trajectories required for quantitative determination of site-specific nuclear PREs. In Figure 1D we show representative measurements of 15N longitudinal relaxation rates (R1) for 28EDTA-Zn2+ and 28EDTA-Cu2+ from series of ten 2D 15N-1H spectra recorded using the pulse scheme in SI Figure S3A with increasing 15N longitudinal relaxation delays (τN) up to 4 s (the complete set of 15N R1 trajectories is shown in SI Figure S4). For the 28EDTA-Cu2+ sample containing ~50 nmol of labeled protein the total measurement time used to acquire these 15N R1 trajectories was only ~3 h. This compares favorably with analogous experiments at 40 kHz MAS based on 2D 15N-13C spectra, where the acquisition of site-specific 15N R1 data of similar quality required roughly an order of magnitude longer measurement times (~24–48 h per paramagnetic mutant) and samples containing ~3 times more labeled protein,13 and bodes well for the broad applications of this paramagnetic solid-state NMR methodology to samples containing even smaller amounts of labeled protein and/or larger-sized proteins. Indeed, the 15N R1 trajectories shown in Figure 1D and SI Figure S4 for the 28EDTA-Zn2+ diamagnetic control sample containing only ~15 nmol of labeled protein, which necessitated the use of 1 s recycle delays (i.e., ~3 times longer relative to those used for 28EDTA-Cu2+), were recorded with a total measurement time of ~16 h, with trajectories of acceptable quality available in as little as ~4 h.
The increased 1H transverse relaxation times characteristic of deuterated and H2O back-exchanged proteins at MAS rates of ~60 kHz and greater26–28 allowed us to also explore the possibility of conducting quantitative measurements of transverse PREs for amide 1H nuclei. Such measurements can, in principle, provide additional protein structural restraints that are complementary to 15N longitudinal PREs. Figure 1E shows representative measurements of 1H transverse relaxation rates (R2) for 28EDTA-Zn2+ and 28EDTA-Cu2+ from series of 18 15N-1H spectra recorded using the pulse scheme in SI Figure S3B with increasing 1H transverse relaxation delays (τH) up to 10 ms (see SI Figure S5 for the complete set of trajectories), where the residue-specific relaxation data for the 28EDTA-Cu2+ sample were recorded with a total measurement time of only ~1.5 h.
2D 15N-1H spectra for proteins larger than GB1 will generally be more crowded due to the larger number of resonances, and, additionally, for non-microcrystalline proteins (e.g., fibrillar or membrane proteins) these spectra often display increased inhomogeneous broadening in the 1H and 15N dimensions further degrading the spectral resolution.28 With these considerations in mind, we investigated the possibility of performing rapid measurements of nuclear PREs via series of 3D 13C-15N-1H correlation spectra, which contain an additional chemical shift dimension for increased resolution. Figure 1C shows representative strips from a 3D 13CO-15N-1H spectrum for 28EDTA-Cu2+ acquired using the pulse scheme shown in SI Figure S1B. This spectrum, which permitted all 55 13CO-15N-1H correlations to be resolved and assigned, was recorded in only ~10 minutes with an average S/N ratio of ~43 ± 14, which is lower than that obtained for the 2D 15N-1H spectrum due to the additional 13C-15N cross-polarization step but nevertheless more than sufficient for quantitative relaxation rate measurements. SI Figure S6 shows representative 15N R1 and 1H R2 trajectories obtained for the 28EDTA-Cu2+ sample from series of 3D 13CO-15N-1H spectra recorded with measurement times of ~14 h and ~6 h, respectively. Also shown in SI Figure S6 are correlation plots for 15N R1 and 1H R2 rates determined via 2D 15N-1H versus 3D 13CO-15N-1H spectra, which illustrate good overall agreement between the different sets of relaxation rates (R2 values of 0.95 and 0.96 were obtained for the 15N R1 and 1H R2 correlations, respectively). Finally, we note that for all relaxation rate measurements the key factor contributing to the overall measurement time is the number of points acquired in the relaxation dimension—this is particularly important for 15N R1 measurements which employ long relaxation delays that extend up to several seconds. We therefore investigated the possibility of extracting quantitative relaxation rates from trajectories containing fewer points in the relaxation dimension. For the 3D 13CO-15N-1H based 15N R1 measurements we show in SI Figure S7A that, for the datasets collected in the present study, trajectories corresponding to only four longitudinal relaxation delays (τN values of 100 μs, 0.5 s, 1.5 s, and 3 s) yield virtually identical R1 rates, within experimental error, as the trajectories containing ten points in the relaxation dimension (c.f., SI Figure S6), while the total measurement time required to record these four-point relaxation trajectories is approximately one-third of that used for the ten-point trajectories (~5 h vs. ~14 h). In analogy, quantitative 1H R2 rates could be extracted from 3D 13CO-15N-1H based trajectories consisting of six relaxation delays (τH values of 66.7 μs, 633.3 μs, 2.0333 ms, 4.0333 ms, 8.0333 ms and 10.0333 ms) instead of 18, with a concomitant reduction in the total experiment time from ~6 h to ~2 h (see SI Figure S7B).
A cursory inspection of the longitudinal 15N and transverse 1H relaxation trajectories (c.f., Figures 1, S4 and S5) reveals significant differences in the site-specific nuclear spin relaxation rates that clearly appear to be correlated with the distance between the paramagnetic Cu2+ center and individual residues, with the helical residues T25-Q32 located in closest proximity to the EDTA-Cu2+ tag at position 28 having the largest paramagnetic contributions to the relaxation rates. For each residue the backbone amide longitudinal 15N and transverse 1H PREs (Γ1N and Γ2H, respectively) were obtained by fitting the experimental relaxation trajectories for the 28EDTA-Cu2+ and 28EDTA-Zn2+ proteins to decaying single exponentials and subtracting the diamagnetic R1 or R2 value from the corresponding paramagnetic one. The experimental PREs are shown in Figure 2 as a function of residue number and mapped onto the crystal structure of GB1. The PRE values ranged between ~0.02 and 0.55 s−1 for Γ1N and between ~1 and 85 s−1 for Γ2H, corresponding to Cu2+ to backbone amide 15N/1H distances in the ~10–20 Å regime according to calculations using the Solomon-Bloembergen equation33–35 with longitudinal relaxation time constant, T1e, of 2.5 ns for the Cu2+ center35–36 (see also Figure 4 and discussion below).
Figure 2.
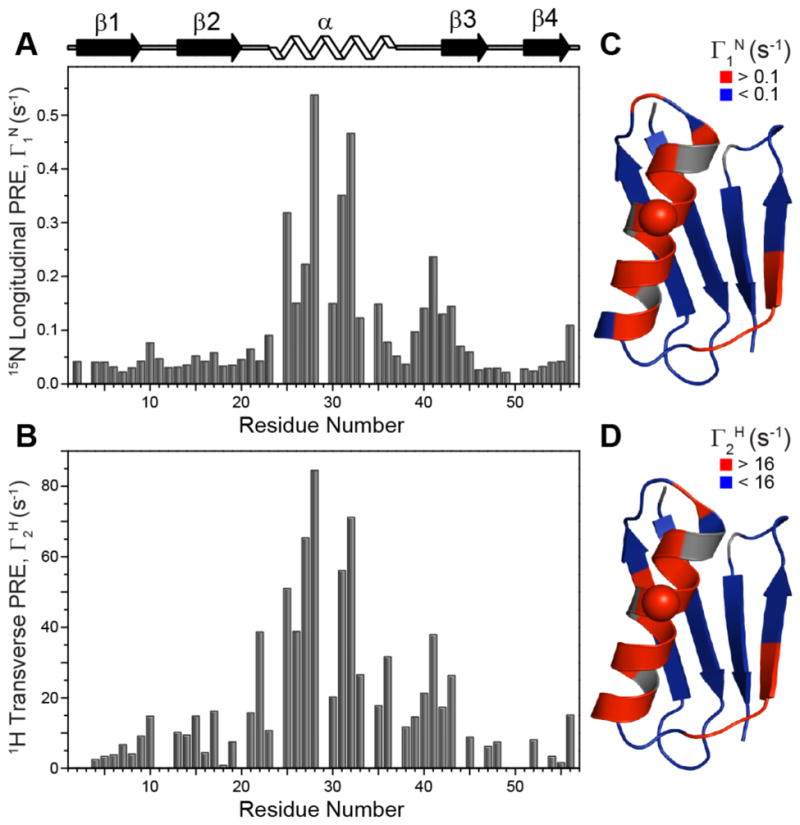
(A, B) Plots of backbone amide (A) 15N longitudinal PREs, Γ1N = R1N (Cu2+) – R1N (Zn2+), and (B) 1H transverse PREs, Γ2H = R2H (Cu2+) – R2H (Zn2+), as a function of residue number extracted from the 2D 15N-1H based datasets. Note that PREs for residues Y3, A24, V29, A34 and K50 could not be determined from these experiments due to spectral overlap. In addition, for several residues the fitted diamagnetic 1H R2 rate slightly exceeded the corresponding paramagnetic relaxation rate resulting in a small negative PRE; for these residues the Γ2H values were set to zero in the plots. (C, D) Ribbon diagrams of GB1 (PDB ID 2GI9) with (C) Γ1N and (D) Γ2H values mapped onto the structure. Residues with Γ1N < 0.1 s−1 and Γ2H < 16 s−1 (corresponding to 15N/1H-Cu2+ distances > ~14 Å) are colored in blue, and those with Γ1N > 0.1 s−1 and Γ2H > 16 s−1 (15N/1H-Cu2+ distances < ~14 Å) are colored in red. Residue 28 corresponding to the location of the EDTA-Cu2+ sidechain is indicated by a sphere.
Figure 4.
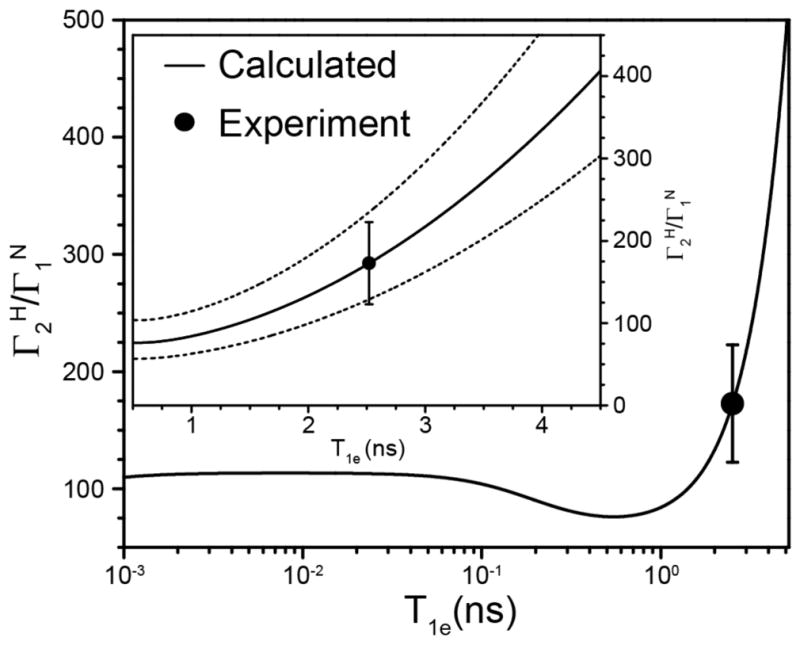
Plot of the calculated ratio (solid line) as a function of T1e in the range of 1 ps to 5 ns, with the inset showing a close-up view of the data for T1e values between 0.5 and 4.5 ns. Also shown in the inset as dashed lines are calculated ratios obtained for distances between the Cu2+ ion and the amide 1H and 15N nuclei that are not identical but differ from each other by ±5% (this corresponds to differences of ±0.5–1.0 Å for distances in the 10–20 Å regime). The experimentally determined average ratio for a subset of 14 residues having the largest PREs (see text for details) is shown as a filled circle. The average ratio corresponds to an estimated T1e value of 2.5 ns for the Cu2+ center, and the range of Cu2+ T1e values consistent with the experimental PRE data, based on the standard deviation of the residue-specific ratios, is ~1.8–3.1 ns.
Figure 3A shows the comparison of the experimental Γ1N and Γ2H values with the corresponding values calculated using a structural model of 28EDTA-Cu2+ generated using the GB1 atomic coordinates within Xplor-NIH.37 Overall the observed and calculated PREs are in good agreement with root-mean-squared deviation (rmsd) of 0.05 s−1 for the 15N longitudinal PREs and 15 s−1 for the 1H transverse PREs, with the most significant relative deviations seen for the smallest calculated Γ1N and Γ2H values (Γ1N < 0.03 s−1 and Γ2H < 5 s−1, corresponding to 15N/1H-Cu2+ distances > ~17.5 Å) most likely due to a combination of the binding of small amounts of excess Cu2+ to endogenous aspartate and glutamate side-chains and residual intermolecular 15N/1H-Cu2+ couplings due to insufficient dilution of the paramagnetic protein in the diamagnetic matrix.31 Comparison of the experimentally determined 15N-Cu2+ and 1H-Cu2+ distances with the distances obtained from the protein structure is shown in Figure 3B for measurements corresponding to calculated Γ1N > 0.03 s−1 and Γ2H > 5 s−1, revealing good agreement between these experimental and calculated distances with rmsd values of 0.9 Å and 1.7 Å for the 15N-Cu2+ and 1H-Cu2+ distances, respectively. Moreover, for individual residues these 15N-Cu2+ and 1H-Cu2+ distances are in reasonably good agreement with each other with a rmsd of 1.4 Å. The precision of these PRE-based distance measurements, as well as their utility for protein structure determination is expected to further improve by using Cu(II)-binding tags that are more compact and rigid relative to the EDTA-type tag employed in the present study,15,22 as well as multiple protein samples containing the paramagnetic tags in several different locations.13,16
Figure 3.

Comparison of the experimental (A) backbone amide 15N longitudinal (filled circles) and 1H transverse (open circles) PREs and (B) 15N-Cu2+ (filled circles) and 1H-Cu2+ (open circles) distances, with the corresponding values calculated from the structural model of 28EDTA-Cu2+.
Notably, the quantitative intramolecular PRE and associated electron-nucleus distance measurements, in particular the 1H transverse PREs, can be performed for the paramagnetic-tagged 2H,13C,15N-labeled protein exchanged in pure H2O and diluted in a diamagnetic matrix consisting of fully protonated protein as opposed to 2H-enriched protein, based on the fact that nearly identical amide 1H R2 values are obtained for diamagnetic reference protein samples irrespective of the degree of deuteration of the matrix (Figures S8 and S9). The latter finding suggests that the experimental amide 1H R2 rates are primarily determined by the local proton density, and has important practical implications related to the ease and cost effectiveness associated with the preparation of protein samples for such studies.
Beyond serving as a source of valuable long-distance restraints on three-dimensional protein structure, the quantitative measurements of amide 15N and 1H PREs also provide an opportunity to estimate, directly from the experimental data, the magnitude of the longitudinal relaxation time constant, T1e, for the paramagnetic metal center in 28EDTA-Cu2+. Assuming that the distances between the Cu2+ ion and the amide 1H and 15N nuclei can be taken to be the same within each residue, the ratio of the residue-specific 1H transverse to 15N longitudinal PREs in the solid state is to a reasonable approximation independent of the 15N/1H-Cu2+ distance and depends only on T1e for the Cu2+ center and constants characteristic of the 1H, 15N and electron spins according to:7
where γH and γN are the 1H and 15N gyromagnetic ratios, respectively, and ωH, ωN and ωe are the Larmor frequencies of the 1H, 15N and electron spins, respectively. Figure 4 shows a plot of the ratio calculated as a function of T1e in the range of 1 ps to 5 ns. Superimposed on the plot as a filled circle is the experimentally determined average ratio, which was found to be 173 ± 50 for a subset of the experimental data corresponding to 14 residues with the largest PREs (i.e., residues with ). The average experimental ratio for these residues corresponds to an estimated T1e value of 2.5 ns for the Cu2+ center, and, taking into account the standard deviation of the residue-specific ratios, the experimental PREs are consistent with Cu2+ T1e values in the ~1.8–3.1 ns range. These estimates of T1e for the Cu2+ center in hydrated microcrystals of 28EDTA-Cu2+ under the conditions employed for our solid-state NMR studies are in remarkable agreement with T1e values that have been reported for several Cu2+ metalloproteins in solution at ambient temperature, ranging between 1.8 and 5.7 ns with an average value of 2.8 ns.36 Furthermore, we note that the 15N-Cu2+ and 1H-Cu2+ distances extracted from the associated PREs are relatively insensitive to the exact value of T1e within the 1.8 to 3.1 ns range. Specifically, the 15N-Cu2+ and 1H-Cu2+ distances calculated with T1e of 1.8 ns or 3.1 ns instead of 2.5 ns are respectively within ca. ±1% and ±5% of the corresponding values obtained by using the best estimate of 2.5 ns for Cu2+ T1e.
In summary, we have shown that quantitative measurements of residue-specific longitudinal and transverse paramagnetic relaxation enhancements for protein backbone nuclei, including most notably 1H transverse PREs, can be carried out at MAS rates of ~60 kHz and above in as little as a few hours of measurement time for samples containing ~10–50 nanomoles of 2H,13C,15N-enriched Cu2+-tagged protein diluted in a diamagnetic matrix consisting of fully protonated protein. In addition to the simplest schemes based on series of proton-detected 2D 15N-1H chemical shift correlation spectra such data can also be recorded without a major increase in the overall measurement time by using 3D 13C-15N-1H based sequences, which offer improved spectral resolution. Collectively, by shifting the experimental bottleneck from data acquisition to preparation of multiple paramagnetic analogs of the protein of interest needed for de novo three-dimensional structure determination,13 the results presented in this study are important for establishing paramagnetic solid-state NMR based on covalent attachment of small molecule tags as a routine tool for the structural analysis of natively diamagnetic proteins. Finally, we illustrate that collection of multiple types of PREs enables the longitudinal relaxation time constant associated with the paramagnetic center to be estimated directly from experimental data, thereby further reducing the uncertainty associated with the conversion of PREs into distances between the protein nuclei and the paramagnetic center.
EXPERIMENTAL METHODS
The 2H,13C,15N-labeled 28EDTA-Cu2+ and 28EDTA-Zn2+ proteins were prepared as described previously.21 Microcrystalline protein samples for solid-state NMR analysis, containing 2H,13C,15N-28EDTA-Cu2+ or 28EDTA-Zn2+ back-exchanged in 100% H2O and natural abundance GB1 in a ~1:3 molar ratio (see text for additional details) were generated as described previously,21 and transferred by ultracentrifugation to 1.3 mm Bruker zirconia rotors.
Solid-state NMR experiments were performed on a three-channel Bruker spectrometer operating at frequencies of 800.3 MHz for 1H, 201.3 MHz for 13C and 81.1 MHz for 15N, equipped with a 1.3 mm HCN MAS probe. The MAS frequency was set to 60 kHz for most experiments and regulated to ca. ± 5 Hz using a Bruker MAS II controller, and the effective sample temperature was regulated at ~30 °C using a variable temperature unit. The pulse sequences and associated parameters are described in detail in the SI. NMR spectra were processed using NMRPipe38 and analyzed using nmrglue.39
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (grants MCB-1243461 and MCB-1715174 to C.P.J.), the National Institutes of Health (grant S10OD012303 to C.P.J.), and the Camille & Henry Dreyfus Foundation (Camille Dreyfus Teacher-Scholar Award to C.P.J).
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website. Figures with pulse scheme diagrams, additional NMR spectra, relaxation trajectories and correlation plots (PDF).
References
- 1.Comellas G, Rienstra CM. Protein structure determination by magic-angle spinning solid-state NMR, and insights into the formation, structure, and stability of amyloid fibrils. Annu Rev Biophys. 2013;42:515–536. doi: 10.1146/annurev-biophys-083012-130356. [DOI] [PubMed] [Google Scholar]
- 2.Loquet A, Habenstein B, Lange A. Structural investigations of molecular machines by solid-state NMR. Acc Chem Res. 2013;46:2070–2079. doi: 10.1021/ar300320p. [DOI] [PubMed] [Google Scholar]
- 3.Wang S, Ladizhansky V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog Nucl Magn Reson Spectrosc. 2014;82:1–26. doi: 10.1016/j.pnmrs.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Meier BH, Bockmann A. The structure of fibrils from ‘misfolded’ proteins. Curr Opin Struct Biol. 2015;30:43–49. doi: 10.1016/j.sbi.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan M, Pinto C, Houben K, Baldus M. Nuclear magnetic resonance (NMR) applied to membrane-protein complexes. Q Rev Biophys. 2016;49:e15. doi: 10.1017/S003358351600010X. [DOI] [PubMed] [Google Scholar]
- 6.Quinn CM, Polenova T. Structural biology of supramolecular assemblies by magic-angle spinning NMR spectroscopy. Q Rev Biophys. 2017;50:e1. doi: 10.1017/S0033583516000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaroniec CP. Solid-state nuclear magnetic resonance structural studies of proteins using paramagnetic probes. Solid State Nucl Magn Reson. 2012;43–44:1–13. doi: 10.1016/j.ssnmr.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Bhaumik A, Luchinat C, Parigi G, Ravera E, Rinaldelli M. NMR crystallography on paramagnetic systems: Solved and open issues. CrystEngComm. 2013;15:8639–8656. [Google Scholar]
- 9.Knight MJ, Felli IC, Pierattelli R, Emsley L, Pintacuda G. Magic angle spinning NMR of paramagnetic proteins. Acc Chem Res. 2013;46:2108–2116. doi: 10.1021/ar300349y. [DOI] [PubMed] [Google Scholar]
- 10.Pintacuda G, Kervern G. Paramagnetic solid-state magic-angle spinning NMR spectroscopy. Top Curr Chem. 2013;335:157–200. doi: 10.1007/128_2011_312. [DOI] [PubMed] [Google Scholar]
- 11.Jaroniec CP. Structural studies of proteins by paramagnetic solid-state NMR spectroscopy. J Magn Reson. 2015;253:50–59. doi: 10.1016/j.jmr.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertini I, Bhaumik A, De Paëpe G, Griffin RG, Lelli M, Lewandowski JR, Luchinat C. High-resolution solid-state NMR structure of a 17.6 kDa protein. J Am Chem Soc. 2010;132:1032–1040. doi: 10.1021/ja906426p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengupta I, Nadaud PS, Helmus JJ, Schwieters CD, Jaroniec CP. Protein fold determined by paramagnetic magic-angle spinning solid-state NMR spectroscopy. Nat Chem. 2012;4:410–417. doi: 10.1038/nchem.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knight MJ, Pell AJ, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Herrmann T, Emsley L, Pintacuda G. Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR. Proc Natl Acad Sci USA. 2012;109:11095–11100. doi: 10.1073/pnas.1204515109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Pilla KB, Li Q, Zhang Z, Su X, Huber T, Yang J. Magic angle spinning NMR structure determination of proteins from pseudocontact shifts. J Am Chem Soc. 2013;135:8294–8303. doi: 10.1021/ja4021149. [DOI] [PubMed] [Google Scholar]
- 16.Tamaki H, Egawa A, Kido K, Kameda T, Kamiya M, Kikukawa T, Aizawa T, Fujiwara T, Demura M. Structure determination of uniformly 13C,15N labeled protein using qualitative distance restraints from MAS solid-state 13C-NMR observed paramagnetic relaxation enhancement. J Biomol NMR. 2016;64:87–101. doi: 10.1007/s10858-015-0010-0. [DOI] [PubMed] [Google Scholar]
- 17.Rovo P, Grohe K, Giller K, Becker S, Linser R. Proton transverse relaxation as a sensitive probe for structure determination in solid proteins. Chemphyschem. 2015;16:3791–3796. doi: 10.1002/cphc.201500799. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Munro RA, Kim SY, Jung KH, Brown LS, Ladizhansky V. Paramagnetic relaxation enhancement reveals oligomerization interface of a membrane protein. J Am Chem Soc. 2012;134:16995–16998. doi: 10.1021/ja308310z. [DOI] [PubMed] [Google Scholar]
- 19.Gustavsson M, Verardi R, Mullen DG, Mote KR, Traaseth NJ, Gopinath T, Veglia G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc Natl Acad Sci USA. 2013;110:17338–17343. doi: 10.1073/pnas.1303006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadaud PS, Helmus JJ, Kall SL, Jaroniec CP. Paramagnetic ions enable tuning of nuclear relaxation rates and provide long-range structural restraints in solid-state NMR of proteins. J Am Chem Soc. 2009;131:8108–8120. doi: 10.1021/ja900224z. [DOI] [PubMed] [Google Scholar]
- 21.Nadaud PS, Helmus JJ, Sengupta I, Jaroniec CP. Rapid acquisition of multidimensional solid-state NMR spectra of proteins facilitated by covalently bound paramagnetic tags. J Am Chem Soc. 2010;132:9561–9563. doi: 10.1021/ja103545e. [DOI] [PubMed] [Google Scholar]
- 22.Sengupta I, Gao M, Arachchige RJ, Nadaud PS, Cunningham TF, Saxena S, Schwieters CD, Jaroniec CP. Protein structural studies by paramagnetic solid-state NMR spectroscopy aided by a compact cyclen-type Cu(II) binding tag. J Biomol NMR. 2015;61:1–6. doi: 10.1007/s10858-014-9880-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ernst M, Meier MA, Tuherm T, Samoson A, Meier BH. Low-power high-resolution solid-state NMR of peptides and proteins. J Am Chem Soc. 2004;126:4764–4765. doi: 10.1021/ja0494510. [DOI] [PubMed] [Google Scholar]
- 24.Wickramasinghe NP, Parthasarathy S, Jones CR, Bhardwaj C, Long F, Kotecha M, Mehboob S, Fung LWM, Past J, Samoson A, Ishii Y. Nanomole-scale protein solid-state NMR by breaking intrinsic 1H T1 boundaries. Nat Meth. 2009;6:215–218. doi: 10.1038/nmeth.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laage S, Sachleben JR, Steuernagel S, Pierattelli R, Pintacuda G, Emsley L. Fast acquisition of multi-dimensional spectra in solid-state NMR enabled by ultra-fast MAS. J Magn Reson. 2009;196:133–141. doi: 10.1016/j.jmr.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 26.Lewandowski JR, Dumez JN, Akbey U, Lange S, Emsley L, Oschkinat H. Enhanced resolution and coherence lifetimes in the solid-state NMR spectroscopy of perdeuterated proteins under ultrafast magic-angle spinning. J Phys Chem Lett. 2011;2:2205–2211. [Google Scholar]
- 27.Asami S, Reif B. Proton-detected solid-state NMR spectroscopy at aliphatic sites: application to crystalline systems. Acc Chem Res. 2013;46:2089–2097. doi: 10.1021/ar400063y. [DOI] [PubMed] [Google Scholar]
- 28.Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G. High-resolution proton-detected NMR of proteins at very fast MAS. J Magn Reson. 2015;253:36–49. doi: 10.1016/j.jmr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Knight MJ, Felli IC, Pierattelli R, Bertini I, Emsley L, Herrmann T, Pintacuda G. Rapid measurement of pseudocontact shifts in metalloproteins by proton-detected solid-state NMR spectroscopy. J Am Chem Soc. 2012;134:14730–14733. doi: 10.1021/ja306813j. [DOI] [PubMed] [Google Scholar]
- 30.Nadaud PS, Helmus JJ, Höfer N, Jaroniec CP. Long-range structural restraints in spin-labeled proteins probed by solid-state nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2007;129:7502–7503. doi: 10.1021/ja072349t. [DOI] [PubMed] [Google Scholar]
- 31.Nadaud PS, Sengupta I, Helmus JJ, Jaroniec CP. Evaluation of the influence of intermolecular electron-nucleus couplings and intrinsic metal binding sites on the measurement of 15N longitudinal paramagnetic relaxation enhancements in proteins by solid-state NMR. J Biomol NMR. 2011;51:293–302. doi: 10.1007/s10858-011-9536-y. [DOI] [PubMed] [Google Scholar]
- 32.Barbet-Massin E, Pell AJ, Retel JS, Andreas LB, Jaudzems K, Franks WT, Nieuwkoop AJ, Hiller M, Higman V, Guerry P, Bertarello A, Knight MJ, Felletti M, Le Marchand T, Kotelovica S, Akopjana I, Tars K, Stoppini M, Bellotti V, Bolognesi M, Ricagno S, Chou JJ, Griffin RG, Oschkinat H, Lesage A, Emsley L, Herrmann T, Pintacuda G. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc. 2014;136:12489–12497. doi: 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solomon I. Relaxation processes in a system of two spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- 34.Bloembergen N, Morgan LO. Proton relaxation times in paramagnetic solutions. Effects of electron spin relaxation. J Chem Phys. 1961;34:842–850. [Google Scholar]
- 35.Bertini I, Luchinat C, Parigi G. Solution NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Models. Elsevier; Amsterdam: 2001. [Google Scholar]
- 36.Banci L, Bertini I, Luchinat C. Electron relaxation. Magn Reson Rev. 1986;11:1–40. [Google Scholar]
- 37.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 38.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 39.Helmus JJ, Jaroniec CP. Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J Biomol NMR. 2013;55:355–367. doi: 10.1007/s10858-013-9718-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.