

Abstract
It is known that dynamin-related protein 1 (Drp1)-mediated mitochondrial fission plays an important role in ischemic injury of myocardial infarction (MI). Apelin, an endogenous ligand for Apelin receptor, acts as a key modulator of cardiovascular diseases. Here, we examined the effects of Apelin on MI injury and underlying mechanisms. Adult male C57BL/6J mice were treated with Apelin for 4 weeks and then subjected coronary artery ligation (LAD) to induce MI and the protective effects of Apelin on MI injury were evaluated at 6 h post LAD. Mitochondrial fission was significantly increased in MI as evidenced by enhanced expression of phosphorylated Drp1 (p-Drp1ser 616) without affecting total Drp-1 level and degenerative transformation of mitochondria into short rods as typical fission. Apelin markedly inhibited p-Drp1ser 616 and preserved mitochondrial morphology in MI. Similar effects of Apelin were consistently observed in primary cultured cardiomyocytes under hypoxia. Apelin decreased hypoxia-induced cardiomyocyte apoptosis as evidenced by decreased TUNEL-positive cells and preserved mitochondrial membrane potential (MMP). Apelin decreased Bax/Bcl-2 ratio and limited the release of cytochrome C and activation of caspase-9 and caspase-3 both in vivo and in vitro. Finally, Apelin diminished the infarct size and normalized the impaired cardiac function as indicated by rescuing of the decreased ejection faction and fractional shortening in MI mice. In conclusion, Apelin prevented mitochondrial fission by inhibiting p-Drp1Ser616, which prevents loss of MMP and inhibits the mitochondria-mediated apoptosis. These results indicate that the inhibition of Drp-1 activation by Apelin is a novel mechanism of cardioprotection against MI injury.
Keywords: apelin, cardiomyocyte apoptosis, mitochondrial fission, hypoxia, myocardial infarction
INTRODUCTION
Myocardial infarction (MI) is still a primary cause of death in the world with an increased risk of morbidity and mortality [1–3]. Many cardioprotective pharmacological agents failed to exert their protective effects [4–6]. While many interventions are effective in reducing MI injury and offering limited protection against ischemia/reperfusion (I/R) injury [7, 8]. Therefore, exploring novel pharmacological agents to help salvage ischemic damages may benefit clinical outcomes for MI patients.
Mitochondria are a vital cell organelle in cardiomyocytes because of their pivotal role in ATP production required for myocardial contraction and survival. Mitochondria undergo cycles of fission (division) and fusion (joining), which play an important role in regulating apoptosis [9–11]. Increased mitochondrial fission in the ischemic heart contributes to apoptosis induction and infarct generation, while inhibiting mitochondrial fission reduces myocardial injury and improves cardiac function following MI [12, 13]. Dynamin-related protein-1 (Drp1) is a cytosolic protein that is recruited over the mitochondrial surface to initiate fission through interaction with binding partners as fission protein 1 (Fis1) [14]. Ding et al. [15] reported that Drp1-mediated mitochondrial fission is increased following I/R under diabetic conditions and its inhibition with Mdivi-1 reduces I/R injury and improves cardiac function. Grohm et al. found that Drp1 siRNA and small molecule inhibitors of Drp1 could prevent mitochondrial fission and loss of mitochondrial membrane potential [16]. Inhibition of Drp1 has been demonstrated to reduce mitochondrial fission and infarct size during ischemic injury in animals [17, 18]. Therefore, manipulating Drp1-dependent mitochondrial fission may be an alternative strategy for the development of new anti-apoptotic drugs.
Apelin is an endogenous ligand for the previously orphaned G-protein-coupled Apelin related receptor (APJ) that is involved in regulating cardiovascular functions. Apelin is known to have potent protective effect on heart and significant therapeutic potential for heart failure [19–23]. Rastaldo et al. found that Apelin can limit infarct size and improve cardiac post-ischemic mechanical recovery only if given after ischemia [24]. Tao et al. reported that Apelin protects the heart against I/R injury through inhibition of the ER (endoplasmic reticulum)-dependent apoptotic pathway [25]. Apelin inactivates GSK-3β resulting in inhibition of the opening of mitochondrial permeability transition pore by activating PI3K/Akt and ERK1/2 involving in the reperfusion injury salvage kinase (RISK) pathway. However, whether Apelin plays a functional role in mitochondrial dynamics in cardiomyocytes remains unknown. The aim of this study was to examine the effects of Apelin on MI injury and explored their underlying mechanisms based on Drp1.
RESULTS
Apelin inhibits mitochondrial fission in MI mice and hypoxic cardiomyocytes
Pronounced changes of mitochondrial morphology were consistently observed in the areas proximal to infarct/ischemic zone in MI mice under a transmission electron microscope. Specifically, ischemia decreased mitochondrial size, indicating an enhanced mitochondrial fission. This damaging effect was attenuated in mice treated with Apelin (Figure 1A & 1B).
Figure 1. Apelin inhibits mitochondrial fission in MI mice and in hypoxic cardiomyocytes.

(A) Representative changes of mitochondrial morphology in MI mice under a transmission electron (B) Quantification of the ratio of fissional mitochondrial and individual mitochondrial. (C) Representative confocal microscopic images of mitochondrial morphology in cultured neonatal mice cardiomyocytes. (D) The representative electron microscopy images of mitochondrial morphology. Data are presented as mean ± SEM, #P < 0.05 vs sham, n = 6; *P < 0.05, **P < 0.01 vs MI, n = 6. P values were analyzed using one-way ANOVA.
Similar alterations of mitochondria were consistently observed in cultured neonatal mice cardiomyocytes. As illustrated in Figure 1C & 1D, confocal images revealed that the mitochondria existed a long and filamentous or tubular-shaped structure in cardiomyocytes under a normoxic condition, whereas they degenerated into short, round-shaped morphology like small spheres or short rods typical of mitochondrial fission in cardiomyocytes under a hypoxic condition (hypoxia caused nearly an eightfold increase in the number of cells with mitochondrial fission). With Apelin treatment, the number of cardiomyocytes with mitochondrial fission induced by hypoxia was substantially decreased (Figure 1C & 1D).
Apelin reduces the activity of Drp1
Drp1 is a critical regulator of mitochondrial fission, which typically resides in an inactive form in the cytosol, and when phosphorylated at Ser616, it can be activated and translocated into mitochondria. To confirm the changes of mitochondrial morphology observed in our models indeed representing the mitochondrial fission, we measured Drp1 activities by determination of the protein level of Ser616-phosphorylated form of Drp1 (p-Drp1Ser616) in cardiomyocytes, which has been commonly used as an indication of mitochondrial fission [26, 27]. Our western blot analysis showed no significant change in the total protein level of Drp1 (t-Drp1) in MI hearts (Figure 2A), but a significant increase in p-Drp1Ser616 protein level in MI compared to sham-operated hearts. Notably, administration of Apelin diminished such an MI-induced increase in p-Drp1Ser616 protein level without affecting t-Drp1 (P < 0.01; Figure 2A & 2B). Consistent with the above in vivo results, the expression of p-Drp1ser616 was also markedly increased in primary cardiomyocytes cultured under a hypoxic condition to mimic the major insult of ischemia at the cellular level and abrogated by Apelin treatment (Figure 2C & 2D), while the expression of t-Drp1 remained unaltered.
Figure 2. Apelin reduces the activity of Drp1.

(A) Expression levels of Drp1 in MI mice. (B) Expression levels of p-Drp1Ser616 in MI mice. (C) Expression levels of Drp1 in cultured neonatal mice cardiomyocytes. (D) Representative western blot band of p-Drp1Ser616 in cultured neonatal mice cardiomyocytes. (E) PKA activity in primary cultured cardiomyocytes under a hypoxic condition. Data are presented as mean ± SEM, #P < 0.05 vs sham, n = 6; *P < 0.05 vs MI, n = 8. P values were analyzed using one-way ANOVA.
It has been reported that PKA stimulates Drp1 phosphorylation and induces mitochondrial fission in hypoxia [28]. We therefore tested the effect of Apelin on PKA activity. As depicted in Figure 2E, Apelin inhibited the PKA activity in primary cultured cardiomyocytes under a hypoxic condition.
Apelin attenuates apoptosis of cardiomyocytes through suppressing the mitochondrial death pathway
Increasing evidence indicates that mitochondrial fission plays a crucial role in inducing apoptosis [27]. Inhibition of mitochondrial fission by Apelin is therefore expected to produce anti-apoptotic effects. To test this hypothesis, we carried out the following experiments.
First, we investigated the effects of varying concentrations of Apelin on cell viability by MTT assay. The results shown that hypoxia decreased cardiomyocyte viability (Figure 3A), and Apelin rescued this damaging effect in a concentration-dependent manner.
Figure 3. Apelin attenuates apoptosis of cardiomyocytes through suppressing the mitochondrial death pathway.

(A) The viability of cells in each groups. (B) TUNEL photomicrographs of cardiomyocyte in different groups (magnification ×100). (C) Percentage of TUNEL-positive cells in each group. Data are presented as mean ± SEM. #P < 0.05 vs sham, n = 6; *P < 0.05 vs MI, n = 6. P values were analyzed using one-way ANOVA.
Second, in order to clarify if the changes of cell viability were attributable to apoptotic cell death, we used TUNEL staining to identify chromosomal fragmentation and quantify apoptosis. As depicted in Figure 3B, hypoxia caused a marked increase in the number of TUNEL positive cells, indicating the induction of apoptosis, and such an increase was mitigated following treatment with Apelin (P < 0.05 Apelin/hypoxia vs hypoxia).
Next, we sought to determine whether the mitochondrial apoptosis pathway was involved in the MI injury in our models. To this end, we determined the changes of expression of two mitochondria-associated signaling molecules, a pro-apoptotic protein Bax and an anti-apoptotic protein Bcl-2 in MI hearts with and without Apelin. While the Bax/Bcl-2 ratio significantly increased in MI mice and attenuated by Apelin treatment (Figure 4A). Qualitatively the same results were consistently observed under in vitro conditions: Bax/Bcl-2 ratio was significantly increased in hypoxia and reversed in the presence of Apelin (Figure 4B).
Figure 4. Apelin attenuates apoptosis of cardiomyocytes through suppressing the mitochondrial death pathway.

(A) Representative Western blot band showing levels of Bax and Bcl-2 in MI mice (B) Representative Western blot band showing levels of Bax and Bcl-2 in cultured neonatal cardiomyocytes. (C) The ratio of red and green influence in the change of mitochondrial membrane high potential (red) and low potential (green). (D) Representative fluorescent images of JC-1 under a hypoxic condition. (E and F) Representative Western blot band showing levels of caspase3, cleaved caspase-3, cytochrome C, caspase9, cleaved caspase-9 in the areas proximal to infarct/ischemic zone in MI mice. (G and H) Quantification of cleaved caspase-3, cytochrome C, cleaved caspase-9 in the areas proximal to infarct/ischemic zone in cultured neonatal mice cardiomyocytes. Data are presented as mean ± SEM. #P < 0.05 vs sham, n = 6; *P < 0.05 vs MI, n = 6. P values were analyzed using one-way ANOVA.
The fate of cells succumbing to the intrinsic pathway of apoptosis is sealed by mitochondrial membrane potential (MMP) and loss of MMP is an important event in the mitochondrial apoptosis pathway [29]. As shown in Figure 4C-4D with the results of JC-1 staining to reflect the MMP status, hypoxia caused a robust decrease in red fluorescence signal, suggesting a loss of MMP in cultured cardiomyocytes and such deleterious change was pronouncedly diminished in a concentration-dependent manner when treated with Apelin for 12 h, indicating the protective effect of Apelin on the integrity of mitochondria.
It is known that rupture of mitochondrial membrane or loss of MMP can eventually result in release of cytochrome C from the inner membrane space of mitochondria to the cytosol and subsequent activation of the caspases-9 that ultimately leads to activation of caspase-3, an executioner of apoptosis [30]. Our data depicted in Figure 4E-4H indeed provide the evidence for such a process in cardiomyocytes exposed to a hypoxic condition: hypoxia increased release of cytochrome C and the levels of cleaved or activated caspase 9 and caspase 3 (P < 0.05 vs sham). These damaging alterations were reversed by Apelin (P < 0.05 vs MI).
Apelin improves cardiac function in MI mice
The results presented above demonstrate that p-Drp1ser616-mediated mitochondrial fission is enhanced in MI mice, and inhibition of p-Drpser616 by Apelin reduces mitochondrial fission and consequently reduces cardiomyocyte apoptosis. These findings suggest that Apelin possesses a cardioprotective effect against MI injury and such a beneficial action could manifest at the level of whole heart and overall cardiac function. To test this notion, we conducted the following experiments.
First, we measured the changes of infarct size with or without Apelin. As demonstrated in Figure 5A, MI mice treated with Apelin showed a significant decrease in infarct size compared with non-treated MI mice (16.9 ± 4.75 vs 37.9 ± 10.1%, n = 8, P < 0.05).
Figure 5. Apelin improves cardiac function in MI mice.
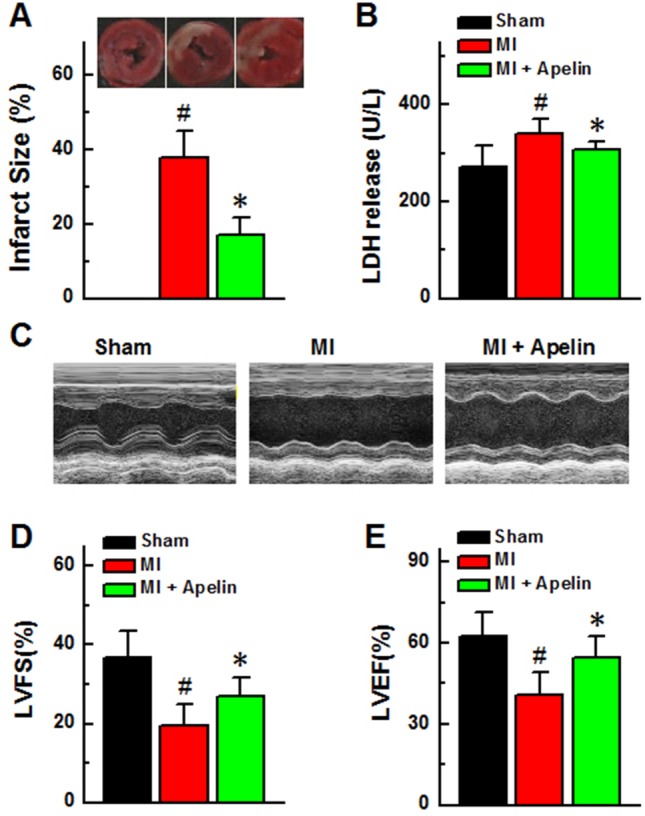
(A) Myocardial infarct sizes in each group and quantification of infarct sizes. Scale bar, 2mm. (B) LDH release. (C) Representative echocardiographic image. (D) Left ventricular fractional shortening. (E) Left ventricular ejection fraction. Data are presented as mean ± SEM. #P < 0.05 vs sham, n = 6; *P < 0.05 vs MI, n = 6. P values were analyzed using one-way ANOVA.
Second, we detected changes of serum LDH levels in MI mice with or without Apelin, as an index of overall cardiac damage. The MI mice received Apelin had considerably lower LDH levels compared with those without Apelin treatment (306 ± 27 vs 341 ± 29 U/L, P < 0.05; Figure 5B).
Finally, we investigated the effects of Apelin on cardiac dysfunction induced by MI using echocardiography. As shown in Figure 5C-5E, administration of Apelin restored the MI-induced decreases in EF and FS (EF: 54.7 ± 7.6% vs 40.9 ± 8.1%; FS: 26.8 ± 5.0 vs 19.5 ± 2.7, P < 0.05).
DISCUSSION
In the present study, we found that Apelin reduced ischemia-induced cardiac injury and improved cardiac function via inhibiting Drp1-mediated mitochondrial fission and its downstream apoptotic signaling, strongly suggesting that the inhibition of Drp1 would be a novel endogenous anti-apoptotic mechanism and promising cardiac protective strategy by which preserves the integrity of mitochondrial membrane and inhibits mitochondria-mediated apoptosis.
Mitochondria are dynamic organelles that undergo cycles of fusion and fission to maintain their morphology and function. Drp1 is one of the proteins proposed to participate in the fission process. Regulation of expression and activity of Drp1 affects mitochondrial fission and mitochondria-dependent apoptosis [31]. Drp1 presents mainly in an unassembled form in the cytosol, but in response to different stimuli such as cardiac ischemia, it can be recruited to the mitochondrial outer membrane and induces mitochondrial fragmentation prior to caspase activation by the release of cytochrome C [32]. It has also suggested that regulation of Drp1 by phosphorylation is important for Drp1 cycling between the cytosol and mitochondria [32]. Two major phosphorylation sites were found in Drp1. When Drp1 was phosphorylated at Ser616 residue, mitochondrial fission occurred, whereas phosphorylation at Ser637 blocked fission and induced fusion and elongation of mitochondria. Drp1 was indicated as one of the PKC-δ substrates and interacting proteins. Recent studies implicated the phosphorylation of Drp1 by CDK5 and PKA promote astrocytes apotosis in the rat hippocampus. Taguchi et al. reported that human Drp1 can be activated by Cdk1/cyclin B-mediated phosphorylation at the site of Ser616 in the variable domain leading to Drp1-dependent mitochondrial fission [33]. In the present study, we found that the level of p-Drp1Ser616 increased after ischemia and under condition of hypoxia accompanied by mitochondrial fission and these alternations could be mitigated dramatically in the presence of Apelin. Moreover, Apelin effectively prevented cardiomyocyte apoptosis by inhibiting the release of cytochrome C and subsequent activation of the caspase-9 - caspase-3 cascade. Furthermore, Apelin markedly improved cardiac function as the result of infarct size as well as serum LDH level reductions in MI mice. To the best of our knowledge, this study represents the first evidence for the cytoprotective action of Apelin in ischemic injury and inhibition of Drp1 as a mechanism underlying its anti-apoptotic property. Our study extends the findings by Ong et al and suggests that inhibition of Drp1 has a therapeutic potential for patients diagnosed with ischemic heart disease.
Several studies about apoptosis revealed the causal relationship between mitochondrial fission and the induction of apoptosis. In our study, we observed that hypoxia induced mitochondrial fission and cardiomyocyte apoptosis, which could be suppressed by Apelin through either downregulating p-Drp1Ser616 level or suppressing Drp-1 activation, or both. This effect resulted in the inhibition of the cytochrome C release and subsequent activation of the caspase-9 - caspase-3 cascade. These results showed that Apelin improved mitochondrial dysfunction and attenuated mitochondria-dependent apoptosis. Likely by acting on this signaling pathway, Apelin reduced infarct size and improved the impaired cardiac function induced by ischemia. This is in agreement with the notion from Rastaldo et al. that Apelin could limit infarct size and improve cardiac post-ischemic mechanical recovery only if given after ischemia [24].
In addition to the mitochondrial death pathway, Apelin has also been documented to act via other singling mechanisms. For instance, Tao et al. reported that Apelin protects the heart against I/R injury through inhibition of the ER (endoplasmic reticulum)-dependent apoptotic pathways [25]. In such a circumstance, Apelin might inhibit the opening of mitochondrial permeability transition pore and stimulate the reperfusion injury RISK pathway by inactivating GSK-3β to activate PI3K/Akt and ERK1/2. It is possible that such a pathway is also involved in our models, and this requires separate studies to verify.
In summary, our data indicated that Apelin decreased p-Drp1Ser616 level likely by suppressing calcineurin activity, leading to inhibition of mitochondria fission. This beneficial effect may play a key role in maintenance of the integrity of mitochondrial membrane and further to protect from the loss of MMP so as to inhibit the mitochondria-mediated apoptosis. Future investigation should be undertaken to address the detailed molecular mechanisms by which Apelin interacts with Drp1 and links between Drp1 and the mitochondrial death pathway.
MATERIALS AND METHODS
Animals
All animal experiments were approved by the Committee on Animal Experimentation of Harbin Medical University (Approval ID: DEC6121) and were conducted in accordance with the international guidelines regarding animal experimentation. Male C57BL6/J mice aged 8-10 weeks and weighing 20-25 g were obtained from the Animal Centre of Harbin Medical University. The mice were maintained in a pathogen-free, temperature-controlled animal house with a 12:12-h light/dark cycle, and given free access to a standard diet and tap water. Mice were randomly divided into four groups: sham-operated rats (control group, n = 8), myocardial infarction group (MI group, n = 8), and MI with Apelin group (MI + Apelin groups (1, 15, and 30 μg/kg, respectively; n = 8).
Chemicals
Apelin was obtained from Sigma Company (A6469, Sigma, china), Apelin stock solution was prepared in double polished distilled water (ddH2O) and to prepared relevant concentration.
Mouse model of myocardial infarction
Adult male C57BL/6J mice were treated with Apelin for 4 weeks (i.p. 1, 15, 30 μg/kg/day, respectively) and then subjected coronary artery ligation (LAD) to induce MI. MI model was carried out by LAD as described elsewhere [34]. In brief, mice were anaesthetized with a mixture of xylazine (5 mg/kg, i.p) and ketamine (100 mg/kg, i.p) and the depth of anesthesia was assessed by monitoring the pedal withdrawal reflex. Mice were then ventilated and subjected to a left-sided thoracotomy and the left coronary artery ligation to induce MI. Sham-operated rats underwent an identical surgical procedure without LAD ligation. After 6 hours of LAD ligation, we evaluate their cardiac function and measure the infarct size using hearts tissue. The heart was quickly excised and weighted in cold (4°C) buffer. The left ventricle tissue was then rapidly frozen in liquid nitrogen and stored at -80°C for subsequent western blot analysis.
Echocardiographic measurements of cardiac function
After 6 h ligation of LAD, mice were anaesthetized with sodium pentobarbital (50 mg/kg, i.p, Merck). We used transthoracic echocardiography with an ultrasound machine (Vivid 7, GE Medical, Horten, Norway) equipped with a 10-MHz phased-array transducer to test left ventricular ejection fraction (EF), and fractional shortening (FS) were calculated from M-mode recording.
Measurement of infarct size by tetrazolium chloride staining
Myocardial infarct size was evaluated by 2, 3, 5-triphenyltetrazolium chloride triazole (TTC) (Solarbio, Beijing, China) staining. Briefly, after 6 h ligation, mice were euthanized and hearts were removed rapidly to measure infarct size and transmission electron microscopy. After wash with phosphate buffer solution (PBS, pH 7.4) and frozen at -20°C for 2 h, the left ventricular (LV) wall was cut into 6 pieces with cross-section of 2-mm thickness. The preparations were incubated in 1% TTC in 0.1 mol/L PBS at 37°C for 15 min, and subsequently fixed in 10% formalin solution (pH 7.4) for 12 h. The infarct area (IS, white color) and area at risk (AAR, red color) were identified.
Transmission electron microscopic study
Mice were evaluated cardiac function after LAD, hearts were cut into pieces and fixed in 0.1 mol/L sodium phosphate buffer containing 2.5% glutaraldehyde for 3 h at 4°C, then osmicated in 1% osmium tetroxide for 1 h at 4°C. After dehydration of the tissues by using ethanol series, the samples were embedded in Epon 812 and sectioned using a Leica EM UC6 (Leica Coviema, Austria) ultra-microtome. Sections were viewed and photographed using a Hitachi 7650 TEM (Hitachi, Tokyo, Japan) at 80 kv.
Western blot analysis
Western blotting was performed as previously described [35]. Briefly, cells were lysed with modified RIPA buffer on ice for 30 min, and cell lysates were subsequently centrifuged at 12,000 g at 4°C for 20 min. The supernatant was transferred to a fresh ice-cold tube, and protein concentrations were determined via a Bio-Rad protein assay. Equal concentrations of protein samples were mixed with SDS sample buffer and denatured at 95°C for 5 min. Samples were resolved in 8% SDS-page gel. Gels were transferred onto nitrocellulose membranes that were then blocked with 5% non-fat dried milk in TTBS (0.1% Tween-20) for 1 h. Following blocking, the blots were incubated with primary antibodies at 4°C overnight. The antibodies against Bax, Bcl-2, total Drp1 protein (t-Drp1; Proteintech Group, Chicago, USA), phosphorylated form of Drp1 at Ser616 (p-Drp1ser616), cytochrome C, caspase 9, and caspase 3, were obtained from Santa Cruz Biotech (Santa Cruz, CA, USA). Following washing with TTBS, the membranes were incubated with HRP-conjugated secondary antibodies (1:5,000, Santa Cruz Biotechnology) for 1 h. Finally, the membranes were washed again with TTBS, and the blots were determined using an enhanced chemiluminescence detection system (ECL; GE Healthcare, Bio-Science AB, Uppsala, Sweden).
Cell culture
Hearts from 2∼3-day old mice were finely minced and digested using type II collagenase (120 units/ml; Worthington Biochemical Corp., Lakewood, NJ) for 10 min, and supernatant was collected. Cardiomyocytes were maintained in DMEM supplemented with penicillin and streptomycin (1%) and fetal bovine serum (FBS; 10%). Cardiomyocytes at the 2nd passage were used in our experiments.
Primary cultures of neonatal mice cardiomyocytes were prepared from 1∼3-day-old Wistar rats by trypsin. Briefly, the ventricular myocardium was minced in DMEM (Hyclone, City, USA). After each of six successive incubations, the cells were suspended in DMEM containing 10% FBS (Hyclone) and centrifuged. Pooled cells were plated onto dishes at a density of 1×105 cells/cm2 and incubated at 37°C in humidified air with 5% CO2 for 72 h, and 0.1 mM bromodeoxyuridine (Sigma, St. Louis, USA) was added into the medium to deplete non-cardiomyocytes. Before experiments, the cells were starved for 24 h and then treated with Apelin at varying concentrations (1, 10 and 100 nM) for 12 h. Then induced by hypoxia for 12h, cell viability was determined by MTT assay.
Measurement of serum LDH levels
Cytotoxicity was measured by lactate dehydrogenase (LDH) release into the blood serum using a commercially available kit (Jiancheng Bioengineering Institute, Nanjing, China), and assessed according to the following equation: LDH release = (LDH activity in the medium/total LDH activity) × 100%.
Measurement of the mitochondrial membrane potential (MMP)
The lipophilic cationic probe JC-1 (Beyotime, Shanghai, China) was employed to measure the mitochondrial membrane potential (ΔΨm) of primary mice cardiomyocytes according to the manufacturer’s directions. Briefly, cells were incubated with JC-1 staining solution (5 μg/mL) at 37°C for 20 min and rinsed twice with JC-1 staining buffer. Then the fluorescence intensity of both mitochondrial JC-1 monomers (excitation wavelength λex 514 nm and emission wavelength λem 529 nm) and aggregates (λex 585 nm and λem 590 nm) was detected using a monochromatic microplate reader (Safire II, Tecan, City, Switzerland). ΔΨm was calculated as the ratio of red to green fluorescence.
TUNEL staining
Cell apoptosis was detected with TUNEL assay (In Situ Cell Death Detection Kit, Roche, Indianapolis, Switzerland). DNA fragmentation of individual cells was further detected in situ by TUNEL. Cells were observed by a fluorescence microscope (Eclipse 80I, Nikon, City, Japan).
Statistical analysis
All values are expressed as the mean ± SEM. Differences between two groups were determined via a Student’s t-test. Comparisons between more than two groups, data were assessed using one-way ANOVA followed by a Bonferroni’s correction. A value of P < 0.05 was considered as statistically significant.
Abbreviations
- (Drp1)
dynamin-related protein 1
- (MI)
myocardial infarction
- (LAD)
coronary artery ligation
- (p-Drp1ser 616)
phosphorylated Drp1
- (MMP)
mitochondrial membrane potential
- (APJ)
apelin related receptor
- (RISK)
salvage kinase
Footnotes
Author contributions
Wei Xu, Hongwei Yu, Rong Zhang, Yuhong Zhou and Hongli Shan contributed to conception and design; Wei Xu, Hongwei Yu, Ruixue Ma, Lina Ma, Changyu Wu, Rong Zhang contributed to acquisition of data, or analysis and interpretation of data; Wei Xu, Hongwei Yu, Yuhong Zhou and Hongli Shan have been involved in drafting the manuscript or revising it critically for important intellectual content; all authors have given final approval of the version to be published.
CONFLICTS OF INTEREST
The authors confirm that there are no conflicts of interest.
FUNDING
This project was supported by the National Natural Science Foundation of China (81673425 and 81573437 and 81400294 and 81470523) and China postdoctoral funding (2014M561377).
REFERENCES
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, et al. Heart disease and stroke statistics-2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.O’Gara PT, Kushner FG, Ascheim DD, Casey DE, Jr, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, et al. American College of Emergency Physicians, and Society for Cardiovascular Angiography and Interventions. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;61:e78–e140. doi: 10.1016/j.jacc.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Collf Cardiol. 2012;59:969–978. doi: 10.1016/j.jacc.2011.07.054. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez-Cascales J. Resveratrol enhances the inotropic effect but inhibits the proarrhythmic effect of sympathomimetic agents in rat myocardium. Peer J. 2017;5:e3113. doi: 10.7717/peerj.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghaboura N, Tamareille S, Ducluzeau PH, Grimaud L, Loufrani L, Croue A, Tourmen Y, Henrion D, Furber A, Prunier F. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3beta signaling. Basic Res Cardiol. 2011;106:147–162. doi: 10.1007/s00395-010-0130-3. [DOI] [PubMed] [Google Scholar]
- 6.Lejay A, Fang F, John R, Van JA, Barr M, Thaveau F, Chakfe N, Geny B, Scholey JW. Ischemia reperfusion injury, ischemic conditioning and diabetes mellitus. J Mol Cell Cardiol. 2016;91:11–22. doi: 10.1016/j.yjmcc.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 7.Yamazaki KG, Romero-Perez D, Barraza-Hidalgo M, Cruz M, Rivas M, Cortez-Gomez B, Ceballos G, Villarreal F. Short- and long-term effects of (-)-epicatechin on myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2008;295:H761–H767. doi: 10.1152/ajpheart.00413.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burn BR, Varner KJ. Environmentally persistent free radicals compromise left ventricular function during ischemia/reperfusion injury. Am J Physiol Heart Circ Physiol. 2015;308:H998–H1006. doi: 10.1152/ajpheart.00891.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 10.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 13.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 14.Tanwar DK, Parker DJ, Gupta P, Spurlock B, Alvarez RD, Basu MK, Mitra K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget. 2016;7:60021–60037. doi: 10.18632/oncotarget.11047. https://doi.org/10.18632/oncotarget.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding M, Dong Q, Liu Z, Liu Z, Qu Y, Li X, Huo C, Jia X, Fu F, Wang X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia-reperfusion injury in diabetic mice. Cardiovasc Diabetol. 2017;16:19. doi: 10.1186/s12933-017-0501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–1458. doi: 10.1038/cdd.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44:532–544. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawamata Y, Habata Y, Fukusumi S, Hosoya M, Fujii R, Hinuma S, Nishizawa N, Kitada C, Onda H, Nishimura O, Fujino M. Molecular properties of apelin: tissue distribution and receptor binding. Biochim Biophys Acta. 2001;1538:162–171. doi: 10.1016/s0167-4889(00)00143-9. [DOI] [PubMed] [Google Scholar]
- 20.Koguchi W, Kobayashi N, Takeshima H, Ishikawa M, Sugiyama F, Ishimitsu T. Cardioprotective effect of Apelin on cardiac performance and remodeling in end-stage heart failure. Circ J. 2012;76:137–144. doi: 10.1253/circj.cj-11-0689. [DOI] [PubMed] [Google Scholar]
- 21.Zhong JC, Zhang ZZ, Wang W, McKinnie SMK, Vederas JC, Oudit GY. Targeting the apelin pathway as a novel therapeutic approach for cardiovascular diseases. Biochim Biophys Acta. 2017;1863:1942–1950. doi: 10.1016/j.bbadis.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Li G, Sun X, Zhao D, He L, Zheng L, Xue J, Wang B, Pan H. A promoter polymorphism in APJ gene is significantly associated with blood pressure changes and hypertension risk in Chinese women. Oncotarget. 2016;7:86257–86265. doi: 10.18632/oncotarget.13370. https://doi.org/10.18632/oncotarget.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang P, Xu C, Wang C, Wu Y, Wang D, Chen S, Zhao Y, Wang X, Li S, Yang Q, Zeng Q, Tu X, Liao Y, et al. Association of SNP Rs9943582 in APLNR with left ventricle systolic dysfunction in patients with coronary artery disease in a Chinese Han GeneID population. PLoS One. 2015;10:e0125926. doi: 10.1371/journal.pone.0125926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rastaldo R, Cappello S, Folino A, Berta GN, Sprio AE, Losano G, Samaja M, Pagliaro P. Apelin limits infarct size and improves cardiac postischemic mechanical recovery only if given after ischemia. Am J Physiol Heart Circ Physiol. 2011;300:H2308–2315. doi: 10.1152/ajpheart.01177.2010. [DOI] [PubMed] [Google Scholar]
- 25.Tao J, Zhu W, Li Y, Xin P, Li J, Liu M, Li J, Redington AN, Wei M. Apelin protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am J Physiol Heart Circ Physiol. 2011;301:H1471–H1486. doi: 10.1152/ajpheart.00097.2011. [DOI] [PubMed] [Google Scholar]
- 26.Chen SD, Zhen YY, Lin JW, Lin TK, Huang CW, Liou CW, Chan SH, Chuang YC. Dynamin-related protein 1 promotes mitochondrial fission and contributes to the hippocampal neuronal cell death following experimental status epilepticus. CNS Neurosci Ther. 2016;22:988–999. doi: 10.1111/cns.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P, Wang P, Liu B, Zhao J, Pang Q, Agrawal SG, Jia L, Liu FT. Dynamin-related protein Drp1 is required for Bax translocation to mitochondria in response to irradiation-induced apoptosis. Oncotarget. 2015;6:22598–22612. doi: 10.18632/oncotarget.4200. https://doi.org/10.18632/oncotarget.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyun HW, Min SJ, Kim JE. CDK5 inhibitors prevent astroglial apoptosis and reactive astrogliosis by regulating PKA andDRP1 phosphorylations in the rat hippocampus. Neurosci Res. 2017;119:24–37. doi: 10.1016/j.neures.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S, Eroglu E, Stokes JA, 3rd, Scissum-Gunn K, Saldanha SN, Singh UP, Manne U, Ponnazhagan S, Mishra MK. Resveratrol induces mitochondria-mediated, caspase-independent apoptosis in murine prostate cancer cells. Oncotarget. 2017;8:20895–20908. doi: 10.18632/oncotarget.14947. https://doi.org/10.18632/oncotarget.14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Zhang X, Yang J, Zhang Y, Zhu D, Zhang L, Zhu Y, Li D, Zhou J. Methylation of BNIP3 in pancreatic cancer inhibits the induction of mitochondrial-mediated tumor cell apoptosis. Oncotarget. 2017;8:63208–63222. doi: 10.18632/oncotarget.18736. https://doi.org/10.18632/oncotarget.18736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang C, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–39. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, Riquelme JA, Pedrozo Z, Chiong M, Sánchez G, Lavandero S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J Cardiovasc Pharmacol. 2014;63:477–87. doi: 10.1097/FJC.0000000000000071. [DOI] [PubMed] [Google Scholar]
- 33.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 34.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 35.Zhou YH, Deng L, Zhao DD, Chen LL, Yao Z, Guo XW, Liu X, Lv LF, Leng B, Xu W, Qiao GF, Shan HL. MicroRNA-503 promotes angiotensin II-induced cardiac fibrosis by targeting Apelin. J Cell Mol Med. 2016;20:495–505. doi: 10.1111/jcmm.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]