

Abstract
There are countless biologically active organic molecules that contain one or more N-containing moieties and broadly applicable and efficient catalytic transformations that deliver them diastereo- and/or enantioselectively are much sought after. Various methods for enantioselective synthesis of α-secondary amines are available (e.g., from additions to protected/activated aldimines), but those involving ketimines are much less common. There are no reported additions of carbon-based nucleophiles to unprotected/unactivated (or N-H) ketimines. Here, we report a catalytic, diastereo- and enantioselective three-component strategy for merging an N-H ketimine, a monosubstituted allene and B2(pin)2, affording products in up to 95% yield, >98% diastereoselectivity and >99:1 enantiomeric ratio. Utility of the approach is highlighted by synthesis of the tricyclic core of a class of compounds that have been shown to possess anti-Alzheimer activity. Stereochemical models, developed with the aid of DFT calculations, which account for the observed trends and levels of enantioselectivity are presented.
Graphical abstract
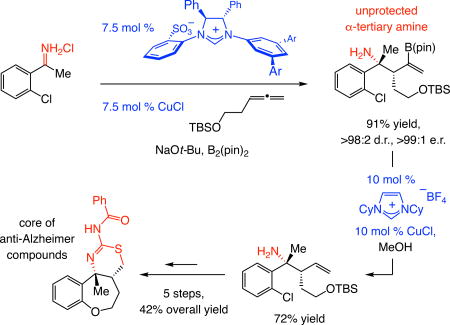
Catalytic enantioselective additions of carbanions to ketimines deliver products with a nitrogen-substituted quaternary stereogenic center (α-tertiary amine) but development of these transformations1,2,3,4 is hardly straightforward. Ketimines are less reactive than aldimines and the reduced size difference between the substituents compared to aldimines makes enantiotopic face differentiation difficult. Catalytic enantioselective additions of allyl moities5 to ketimines, while much sought after, remain scarce. One study shows that reactions of allyl–B(pin) (pin = pinacolato) with acyclic N-benzyl ketimines may be promoted by a chiral bis-phosphine–Cu complex (Fig. 1a)6, and another deals with reactions of functionalized allylsilanes and tosyl-protected ketimines catalyzed by phosphoramidite–Pd complexes (Fig. 1a)7. Other disclosures cover highly activated ketimines, including cyclic sulfonylimines and their reaction with potassium allyltrifluoroborates (with Rh-based catalysts)8, 9 and isatin-derived N-Boc-ketimines and their reaction with allylsilanes (with Pd-based complexes and stoichiometric silver fluoride)10. Other approaches involve either enantiomerically pure ketimines11, 12 or enantiomerically pure allyl reagents13,14,15. Our goal was to develop a method that would not require ketimine activation/protection and subsequent unmasking (Fig. 1b). The absence of a protecting group would bypass the intermediacy of E and Z mixtures of ketimine isomers, which can lead to lowering of enantioselectivity. Although preparation of N-H ketimines by condensation of ketones with ammonia followed by reaction with allylboron reagents is known16, as far as we are aware, diastereo- and/or enantioselective variants have not been introduced.
Figure 1. State-of-the-art in allyl additions to ketimines and goals of this study.

There are significant exisiting limitations and a number of compelling issues remain unaddressed. a, There are only a small number of methods for catalytic enantioselective addition of an allyl group to a ketimine. The substrate is typically equipped with an activating/protecting group, which might prove difficult to remove in the presence of similar functional groups within a product structure (e.g., another N-benzylamine). b, A direct approach to synthesis of α-tertiary amines may entail preparation of the requisite unprotected N-H ketimine through alkylation of readily available nitriles followed by catalytic site-, diastereo- and enantioselective multicomponent addition of 2-boryl-substituted allyl groups. One application relates to synthesis of the core tricyclic structure of a set of heterocyclic molecules that exhibit strong anti-Alzheimer activity. Bn, benzyl; Ts, tosyl; Ac, acyl; pin, pinacolato; G, R, L, functional groups; Pg, protecting group.
Based on the earlier investigations regarding enantioselective additions to aldehydes or ketones17,18,19, which were recently extended to N-anisidyl aldimines20,21, we envisioned the sequence in Fig. 1b. N-H ketimines would be accessed by addition of an organolithium or a Grignard reagent to a nitrile, many of which are commercially available22,23. The ensuing catalytic process would combine an N-H ketimine, a monosubstituted allene and B2(pin)2 to generate homoallylic amines containing a pair of stereogenic centers and an alkenyl–B(pin) group. A number of biologically active organic molecules would thus become more readily accessible in enantiomerically enriched form; an example would be of the core structure of class of polycyclic compounds shown to possess the ability to reduce beta-amyloid production (Fig. 1b)24, 25. Complications typically associated with site-selective removal of protecting/activating units would thus be obviated, particularly when relatively strong reducing (e.g., for an N-benzyl or an N-tosyl group, Fig. 1a) or oxidizing conditions20 are needed and a stereogenic benzylic C–N bond is present.
Results
Establishing feasibility
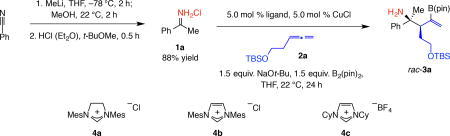
We began with the reaction of allene 2a with N-H ketimine 1a, obtained from the reaction of benzonitrile with methyllithium (MeLi; 88% yield; Table 1). Diastereoselectivity was complete in every case [>98:2 diastereomeric ratio (d.r.)] but efficiency was catalyst-dependent. There was 60–75% conversion to rac-3a with Cu complexes derived from triphenylphosphine, tricyclohexylphosphine or racemic 2,2’-bis(diphenylphosphino)-1,1’-binaphthalene (rac-binap) (entries 1–3). Evaluation of N-heterocyclic carbene (NHC) complexes (entries 4–6) showed that the combination of cyclohexyl-substituted imidazolium salt 4c and CuCl is the most effective: rac-3a was obtained in 90% yield and >98:2 d.r. (Table 1, entry 6).
Table 1.
Probing efficiency and diastereoselectivity with different (achiral) catalyst types.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Ligand | Conv. (%)* | Yield (%)† | d.r.* |
| 1 | PPh3 | 60 | 49 | >98:2 |
| 2 | PCy3 | 71 | 52 | >98:2 |
| 3 | rac-binap | 75 | 72 | >98:2 |
| 4 | 4a | 80 | 66 | >98:2 |
| 5 | 4b | 55 | 53 | >98:2 |
| 6 | 4c | >98 | 90 | >98:2 |
Reactions were carried out under N2 atmosphere; 1.2:1 ratio of ketimine:allene was used.
Conversion (based on allene consumption; (includes desired and decomposition products) and diastereomeric ratio (d.r.) values were measured by analysis of 400 MHz 1H NMR spectra of unpurified mixtures; variance of values is estimated to be <±2%.
Yield of isolated and purified product (<±5%). See the Supplementary Information for details).
Abbreviations: TBS, tert-butyldimethylsilyl; pin, pinacolato; Mes, 2,4,6-Me3C6H2; Cy, cyclohexyl; rac, racemic; binap, 2,2’-bis(diphenylphosphino)-1,1’-binaphthalene.
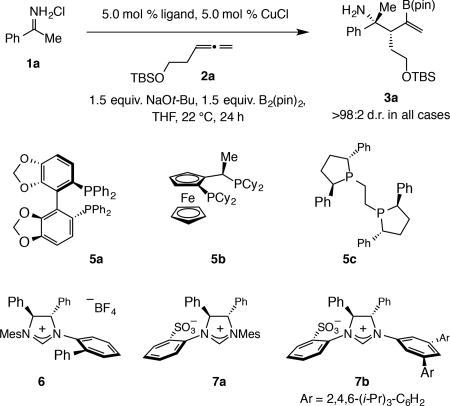
Identifying a chiral catalyst
Several types of Cu complexes were examined (Table 2). Enantioselectivity was minimal with binap [(2,2'-bis(diphenylphosphino)-1,1'-binaphthyl); entry 1, 44:56 enantiomeric ratio (e.r.)]. The desired product was isolated in appreciable yield and e.r. with segphos (5a, 51% yield, 18:82 e.r.; entry 2) or josiphos (5b, 51% yield, 19.5:80.5 e.r.; entry 3), but less so with the more conformationally flexible 5c (27%, 69:31, respectively; entry 4).
Table 2.
Studies to identify an effective chiral catalyst.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Ligand | Conv. (%)* | Yield (%)† | e.r.†† |
| 1 | R-binap | 70 | 65 | 44:56 |
| 2 | 5a | 55 | 51 | 18:82 |
| 3 | 5b | 65 | 51 | 19.5:80.5 |
| 4 | 5c | 29 | 27 | 69:31 |
| 5 | 6 | 80 | 51 | 40:60 |
| 6 | 7a | >98 | 65 | 55:45 |
| 7 | 7b | 54 | 51 | 95:5 |
Reactions were carried out under N2 atmosphere; 1.2:1 ratio of ketimine:allene was used.
Conversion (consumption of allene; (includes desired and decomposition products) and d.r. was measured by analysis of 400 MHz 1H NMR spectra of unpurified mixtures; variance of values is estimated to be <±2%.
Yield of isolated and purified products (<±5%).
Enantiomeric ratio (e.r.) values were determined by HPLC analysis (<±1%) (see the Supplementary Information for details).
Abbreviations: TBS, tert-butyldimethyl silyl; pin, pinacolato; Mes, 2,4,6-Me3C6H2; Cy, cyclohexyl; binap, 2,2’-bis(diphenylphosphino)-1,1’-binaphthalene.
With imidazolinium salt 6 or sulfonate-bearing 7a enantioselectivity remained low (entries 5–6). However, when the mesityl (Mes) moiety of 7a was replaced by a 3,5-diaryl-substituted phenyl moiety (7b, entry 7) enantioselectivity increased dramatically: 3a was obtained in 95:5 e.r. (for X-ray structure of the derived alcohol, see Fig. 3a). The effectiveness of the 7b-derived catalyst was surprising for several reasons. Enantioselectivity was considerably higher compared to the closely related N-mesityl-substituted variant (7a; see below for mechanistic analysis). Additionally, while sulfonate-containing NHC–Cu catalysts have been used for enantioselective allylic substitutions26,27 and conjugate additions28,29,30, none emerged as optimal for a 1,2-addition.
Figure 3. Stereochemical models.

DFT calculations shed light on the origins of enantioselectivity. a, Transition states with a N→Na interaction account for high e.r.; I represents the preferred mode. b, The model suggests that disruption of the N→Na coordination by the long, flexible alkyl ketimine chain (3w,x Figure 2) in V might render mode VI competitive, leading to lower e.r. Free energy values relative to the major pathway refer to the MN12SX/Def2TZVPPTHF(PCM) level after geometry optimization performed with either MN12SX/Def2SVPTHF(PCM) (for a) or M06L/Def2SVPTHF(PCM) (for b and c). For details, see Sections 4 and 5 of the the Supplementary Information. Abbreviations: NHC, N-heterocyclic carbene; THF = tetrahydrofuran.
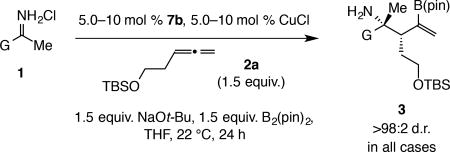
Applicability
The method has considerable scope (Table 3). At times higher catalyst loading was necessary for high conversion (e.g., 3a, entry 1 Table 3 vs. entry 7, Table 2) and ketimine:allene ratio was changed to 1:1.5 for better yield (from 1.2:1). Regardless of whether the N-H ketimine had an ortho aryl substituent that is electron donating (3b), electron withdrawing (3c) or relatively sizeable (3f–g), products were isolated in 59–95% yield, >98:2 d.r. and 98.5:1.5–99.5:0.5 e.r. Reactions with meta-substituted N-H ketimines were similarly efficient and selective (entries 8–10, Table 3). In reactions of ketimines with different para-substituted aryl units (see 3k–3m) e.r. ranged from 92.5:7.5 to 95:5. Products 3n–o (Table 3, entries 11–12), from reactions with the less electrophilic alkyl-substituted N-H ketimines, were isolated in 38% and 48% yield and 91:9 and 95:5 e.r., respectively. Fluoroaryl-substituted amines 3d–e (entries 4–5), 3h (entry 8), and 3l (entry 12) were obtained in 64–91% yield and 94:6–98:2 e.r. (>98:2 d.r.); the high yield and e.r. in these transformations, regardless of the position of the fluorine atom, shows that fluorine-metal interaction31,32,33 does not exert an adverse influence. Diastereoselectivity was exceptional throughout (>98:2 d.r.).
Table 3.
Catalytic diastereo- and enantioselective additions to N-H ketimines.
| |||
|---|---|---|---|
|
| |||
| Entry | G | Mol %; Yield (%)† | e.r.†† |
| 1 | C6H5 (a) | 7.5; 76 | 95:5 |
| 2 | o-MeOC6H4 (b) | 10; 95 | 98.5:1.5 |
| 3 | o-ClC6H4 (c) | 7.5; 91 | 99.5:0.5 |
| 4 | o-FC6H4 (d) | 7.5; 72 | 97.5:2.5 |
| 5 | o,o-F2C6H3 (e) | 7.5; 91 | 98:2 |
| 6 | 1-naphthyl (f) | 10; 66 | 98.5:1.5 |
| 7 | o-MeC6H4 (g) | 7.5; 59 | 99.5:0.5 |
| 8 | m-FC6H4 (h) | 5.0; 64 | 94:6 |
| 9 | m-MeOC6H4 (i) | 10; 57 | 98.5:1.5 |
| 10 | 2-naphthyl (j) | 7.5; 81 | 93:7 |
| 11 | p-ClC6H4 (k) | 7.5; 56 | 92.5:7.5 |
| 12 | p-FC6H4 (l) | 5.0; 71 | 95:5 |
| 13 | p-MeOC6H4 (m) | 7.5; 39 | 92.5:7.5 |
| 14 | CyCH2 (n) | 10; 38 | 91:9 |
| 15 | Cy (o) | 10; 48 | 95:5 |
Reactions were carried out under N2 atmosphere; >98% disappearance of ketimine in all cases (includes desired and decomposition products).
Yield of isolated and purified products (<±5%).
Enantiomeric ratios determined by HPLC analysis (<±1%; see the Supplementary Information for details).
Abbreviations: TBS, tert-butyldimethylsilyl; pin, pinacolato; Cy, cyclohexyl.
Experiments were performed at least in triplicate. See the Supplementary Information for details and the results with achiral imidazolinium salt 4c.
Additions to heterocyclic N-H ketimines afforded products in high e.r. (cf. 3p–3r, Fig, 2a), although efficiency was slightly lower. Synthesis of amines 3s–3u shows that different monosubstituted allenes may be used, but the size of the allene substituent did impact efficiency (41–78% yield, >98:2 d.r., up to >99:1 e.r.).
Figure 2. Further exploration of scope and illustration of utility.

A variety of desirable products can be synthesised. a, The method is applicable to a variety of heterocyclic substrates and allenes. Products derived from ketimines containing n-alkyl or iso-alkyl substituents (vs. methyl) can be obtained efficiently, in >98:2 d.r. and 85.5:14.5–96:4 e.r., depending on the substituent identity. For results with achiral imidazolinium salt 4c, see the Supplementary Information Table 1. b, Oxidation of the alkenylboronate moiety within the products derived from the NHC–Cu-catalyzed multicomponent reactions proceed efficiently to deliver the corresponding β-amino ketones (e.g., 9a), which represent the products of diastereo- and enantioselective Mannich-type additions. c, The method may be applied to the synthesis of the polycyclic core of compounds recently implicated in the fight against Alzheimer’s disease. Conversion of the C–B(pin) to a C–H bond promoted by a readily accessible NHC–Cu complex afforded 10. Formation of the derived thiourea and another NHC–Cu-catalyzed reaction generated the oxepane ring of 12. A two-step procedure involving oxidative cleavage/reduction and activation of the resulting primary alcohol delivered the desired aminothiazine ring and the strained tricyclic 13. Reactions were performed under N2; there was >98% disappearance of ketimine in all cases (might include decompositiopn products). Yields correspond to isolated and purified products and represent an average of at least three runs (±5%). Diastereomeric ratios were determined by analysis of the 400 MHz 1H NMR spectra of unpurified product mixtures (±2%). Enantiomeric ratios were determined by HPLC analysis (± 1%). See the Supplementary Information for experimental details and spectroscopic analyses.
The method extends beyond methyl-substituted substrates. Ketimines bearing an n-alkyl or unsaturated alkyl group (e.g., 3v–x, Fig. 2a) and the iso-propyl-containing ketimine precursor to 3y were converted to the desired amines efficiently and with exceptional diastereoselectivity. Nevertheless, e.r. varied depending on the substituent. Whereas 3v was formed in 94:6 e.r., there was gradual diminution in enantioselectivity as the side chain became longer (e.g., 88:12 e.r. for n-butyl-substituted 3w, 85.5:14.5 e.r. for pentenyl-substituted 3x). More enantioselective was the reaction that afforded iso-poropyl-containing 3y (96:4 e.r.); the X-ray structure secured for the N-acetyl derivative of 3y (Fig. 2a) indicates that there is no reversal in stereochemical induction (see Fig. 3a for corroborative X-ray data). A possible rationale for these selectivity trends will be provided below. Various compounds of interest, such as those derived from intramolecular hydroamination that afford heterocyclic derivatives34, 35, may be accessed through functionalization of compounds such as 3x. Nonetheless, there are limitations. Reactions of ketimines that contain an α- or β-alkoxy or a benzyl group are inefficient, likely due to facile decomposition (enamine formation and β-elimination, respectively). The same applies to additions to trifluoromethyl-substituted ketimines (decomposition to unidentified products). There was no reaction with phenyl-tert-butyl N-H ketimine.
Utility
Catalytic enantioselective addition to ketimine 8 followed by oxidation of the C–B bond gave β-amino ketone 9a, the product of a Mannich-type addition, in 70% overall yield, yield and without any loss in d.r. (Fig. 2b). The efficiency with which 9a was obtained is higher than those shown in Table 3 and Fig. 2a, indicating that there might be some decomposition during purification and that yields may be improved if the alkenyl–B(pin) moiety is modified. The absolute stereochemistry of the product was confirmed through X-ray structure of primary alcohol 9b. It merits note that catalytic enantioselective enolate additions to N-activated ketimines (e.g., N-phosphinoylketimines36,37 or those derived from α-ketoesters38 or diethyl ketomalonate39) are limited in scope (see the Supplementary Information for additional references).
We then investigated the possibility of application to enantioselective synthesis of the core structure of the aforementioned anti-Alzheimer compounds (Fig. 2c). NHC–Cu-catalyzed protodeboration17 of enantiomerically enriched alkenyl–B(pin) amine 3c furnished 10 in 72% yield. Synthesis of α-olefin 10 by a related route and with a sterically less demanding Cu–H complex [vs. Cu–B(pin) addition/protodeboration] would present a chemoselectivity issue (competitive reaction with ketimine40); moreover, we find that the presence of a B(pin) group is critical to high enantioselectivity (see Fig. 3). Thiourea generation and removal of the silyl group afforded alcohol 11 in 71% overall yield. The cyclic ether was formed by treatment of 11 with 10 mol % CuI and 20 mol % 8-hydroxyquinoline (110 °C, 24 h)41, affording oxepane 12 in 78% yield. Oxidative cleavage of the vinyl group, reduction of the resulting aldehyde and subjection of the resulting primary alcohol to triflic anhydride (−20 °C, 2 h)25 delivered 13 in 75% yield after recrystallization (this compound is unstable towards a variety of chromatography procedures). The aryl ring in 13 may be functionalized site selectively according to formerly reported procedures42,43,44 (see the Supplementary Information for extended bibliography).
Stereochemical models
The results of DFT calculations are in agreement with the high diastereoselectivities (see the Supplementary Information for details). We then evaluated the role of the chiral NHC ligand that contains a pendant sulfonate moiety on enantioselectivity (Fig. 3a). The computational errors for modeling a charged species notwithstanding, we propose a similar steric and electronic environment as suggested formerly vis-à-vis enantioselective allylic substitutions effected by the same catalyst class27. The sulfonate group is probably situated in the rear (I–II); this would allow for the large 3,5-bis-(2,4,6-(i-Pr)3-phenyl)phenyl moiety to obstruct the right side of the complex, and causes the sizeable B(pin) moiety to be situated in the less occupied left/front quadrant in I (Fig. 3a). In II, which would lead to the minor enantiomer, there is steric repulsion between the B(pin) group and the chiral ligand’s N-aryl group, and thus the energy barrier would be higher (7.4 kcal/mol less favored than I). Consistent with the above analysis (Fig. 3a) the high energy of II and the steric pressure involving the B(pin) moiety is reflected in a considerably widened CNHC–Cu–C1–C2 dihedral angle (177.2° vs. 151.3° in I). Calculations on the system containing the more diminutive NHC–Cu complex derived from 7a point to energetically similar processes (energy difference of 0.6 kcal/mol between III and IV; Fig. 3b), which is in agreement with the lower e.r. obtained when the NHC–Cu complex derived from 7a is involved (55:45 vs. 95:5 e.r., Table 2).
The stereochemical model offers a rationale for why reactions with imines containing longer linear alkyl groups (e.g., n-butyl or 4-pentenyl) are less enantioselective (Fig. 2c); these lower e.r. might partly arise from an increase in attractive London dispersion forces between the 3,5-bis-(2,4,6-(i-Pr)3-phenyl)phenyl group and the substrate’s alkyl chain45,46,47. It is however more plausible that higher conformational mobility of the alkyl chains disrupt N→Na chelation [less optimal CNHC-Cu-C1-C2 dihedral angle in I (151.3°) vs. in I (131.8°)]. The smaller energy difference between anionic structures V and VI (2.2 kcal/mol, Fig. 2a) supports the notion that enantioselectivity would probably be lower without a sodium bridge, and that reaction via VI is likely the most competitive pathway versus the involvement of the most favored I. The CNHC–Cu–C1–C2 and N–Cu–C1–CNHC dihedral angles of V and VI are close to the optimal parameters in I, implying that some strain induced by N→Na association in I is released in V. With the less flexible isopropyl substituent in 3y the aforementioned chelation may remain intact, allowing for higher enantioselectivity (96:4 e.r.).
Conclusions
The catalytic method introduced here puts forth an expeditious strategy for synthesis of α-tertiary amines in high diastereo- and enantiomeric purity, thus providing a solution to an important and persisting problem in catalytic enantioselective synthesis. There are no more than a small number of catalytic enantioselective protocols that allow access to such coveted N-containing compounds, yet none involves an unprotected/unactivated imine. These investigations provide the first step towards development of a series of catalytic enantioselective reactions involving N-H ketimines and other types of readily available and versatile carbon-based nucleophiles, protocols that render a range of chiral drug candidates with one or more α-tertiary amine moieties much more accessible. Finally, this study further expands the utility of sulfonate-containing chiral NHC ligands, previously utilized in catalytic enantioselective conjugate additions48, allylic substitutions49 as well as copper-boryl additions to alkenes50 and allenes51 and copper–hydride additions to alkenes52, to include allyl additions to ketimines.
Supplementary Material
Acknowledgments
This research was supported by a grant from the National Institutes of Health (GM-57212). H. J. was supported as a LaMattina Graduate Fellow in Chemical Synthesis. We thank F. Meng and J. Lee for helpful discussions.
Footnotes
Data availability
X-ray crystallographic data for compounds rac-3a, N-acetyl derivative of 3y and 9b are freely available from the Cambridge Crystallographic Data Centre (CCDC 1547738, 1547736 and 1547737, respectively).
Author Contributions
H. J. and F. R. developed the catalytic method and its various applications. S. T. designed and performed the DFT calculations. A. H. H. directed the investigations and composed the manuscript with revisions provided by the other authors.
The authors declare competing financial interests.
References
- 1.Riant O, Hannedouche J. Asymmetric catalysis for the construction of quaternary carbon centres: nucleophilic addition on ketones and ketimines. Org. Biomol. Chem. 2007;5:873–888. doi: 10.1039/b617746h. [DOI] [PubMed] [Google Scholar]
- 2.Shibasaki M, Kanai M. Asymmetric synthesis of tertiary alcohols and α-tertiary amines via Cu-catalyzed C–C bond formation to ketones and ketimines. Chem. Rev. 2008;108:2853–2873. doi: 10.1021/cr078340r. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi S, Mori Y, Fossey JS, Salter MM. Catalytic enantioselective formation of C–C bonds by addition to imines and hydrazones: A ten-year update. Chem. Rev. 2011;111:2626–2704. doi: 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]
- 4.Clayden J, Donnard M, Lefranc J, Tetlow DJ. Quaternary centres bearing nitrogen (α-tertiary amines) as products of molecular rearrangements. Chem. Commun. 2011;47:4624–4639. doi: 10.1039/c1cc00049g. [DOI] [PubMed] [Google Scholar]
- 5.Yus M, González-Gómez JC, Foubelo F. Catalytic enantioselective allylation of carbonyl compounds and imines. Chem. Rev. 2011;111:7774–7854. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- 6.Wada R, Shibuguchi T, Makino S, Oisaki K, Kanai M, Shibasaki M. Catalytic enantioselective allylation of ketoimines. J. Am. Chem. Soc. 2006;128:7687–7691. doi: 10.1021/ja061510h. [DOI] [PubMed] [Google Scholar]
- 7.Trost BM, Silverman SM. Enantioselective construction of highly substituted pyrrolidines by palladium- catalyzed asymmetric [3+2] cycloaddition of trimethylenemethane with ketoimines. J. Am. Chem. Soc. 2010;132:8328–8238. doi: 10.1021/ja102102d. [DOI] [PubMed] [Google Scholar]
- 8.Luo Y, Hepburn HB, Chotsaeng N, Lam HW. Enantioselective rhodium-catalyzed nucleophilic allylation of imines with allylboron reagents. Angew. Chem. Int. Ed. 2012;51:8309–8313. doi: 10.1002/anie.201204004. [DOI] [PubMed] [Google Scholar]
- 9.Hepburn H, Lam HW. The isomerization of allylrhodium intermediates in the rhodium-catalyzed nucleophilic allylation of cyclic imines. Angew. Chem. Int. Ed. 2014;53:11605–11610. doi: 10.1002/anie.201407233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura S, Hyodo K, Nakamura M, Nakane D, Masuda H. Catalytic enantioselective allylation of ketimines by using palladium pincer complexes with chiral bis(imidazoline)s. Chem. Eur. J. 2013;19:7304–7309. doi: 10.1002/chem.201300685. [DOI] [PubMed] [Google Scholar]
- 11.Tang TP, Ellman JA. Asymmetric synthesis of β-amino acids derivatives incorporating a broad range of substitution patterns by enolate additions to tert-butanesulfinyl imines. J. Org. Chem. 2002;67:7819–7832. doi: 10.1021/jo025957u. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y-S, Liu Q, Tian P, Tao J-C, Lin G-Q. Copper-catalyzed asymmetric allylation of chiral N-tert-butanesulfinyl imines: dual stereocontrol with nearly perfect diastereoselectivity. Org. Biomol. Chem. 2015;13:4174–4178. doi: 10.1039/c5ob00322a. [DOI] [PubMed] [Google Scholar]
- 13.Chen JL-Y, Aggarwal VK. Highly diastereoselective and enantiospecific allylation of ketones and imines using borinic esters: Contiguous quaternary stereogenic centers. Angew. Chem. Int. Ed. 2014;53:10992–10996. doi: 10.1002/anie.201407127. [DOI] [PubMed] [Google Scholar]
- 14.Rabbat PMA, Valdez SC, Leighton JL. Phenol-directed enantioselective allylation of aldimines and ketimines. Org. Lett. 2006;8:6119–6121. doi: 10.1021/ol062589y. [DOI] [PubMed] [Google Scholar]
- 15.Perl NR, Leighton JL. Enantioselective imidazole-directed allylation of aldimines and ketimines. Org. Lett. 2007;9:3699–3701. doi: 10.1021/ol701723w. [DOI] [PubMed] [Google Scholar]
- 16.Dhudshia B, Tiburcio J, Thadani AN. Diastereoselective allylation and crotylation of N-unsubstituted imines derived from ketones. Chem. Commun. 2005:5551–5553. doi: 10.1039/b511411j. [DOI] [PubMed] [Google Scholar]
- 17.Meng F, Jang H, Jung B, Hoveyda AH. Cu-catalyzed chemoselective preparation of 2-(pinacolato)boron-substituted allylcopper complexes and their in situ site-, diastereo-, and enantioselective additions to aldehydes and ketones. Angew. Chem. Int. Ed. 2013;52:5046–5051. doi: 10.1002/anie.201301018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng F, Haeffner F, Hoveyda AH. Diastereo- and enantioselective reactions of bis(pinacolato)diboron, 1,3-enynes, and aldehydes catalyzed by an easily accessible bisphosphine–Cu complex. J. Am. Chem. Soc. 2014;136:11304–11307. doi: 10.1021/ja5071202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng F, McGrath KP, Hoveyda AH. Multifunctional organoboron compounds for scalable natural product synthesis. Nature. 2014;513:367–374. doi: 10.1038/nature13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung K, Ruscoe RE, Rae J, Pulis AP, Procter DJ. Enantioselective generation of adjacent stereocenters in copper-catalyzed three-component coupling of imines, allenes and diboranes. Angew. Chem. Int. Ed. 2016;55:11912–11916. doi: 10.1002/anie.201606710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu RY, Yang Y, Buchwald SL. Regiodivergent and diastereoselective CuH-catalyzed allylation of imines with terminal alkenes. Angew. Chem. Int. Ed. 2016;55:14077–14080. doi: 10.1002/anie.201608446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou G, Gosselin F, Li W, McWilliams JC, Sun Y, Weisel M, O’Shea PD, Chen C, Davies IW, Zhang X. Enantioselective hydrogenation of N–H imines. J. Am. Chem. Soc. 2009;131:9882–9883. doi: 10.1021/ja903319r. [DOI] [PubMed] [Google Scholar]
- 23.Tran DN, Cramer N. syn-Selective rhodium(I)-catalyzed allylation of ketimines proceeding through a directed C–H activation/allene addition sequence. Angew. Chem. Int. Ed. 2010;49:8181–8184. doi: 10.1002/anie.201004179. [DOI] [PubMed] [Google Scholar]
- 24.Thompson LA, et al. Compounds for the reduction of beta-amyloid production. 2013/0131051 A1 United States Patent US.
- 25.Butler CR, et al. Discovery of a series of efficient, centrally efficacious BACE1 inhibitors through structure- based drug design. J. Med. Chem. 2015;58:2678–2702. doi: 10.1021/jm501833t. [DOI] [PubMed] [Google Scholar]
- 26.Gao F, Carr JL, Hoveyda AH. A broadly applicable NHC–Cu-catalyzed approach for efficient, site- and enantioselective coupling of readily accessible (pinacolato)alkenylboron compounds to allylic phosphates and applications to natural product synthesis. J. Am. Chem. Soc. 2014;136:2149–2161. doi: 10.1021/ja4126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Jung B, Torker S, Hoveyda AH. N-Heterocyclic carbene–copper-catalyzed group-, site-, and enantioselective allylic substitution with a readily accessible propargyl(pinacolato)boron reagent: Utility in stereoselective synthesis and mechanistic attributes. J. Am. Chem. Soc. 2015;135:8948–8964. doi: 10.1021/jacs.5b05805. [DOI] [PubMed] [Google Scholar]
- 28.Brown KM, May TL, Baxter CA, Hoveyda AH. All-carbon quaternary stereogenic centers by enantioselective Cu-catalyzed conjugate additions promoted by a chiral N-heterocyclic carbene. Angew. Chem. Int. Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]
- 29.Dabrowski JA, Villaume MT, Hoveyda AH. Enantioselective synthesis of quaternary carbon stereogenic centers through copper-catalyzed conjugate additions of aryl- and alkylaluminum reagents to acyclic trisubstituted enones. Angew. Chem. Int. Ed. 2013;52:8156–8159. doi: 10.1002/anie.201304035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peese KM, Gin DY. Asymmetric synthetic access to the hetisine alkaloids: Total synthesis of (+)-nominine. Chem. Eur. J. 2008;14:1654–1665. doi: 10.1002/chem.200701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takemura H, Nakashima S, Kon N, Yasutake M, Shinmoyozu T, Inazu T. A study of C–F•••M+ interaction: Metal complexes of fluorine containing cage compounds. J. Am. Chem. Soc. 2001;123:9293–9298. doi: 10.1021/ja0043587. [DOI] [PubMed] [Google Scholar]
- 32.Yamazaki T, Kawashita S, Kitazume T, Kubota T. Diastereoselective alkylation of glycinates by assistance of intramolecular potassium•••fluorine interactions. Chem. Eur. J. 1999;15:11461–11464. doi: 10.1002/chem.200901984. [DOI] [PubMed] [Google Scholar]
- 33.Sazarin Y, Liu B, Maron L, Carpentier J-F. Discrete, solvent-free alkaline-earth metal cations: Metal•••fluorine interactions and ROP catalytic activity. J. Am. Chem. Soc. 2011;133:9069–9087. doi: 10.1021/ja2024977. [DOI] [PubMed] [Google Scholar]
- 34.Julian LD, Hartwig JF. Intramolecular hydroamination of unbiased and functionalized primary aminoalkenes catalyzed by a rhodium aminophosphine complex. J. Am. Chem. Soc. 2010;132:13813–13822. doi: 10.1021/ja1052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musacchio AJ, Nguyen LQ, Beard GH, Knowles RR. Catalytic olefin hydroamination with aminium radical cations: A photoredox method for direct C–N bond formation. J. Am. Chem. Soc. 2014;136:12217–12220. doi: 10.1021/ja5056774. [DOI] [PubMed] [Google Scholar]
- 36.Du Y, Xu L-W, Shimizu Y, Oisaki K, Kanai M, Shibasaki M. Asymmetric reductive Mannich reactions to ketimines by a C(I) complex. J. Am. Chem. Soc. 2008;130:16146–16147. doi: 10.1021/ja8069727. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi M, Iwanaga M, Shiomi N, Nakane D, Masuda H, Nakamura S. Direct asymmetric Mannich-type reaction of α-isocyanoacetates with ketimines using cinchona alkaloid/copper(II) catalysts. Angew. Chem. Int. Ed. 2014;53:8411–8415. doi: 10.1002/anie.201404629. [DOI] [PubMed] [Google Scholar]
- 38.Wieland LC, Vieira EM, Snapper ML, Hoveyda AH. Ag-catalyzed diastereo- and enantioselective vinylogous Mannich reactions of α-ketoimine esters. Development of a method and investigation of its mechanism. J. Am. Chem. Soc. 2009;131:570–576. doi: 10.1021/ja8062315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kano T, Song S, Kubota Y, Maruoka K. Highly diastereo- and enantioselective Mannich reactions of synthetically flexible ketimines with secondary amine organocatalysts. Angew. Chem. Int. Ed. 2012;51:1191–1194. doi: 10.1002/anie.201107375. [DOI] [PubMed] [Google Scholar]
- 40.Lipshutz BH, Shimizu H. Copper(I)-catalyzed asymmetric hydrosilylations of imines at ambient temperature. Angew. Chem. Int. Ed. 2004;43:2228–2230. doi: 10.1002/anie.200353294. [DOI] [PubMed] [Google Scholar]
- 41.Niu J, Guo P, Kang J, Li Z, Xu J, Hu S. Copper(I)-catalyzed aryl bromides to form intermolecular and intramolecular carbon–oxygen bonds. J. Org. Chem. 2009;74:5075–5078. doi: 10.1021/jo900600m. [DOI] [PubMed] [Google Scholar]
- 42.Wang JL, et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010;20:7159–7163. doi: 10.1016/j.bmcl.2010.07.054. [DOI] [PubMed] [Google Scholar]
- 43.Shavnya A, Coffey SB, Smith AC, Mascitti V. Palladium-catalyzed sulfination of aryl and heteroaryl halides: Direct access to sulfones and sulfonamides. Org. Lett. 2013;15:6226–6229. doi: 10.1021/ol403072r. [DOI] [PubMed] [Google Scholar]
- 44.Ye X-Y, et al. Synthesis and structure–activity relationship of dihydrobenzofuran derivatives as novel human GPR119 agonists. Bioorg. Med. Chem. Lett. 2014;24:2539–2545. doi: 10.1016/j.bmcl.2014.03.096. [DOI] [PubMed] [Google Scholar]
- 45.Wagner JP, Schreiner PR. London Dispersion in Molecular Chemistry – Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015;54:12274–12296. doi: 10.1002/anie.201503476. [DOI] [PubMed] [Google Scholar]
- 46.Chen L, Ren P, Carrow BP. Tri(1-adamantyl)phosphine: Expanding the Boundary of Electron-Releasing Character Available to Organophosphorus Compounds. J. Am. Chem. Soc. 2016;138:6392–6395. doi: 10.1021/jacs.6b03215. [DOI] [PubMed] [Google Scholar]
- 47.Albers L, Rathjen S, Baumgartner J, Marschner C, Müller T. Dispersion-Energy-Driven Wagner-Meerwein Rearrangements in Oligosilanes. J. Am. Chem. Soc. 2016;138:6886–6892. doi: 10.1021/jacs.6b03560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slutskyy Y, Jamison CR, Lackner GL, Müller DS, Dieskau AP, Untiedt NL, Overman LE. Short enantioselective total syntheses of trans-clerodane diterpenoids: Convergent fragment coupling using a trans-decalin tertiary radical generated from a tertiary alcohol precursor. J. Org. Chem. 2016;81:7029–7035. doi: 10.1021/acs.joc.6b00697. [DOI] [PubMed] [Google Scholar]
- 49.Shi Y, Jung B, Torker S, Hoveyda AH. N-Heterocyclic carbene–copper-catalyzed group-, site-, and enantioselective allylic substitution with a readily available propargyl(pinacolato)boron reagent: Utility in stereoselective synthesis and mechanistic attributes. J. Am. Chem. Soc. 2015;137:8948–8964. doi: 10.1021/jacs.5b05805. [DOI] [PubMed] [Google Scholar]
- 50.Lee Y, Hoveyda AH. Efficient boron–copper additions to aryl-substituted alkenes promoted by NHC-based catalysts. Enantioselective Cu-catalyzed hydroboration reaction. J. Am. Chem. Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jang H, Jung B, Hoveyda AH. Catalytic enantioselective protoboration of disubstituted allenes. Access to alkenylboron compounds in high enantiomeric purity. Org. Lett. 2014;16:4658–4661. doi: 10.1021/ol5022417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J, Torker S, Hoveyda AH. Versatile homoallylic boronates by chemo-, SN2’-, diastereo- and enantioselective catalytic sequence of Cu–H addition to vinyl-B(pin)/allylic substitution. Angew. Chem. Int. Ed. 2017;56:821–826. doi: 10.1002/anie.201611444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.