

Abstract
Gene duplication is an important source of novelties and genome complexity. What genes are preserved as duplicated through long evolutionary times can shape the evolution of innovations. Identifying factors that influence gene duplicability is therefore an important aim in evolutionary biology. Here, we show that in the yeast Saccharomyces cerevisiae the levels of gene expression correlate with gene duplicability, its divergence, and transcriptional plasticity. Genes that were highly expressed before duplication are more likely to be preserved as duplicates for longer evolutionary times and wider phylogenetic ranges than genes that were lowly expressed. Duplicates with higher expression levels exhibit greater divergence between their gene copies. Duplicates that exhibit higher expression divergence are those enriched for TATA-containing promoters. These duplicates also show transcriptional plasticity, which seems to be involved in the origin of adaptations to environmental stresses in yeast. While the expression properties of genes strongly affect their duplicability, divergence and transcriptional plasticity are enhanced after gene duplication. We conclude that highly expressed genes are more likely to be preserved as duplicates due to their promoter architectures, their greater tolerance to expression noise, and their ability to reduce the noise-plasticity conflict.
Keywords: gene expression, gene duplication, transcriptional plasticity, duplicability, Saccharomyces cerevisiae
1. Introduction
Gene duplication is believed to be a rich source of novel functions and adaptations.1–3 This belief is supported by evidence coming from innovations following gene duplications in yeast, plants and animals. Indeed, protein families expanded after whole genome and small-scale duplications yielding an unprecedented morphological diversity in plants.4–11 Other major innovations in animals have also been achieved through gene duplication,12 including increased synapse and behaviour complexity13 and the neural crest formation and plasticity in vertebrates.14 In yeast, gene duplication has contributed to metabolic innovation through the alteration of regulatory and transcriptional networks15 or the increased glycolytic fluxes.16 However, it remains unclear why certain duplicates have been preferred over others to persist in the genomes and be the source of innovations.
Since duplication is immediately followed by relaxed selection constraints on one or the two gene copies, the survival time of each gene copy is a limiting factor in the determination of its functional fate. In the majority of cases, duplication is resolved by the non-functionalization of one of the gene copies and its subsequent erosion from the genome.1,3 Accordingly, 92% of all genes that were duplicated through whole-genome duplication (WGD) > 100 MYA in Saccharomyces returned to single copy genes ‘shortly’ after duplication.17 Nonetheless, in many species, including yeast, the number of duplicated genes is larger than predicted by theory ranging between 30% of the genes in yeast18 and more than 50% in plants.6,19 Determining what genes remain in the genome as duplicates, and consequently lead to evolutionary leaps, is an important aim in evolutionary biology. However, this objective remains to be achieved.
A number of hypotheses have been proposed to explain the persistence of certain genes in duplicate. Rapid sequence divergence between gene copies can lead to their functional divergence followed by strong selective constraints on each copy, which could contribute to the preservation of duplicates in the genomes.20–23 Functional divergence requires, nevertheless, long evolutionary times and given that selection relaxes after gene duplication, selective pressures are unlikely to retain both gene copies during the first million years following duplication. Preservation of duplicates can also be selectively favoured by the need to maintain gene–dosage balance,5,21,24,25 or provide genetic robustness against deleterious mutations.26,27 However, all these scenarios do not provide a general mechanistic explanation for what makes duplicates persist or alternatively perish.
Recently, it has been proposed dosage sub-functionalization as a plausible hypothesis to explain the fate of whole-genome duplicates.28 According to this hypothesis, highly expressed genes are more likely to be preserved as duplicates than lowly expressed genes. This is because stochastic variations in the levels of expression of the gene copies of highly expressed duplicates would not lead to copies with a lower expression level than that required for purifying selection to act upon them. Therefore, highly expressed duplicates are less likely to return to single copy genes by drift. Whether this hypothesis could be applied to all duplicates regardless of the mechanism that originated them and whether such dosage sub-functionalization could also determine the patterns of divergence between gene copies has not been explored before.
Here, we present evidence that the levels of gene expression are correlated with the fates of whole-genome and small-scale duplicates, with highly expressed genes being more likely to be retained in double copy after duplication for longer periods of time than lowly expressed genes. Such duplicates are also more phylogenetically stable. We also show that the ancestral levels of gene expression are correlated with the evolution of duplicates expression. Retained duplicated genes evolve strong patterns of transcriptional (also known as phenotypic) plasticity, which are also correlated with the levels of gene expression. Finally, while the levels of gene expression are correlated with the duplicability of genes, duplicates phenotypic plasticity is manifested only after gene duplication; and this plasticity is proportional to the expression divergence between the copies of duplicated genes.
2. Material and methods
2.1. Identification of duplicated genes
Paralogs pairs of duplicated genes were identified as the resulting best reciprocal hits from all-against-all BLAST searches using BLASTP with an E-value cutoff of 1E−5 and a 50 bit score.29 Paralogs were then divided into two groups according to the mechanism of their origin: WGDs and SSDs. WGDs are those extracted from the reconciled list provided by the Yeast Gene Order Browser (YGOB, http://wolfe.gen.tcd.ie//ygob30) (555 pairs of genes), and these were not subjected to subsequent SSD. All other paralogs were considered to belong to the category of SSDs (560 pairs of genes).
2.2. Growth of S. cerevisiae and gene expression analyses
The transcriptomic profiling was performed in the S. cerevisiae Y06240 haploid msh2 deletion strain (BY4741; Mata his3D1 leu2DO met15DO ura3DO msh2::kanMX4), with three technical replicates for each biological stress condition (3% lactic acid (YPL), 3% ethanol (YPE), 3% glycerol (YPG), 0.25mM H2O2 + 1.5% dextrose (YPOxD)) in comparison with the normal growth condition (YPD media) (Fig. 1). Total RNA extractions were performed with RNeasy kit (Qiagen) following manufacturer instructions. Ribosomal RNA was removed by using Ribo-Zero Gold rRNA removal yeast (Illumina) depletion kit. Stranded RNA libraries were constructed using TruSeq stranded mRNA (Illumina) from oligo-dT captured mRNAs from depleted samples. Libraries were run in NextSeq 500 (Illumina) at 75nt single read by using High Output 75 cycles kit v2.0 (Illumina).
Figure 1.

Growth experiments of S. cerevisiae under stress conditions. An initial isogenic population of S. cerevisiae (strain Y06240) was grown for 24 h in Yeast extract, peptone, dextrose medium (YPD) at 28 °C. This grown population was then subjected to four different stress conditions: ethanol, glycerol, lactate and oxidative stress in a medium supplemented with dextrose (each stress in indicated in a different colour). Growth experiments were performed in triplicate. Samples of each of the populations, in addition to the control population grown in YPD, were processed for RNA sequencing. The technical steps for the extraction and analysis of RNA sequences are also shown.
RNA libraries were sequenced at Genomic core facility at Servicio Central de Soporte a la Investigación Experimental (SCSIE) from University of Valencia, Spain. Raw reads were analyzed using FastQC report and cleaned with CutAdapt as implemented in RobiNA software package v 1.2.4.31 Low-quality reads were filtered and trimmed (Phred score inferior to 20 and size less than 40 nt were discarded). The reads were then aligned with Bowtie (up to two mismatches accepted) to the reference transcriptome (PRJNA290217) from the reference S288c strain. The normalization and statistical evaluation of differential gene expression has been performed using edgeR32 or DESeq33 with a P-value cut-off of 0.05, using the Benjamini–Yekutieli34 method for multiple testing correction of P-value, and setting the Log-fold change at min = 1 to determine differential expression. The raw data (reads counts) was normalized according to the default procedure of the differential expression analysis package used (edgeR or DESeq), being the dispersion estimated using the pooled setting, and RPKM (Reads Per Billion) expression values estimated as implemented in RobiNA software.31 All newly sequenced RNA sequences are available from the Sequence Read Archive with the following accession number (SRP074821). Expression data for each of the S. cerevisiae genes under YPD and each of the four stress conditions as well as the adjusted probabilities to identify significant fold changes are available in Supplementary Tables S1–S4.
2.3. Expression data for Lachancea kluyveri
Growth conditions, RNA extraction and sequencing are specified in a previous study.35 Briefly, authors performed the analyses on L. kluyveri reference strain CBS 3082_a (MATa). Transcriptomic data were obtained from growth cultures at mid exponential phase and for 20 different media, including YPD and 19 other stress conditions (listed in Ref. [35]). RNA sequencing was performed using Illumina HiSeq2000 platform with 50-base pair non-oriented single reads (Supplementary Table S5).
2.4. Expression data for Candida glabrata
Candida glabrata ATC2001 strain expression data (in the form of RPKMs) were obtained from a previous study.36 Briefly, authors grown C. glabrata in normal YPD media and in the M199 medium at different pH values for pH shift. Total RNA was isolated by hot phenol–chlorophorm method. Libraries were subjected to ribosomal RNA depletion, and sequenced using Illumina HiSeq2000 platform with 100-bp paired-end strand-specific. Reads were mapped with TOPHAT2, and counted using htseq (-m union, -t exon conditions) and normalized by the number of reads per kilobase of exon per million mapped reads (Supplementary Table S6). Differentially expressed genes were identified with raw counts by DeSeq and EdgeR, using the same cut-off parameters than the ones used in this study.
2.5. Software
Calculations and statistics were performed using MS Excel and R 3.2.1. Data management was possible using in-house built PERL scripts.
3. Results
3.1. Duplicates preservation and phylogenetic stability are correlated with the levels of gene expression
Highly expressed genes are more likely to be preserved as duplicates after whole-genome duplication (WGD) than lowly expressed genes in S. cerevisiae.37,28,38–41 We searched for the orthologs of S. cerevisiae genes in L. kluyveri (strain CBS 3082; synonymous of Saccharomyces kluyveri) and their expression in Yeast Extract Peptone containing dextrose medium (YPD)35 (Supplementary Table S5). Lachancea kluyveri is a respiratory yeast species pre-dating the WGD that took place in S. cerevisiae > 100 million years ago.17 We found L. kluyveri orthologs for 5643 S. cerevisiae genes. Of the 5643 genes, 1469 genes were orthologs of S. cerevisiae duplicates, including WGDs and small-scale duplicates (SSDs), and 4174 were orthologs of S. cerevisiae singletons (Supplementary Table S5). The expression of L. kluyveri orthologs of S. cerevisiae duplicates (Median: 10.43; measured as the log2-transformed Reads Per billion, RPKM) was significantly greater than that of L. kluyveri orthologs of S. cerevisiae singletons (Median: 9.52) (Wilcoxon rank test: P < 2.2 × 10−16). We compared the transcription levels of genes in S. cerevisiae obtained in our study with those from another study42 that used ribosomal profiling, a technique that measures ribosome occupancy and translation genome wide and provides an accurate measure of the translatable mRNA. RPKMs correlated strongly and significantly with the data of ribosome profiling (Spearman’s correlation: ρ = 0.77, P < 2.2 × 10−16, Supplementary Data S1), indicating that RPKMs are indicative of the levels of gene expression and also the translatable mRNAs.
Previous studies concluded that highly expressed genes were more likely to be preserved as duplicates after WGDs because of absolute dosage constraints and constraints on dosage balance.28,37,39,43–46 Indeed, we found that this trend is true for L. kluyveri orthologs of S. cerevisiae WGDs (N = 561, Median expression: 10.64, Wilcoxon rank test: P < 2.2 × 10−16) and also for orthologs of S. cerevisiae SSDs (N = 908, Median expression: 10.28, Wilcoxon rank test: P < 2.2 × 10−16). The level of expression of orthologs of WGDs was, nevertheless, higher than that for SSDs (Wilcoxon rank test: P = 7.79 × 10−5).
The higher expression of duplicates compared to singletons can be due to a greater presence of genes encoding protein-complex proteins among duplicates than singletons. We extracted the list of protein complexes from a previous study, using the table of annotated yeast high-throughput complexes available at (http://wodaklab.org/cyc2008/downloads).47 Genes encoding proteins that are part of protein complexes (N = 1913) (Supplementary Table S7) do exhibit greater expression (Median expression: 11.56) than genes encoding complex-free proteins (N = 3960) (Median expression: 10.61, Wilcoxon rank test: P < 2.2 × 10−16). However, neither WGDs were more enriched for complex-encoding genes than singletons in S. cerevisiae (Fisheŕs exact test: F = 1.02, P = 0.76) nor SSDs showed significant difference in terms of enrichment for complex-encoding genes when compared to singletons (Fisheŕs exact test: F = 1.11, P = 0.15).
One caveat in this analysis is that gene expression in L. kluyveri may not reflect gene expression immediately after WGD. Against this prediction, gene expression in L. kluyveri, a species predating the WGDs and SSDs used in this study, was strongly and significantly correlated with gene expression in S. cerevisiae (Spearman correlation: ρ = 0.59, P < 2.2 × 10−16, Fig. 2a).
Figure 2.

The levels of gene expression determine gene duplicability. (a) Expression of genes in the pre-WGD species Lachancea kluyveri correlate positively with genes expressions in the post-WGD species Saccharomyces cerevisiae. Axes represent the transcription levels of genes as the logarithmic transformation of the Fraction of Reads Per billion. (b) Estimation of the phylogenetic stability (i.e. number of species) of duplicated genes after the whole genome duplication in yeast. Black and gray circles refer to gene copies. A missing circle in one of the post-WGD species represent a gene copy loss, hence return of that duplicate to single-copy gene in that species. The phylogenetic stability (PS) of that duplicate is calculated as the mean number of species in which gene copies are present. (c) Phylogenetic stability correlates with the levels of gene expression of duplicates. (d) Correlation of PS of WGDs and their expression levels. (e) Correlation of PS of SSDs and their expression levels.
Duplicated genes exhibit different patterns of gene retention and phylogenetic stability in the different post-WGD Saccharomyces species.48 We classified S. cerevisiae duplicated genes according to the presence of the two copies in each of the twelve available species post-dating the WGD (Fig. 2b). We first asked whether the expression of duplicates generated before Saccharomycetales speciation (including WGDs and SSDs) correlates with their phylogenetic stability, measured as the mean number of post-WGD species in which each copy was present (Fig. 2b). To determine the number of species postdating WGD in which each gene copy is present we used the Pillars information available from the Yeast Gene Order Information,30 which provides gene order and annotation for 12 post-WGD yeast species (Supplementary Table S8). For each gene copy, we counted the number of species in which it is found and averaged this number for the two sister gene copies in a duplicated pair (Fig. 2b). There was a positive and significant correlation between the mean gene copies expression in YPD and their phylogenetic stability (Spearman correlation: ρ = 0.27, P < 2.2 × 10−16, Fig. 2c). We then repeated the analysis for WGDs and SSDs separately. WGDs exhibited positive weak but significant correlation between gene expression and phylogenetic stability (Spearman correlation: ρ = 0.13, P = 1.15 × 10−5, Fig. 2d). In contrast, SSDs showed strong and significant correlation between gene expression and phylogenetic stability (Spearman correlation: ρ = 0.40, P < 2.2 × 10−16, Fig. 2e).
3.2. The magnitude of divergence of duplicates expression correlates with the level of gene expression
A pivotal hypothesis to the dosage sub-functionalization proposed by Gout and Lynch28 is that highly expressed duplicates should exhibit more expression variation despite the action of purifying selection than lowly expressed genes. This is because noise in the expression of highly expressed genes is unlikely to compromise the selective constraints on these genes. That is, genes with higher expression levels should be more ‘noisy’ in their expression when duplicated, and thus they should generate more expression polymorphism in the population than lowly expressed genes. Accordingly, highly expressed duplicates are more likely to yield gene copies with diverged expressions than lowly expressed duplicates. To test this hypothesis, we first measured the fold expression difference (D) between the gene copies i and j when S. cerevisiae was grown under YPD as
with E referring to the expression of the gene under normal conditions (Supplementary Table S9). Di,j is normalized by the level of gene expression (i.e. the value is 0 ≤ Di,j ≤ 1), and thus it is an unbiased measure of the expression divergence between the gene copies. In support of our hypothesis, there was a weak but very significant correlation between the average expression of the gene copies of duplicates and Di,j (Spearman correlation: ρ = 0.18, P = 4.23 × 10−8). This correlation was also maintained when we analyzed separately WGDs (Spearman correlation: ρ = 0.18, P = 6.98 × 10−5) and SSDs (Spearman correlation: ρ = 0.17, P = 2.89 × 10−4).
3.3. The expression levels and promoter architecture correlate with patterns of expression divergence of duplicates and their transcriptional plasticity
Because higher expression can increase the chance for expression divergence after gene duplication, we sought to investigate if genes with higher expression can also evolve greater transcriptional plasticity under stress. Transcriptional plasticity is defined here as the ability of the gene to change its expression, while keeping its genotype, when the environment changes. For all four stresses with which S. cerevisiae was challenged (see section Material and methods), transcriptionally altered duplicates belonged to a set of genes with significantly higher expression in YPD growth media than transcriptionally unaltered duplicates (Fig. 3a–d). This was also true, with the exception of ethanol-induced stress, for singletons, albeit the effect was more pronounced in duplicates than it was in singletons. Noticeably, for all four-stress conditions in S. cerevisiae, the levels of expression of duplicates with no altered transcription under stress were significantly higher than that for transcriptionally altered singletons (Fig. 3a–d).
Figure 3.

The levels of gene expression determine the transcriptional plasticity of genes. We present the levels of expression as the logarithmic transformation of the Reads Per billion (RPKM) for duplicates (red boxes) and singletons (blue boxes) with transcriptional alterations and duplicates (orange boxes) and singletons (white boxes) without altered expressions when S. cerevisiae is faced with four different stress conditions (a) Ethanol, (b) glycerol, (c) lactate and (d) oxidative stress in a medium supplemented with dextrose. Significant differences in the expression levels of sets of genes are indicated as *, **, ***, when the probabilities are P < 0.05, P < 0.01 and P < 10−6, respectively, using a Wilcoxon rank test.
We explored other mechanistic explanations for this expression difference between unaltered duplicates and altered singletons. One important factor that contributes to transcriptional plasticity is the existence of the TATA-box motif in the gene promoter, with TATA-containing genes being more sensitive to regulatory changes than TATA-less genes.49 Importantly, the level of expression of TATA-containing genes was higher than that of TATA-less genes, and this was true for duplicates and singletons in S. cerevisiae (Fig. 4a). The set of transcriptionally altered genes under stress conditions was enriched for TATA-containing genes when compared to the set of genes with no transcriptional plasticity, being this true for duplicates (Fig. 4b) and singletons (Fig. 4c). Generally, TATA-containing genes also exhibit expression noise, which can be coupled with transcriptional plasticity provided that noise and plasticity are not in conflict.50 Since gene duplication relaxes noise-plasticity conflict,50 we expected duplicated genes to be enriched for TATA motifs when compared to singletons. Of the 1090 genes containing TATA-motifs, 558 belonged to duplicates (281 were WGDs and 277 were SSDs) (25% of all duplicates) and 532 to singletons (12% of all singletons) (Supplementary Table S10). Indeed, duplicated genes were more enriched for TATA-containing genes than singletons (Fisheŕs exact test: F = 2.52, P < 2.2 × 10−16), and this was the case for both transcriptionally plastic genes and genes with no transcriptional plasticity (Supplementary Fig. S1a). Remarkably, duplicates with no transcriptional plasticity were slightly more enriched for TATA-containing genes than singletons with transcriptional plasticity, with the difference being significant in the case of S. cerevisiae grown under oxidative stress (Supplementary Fig. S1b).
Figure 4.

TATA-containing genes exhibit greater expression levels and transcriptional plasticity than TATA-less genes. (a) Comparison of the expression levels (measured as the logarithmic transformation of the Fragment Reads Per billion) between TATA-containing and TATA-less genes for duplicates (red and orange boxes, respectively) and singletons (blue and white boxes, respectively). Differences were tested using the Wilcoxon rank test and significant differences are identified as * and **, for probabilities of P < 0.05 and P < 0.01, respectively. The percentage of genes with TATA-containing promoters for transcriptionally plastic and transcriptionally unaltered genes when S. cerevisiae is growing under four different stress conditions (ethanol, glycerol, lactate and oxidative stress in a medium supplemented with dextrose) was compared for duplicates (b) and for singletons (c). Significant differences between plastic and unaltered genes were identified using Fisheŕs exact test and are identified as ***, to indicate a probability of P < 2.2 × 10−16.
Finally, the magnitude of expression divergence between duplicates gene copies was correlated with the magnitude of transcriptional plasticity (measured as the fold change in expression of the most altered gene copy between YPD and stress) (Table 1), both of which are in turn correlated with the levels of gene expression.
Table 1.
Expression divergence between gene copies correlates with transcriptional plasticity of duplicates
| Stress source | Correlation (Spearman) | Probability |
|---|---|---|
| Ethanol | 0.17 | 4.22 × 10−8 |
| Glycerol | 0.20 | 1.78 × 10−10 |
| Lactate | 0.16 | 4.13 × 10−7 |
| Oxidative + dextrose | 0.15 | 7.11 × 10−7 |
3.4. The levels of gene expression correlate with the patterns of duplicates transcriptional plasticity
A prediction of the dosage sub-functionalization hypothesis is that the patterns of duplicates transcriptional plasticity should be dependent on the levels of gene expression. For instance, transcriptionally plastic genes that are lowly expressed under normal conditions should only be able to over-express under stress because a decline in their expression could drive one of the gene copies to non-functionalization due to relaxed selective constraints. Because plasticity is often correlated with expression noise50 and, since surviving duplicates are those whose expression noise falls within the range of expression detectable by selection, expression noise should depend on the levels of duplicates expression. We divided duplicated genes according to the patterns of transcriptional plasticity they show when S. cerevisiae is grown under stress: (i) up-regulated: when the two gene copies were up-regulated under stress; (ii) down-regulated: when the two gene copies were down-regulated under stress; (iii) discordant: when one copy was up-regulated and the other was down-regulated under stress; and (iv) one-altered: when one copy was not altered but its paralogous copy was either up-regulated or down-regulated under stress (Supplementary Tables S11–S26). In all four stress conditions, duplicates that were down-regulated were also those that exhibited the highest expression levels under normal conditions, being these followed by duplicates in which only one copy exhibits transcriptional plasticity, then discordant duplicates and finally up-regulated duplicates (Fig. 5a–d).
Figure 5.

The expression level of duplicates correlated with their patterns of expression divergence. Expression was measured as the logarithmic transformation of the Reads Per billion (RPKM). Significant differences in the expression levels of gene sets are indicated as *, ** and ***, referring to probabilities of P < 0.05, P < 0.01 and P < 10−6, using a Wilcoxon rank test. (a) Expression under ethanol stress, (b) expression under glycerol stress, (c) expression under lactate stress and (d) expression under oxidative stress.
Interestingly, the mean level of expression under normal conditions of duplicates gene copies was correlated with the level of expression divergence of the gene copies under normal conditions in those duplicates that belong to the category discordant and one-altered, those categories with the highest expression divergence between gene copies, but not in those pairs in which both copies were either up-regulated or down-regulated (Fig. 6).
Figure 6.

The levels of gene expression correlate with the level of expression divergence in duplicates in which gene copies are either discordant (one copy up-regulated and the other copy down-regulated) or one copy only altered under stress. Transcriptional alteration patterns of duplicates were measured in four stress conditions (ethanol, glycerol, lactate and oxidative stress in a medium supplemented with dextrose). Expression divergence of duplicates was measured as 1 – (the expression of the lowest expressed copy divided by the expression of the highest expressed copy). Spearman correlation (ρ) between the levels of gene expression (measured as the logarithmic transformation of the Reads Per billion (RPKM)) and the levels of expression divergence was tested for significance. Significant plots are indicated in bold. We found significant associations between the levels of gene expression and the expression divergence between gene copies of discordant duplicates and those with one copy altered but not for up-regulated or down-regulated duplicates.
3.5. Gene duplication has contributed to increased transcriptional plasticity in yeast
We examined the link between gene duplication and phenotypic plasticity by comparing the transcriptomes of S. cerevisiae grown under a number of key stress conditions that this species faces in nature to those transcriptomes of S. cerevisiae grown under normal YPD media (section Material and methods). Transcriptionally altered genes were more enriched for duplicates than for singleton genes over all stress conditions (Fig. 7). This trend was also true when we compared WGDs to singletons and SSDs to singletons (Fig. 7). To determine whether transcriptional plasticity is directly linked to gene duplication, we examined the transcriptional plasticity of S. cerevisiae duplicates and singletons orthologs in the pre-WGD species L. kluyveri. The transcription of L. kluyveri genes was previously assessed in 19 different stress conditions.35 For each condition, we sought the percentage of genes that were orthologs to S. cerevisiae duplicates (N = 1469) and singletons (N = 4174) that exhibited transcriptional alteration. In 18 of the 19 conditions, there was no significant difference in the percentage of transcriptionally altered genes under stress between the orthologs of S. cerevisiae duplicates and those of singleton (Table 2). The only exception was SDS stress, in which the percentage of transcriptionally altered orthologs for S. cerevisiae duplicates was higher than that for transcriptionally altered singleton orthologs (Fisheŕs exact test: odds ratio F = 1.22, P = 5 × 10−3, Table 2). In all other stresses that were equivalent to the ones used in our experiments (e.g. glycerol, ethanol), there was no significant difference in the number of transcriptionally altered genes between orthologs of S. cerevisiae duplicates and singletons (Table 2). These data indicate that the high transcriptional plasticity of duplicates in S. cerevisiae was acquired after gene duplication.
Figure 7.
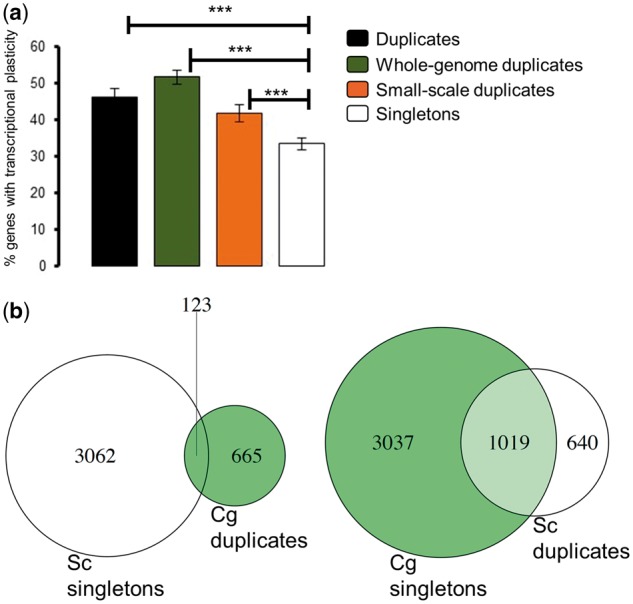
Duplicates are more enriched for transcriptional plasticity than singletons, regardless of the mechanism of duplication. (a) Transcriptional plasticity of duplicates and singletons in S. cerevisiae. Y-axis represents the mean percentage of genes with altered transcription across four stress conditions (ethanol, glycerol, lactate and oxidative stress in a medium supplemented with dextrose). We also compared WGDs and SSDs to singletons. Significant differences were found using Fisher’s exact test, with *** indicating a probability of P < 2.2 × 10−16. (b) Venn diagram representing the overlap in the number of duplicates and singletons between C. glabrata and S. cerevisiae.
Table 2.
Transcriptional alterations of L. kluyveri genes orthologs of S. cerevisiae under nineteen different stress conditions
| Stress | # duplicates orthologs (%) | # singletons orthologs (%) | Odds’ ratio F | P |
|---|---|---|---|---|
| Galactose | 588 (40.1) | 1586 (37.6) | 0.93 | 0.24 |
| Glycerol | 865 (58.8) | 2378 (56.9) | 1.08 | 0.21 |
| 23 °C | 178 (12.1) | 540 (12.9) | 0.92 | 0.44 |
| 37 °C | 458 (31.2) | 1282 (30.7) | 1.02 | 0.74 |
| YNB | 487 (33.2) | 1366 (32.7) | 1.02 | 0.77 |
| Ethanol | 609 (41.5) | 1645 (39.4) | 1.09 | 0.17 |
| Methanol | 291 (19.8) | 865 (20.7) | 0.95 | 0.49 |
| SDS | 410 (27.9) | 1008 (24.1) | 1.22 | 0.005 |
| DMSO | 654 (44.5) | 1758 (42.1) | 1.10 | 0.11 |
| NaCl | 234 (15.9) | 757 (18.1) | 0.86 | 0.06 |
| CaCl2 | 874 (59.5) | 2499 (59.9) | 0.98 | 0.80 |
| NiSO4 | 129 (8.8) | 332 (7.9) | 1.11 | 0.32 |
| LiCl | 253 (17.2) | 737 (17.7) | 0.97 | 0.72 |
| CoSO4 | 825 (56.2) | 2362 (56.6) | 0.99 | 0.85 |
| BME | 375 (25.6) | 1102 (26.4) | 0.96 | 0.53 |
| 5FU | 298 (20.3) | 922 (22.1) | 0.89 | 0.15 |
| Arsenic | 127 (8.6) | 343 (8.2) | 1.06 | 0.62 |
| 6AU | 230 (15.7) | 674 (16.1) | 0.96 | 0.68 |
| Fluconazole | 437 (29.7) | 1195 (28.6) | 1.06 | 0.42 |
To shed more light on the role of gene duplication in the acquisition of transcriptional plasticity, we examined the transcriptional patterns of a post-WGD species, Candida glabrata, in which some orthologs of S. cerevisiae duplicates are in single gene copy in C. glabrata, while others are preserved as duplicates and for which we had transcriptional information under acidic stress similar to our lactate stress dataset.36Saccharomyces cerevisiae orthologs in C. glabrata were identified using synteny information available in the Pillars of the Yeast Gene Order Browser (YGOB).30 In total, we identified 4844 reliable S. cerevisiae: C. glabrata orthologs. Of these 4844 genes, 788 genes were duplicated in C. glabrata, of which 123 were orthologs of S. cerevisiae singletons. These 4844 genes included 1659 out of the 2240 duplicated S. cerevisiae genes and 3185 singletons. Of the 2240 S. cerevisiae duplicates, orthologs for 1019 of them were in single gene copy in C. glabrata (Fig. 7b). Importantly, these 1019 genes exhibited as much transcriptional plasticity under acidic stress in C. glabrata (N = 599, 58.7% of the C. glabrata singletons) as C. glabrata singletons that had no duplicates orthologs in S. cerevisiae (1725 out of 3062 singletons, 56.3%) (Fisher’s exact test: odd’s ratio F = 1.10, P = 0.17). In conclusion, transcriptional plasticity seem to have been acquired after gene duplication because orthologs of duplicates that are in single copy genes in other species exhibit no evidence for transcriptional plasticity in these species.
3.6. Transcriptionally plastic duplicates contribute to the response of S. cerevisiae to stress
Among the transcriptionally plastic duplicates, many had a significant biological role in the response to stress. For instance, analyses of duplicated genes up-regulated when S. cerevisiae is grown in ethanol identified a number of genes as largely up-regulated that are involved in ethanol metabolism (Supplementary Table S27). Heading the list of up-regulated duplicates is the one encoding the alcohol dehydrogenase ADH2 and ADH1, directly involved in the quick metabolism of ethanol inside the cell into acetaldehyde (Fig. 8). A number of other duplicated genes that are essential in the metabolism of Ethanol, including transmembrane transporters such as YAT1 to start the tricarboxylic cycle or the enzyme MSL1 that is essential for malate production, among others (Fig. 8), they are all listed as duplicates with the highest up-regulation. This also applies to other stress conditions used in this study (Fig. 8).
Figure 8.

Schematic representation of S. cerevisiae central metabolic pathways for the utilization of non-fermentable carbon sources (glycerol, lactate and ethanol). Solid arrows correspond to enzymatic reactions while dashed arrows correspond to regulatory steps. Proteins encoded by duplicated genes that are transcriptionally altered and are involved in each process are indicated (transporters are shown with a can form, and enzymes exhibit heptagonal forms).
4. Discussion
A number of factors have been considered key players in determining the fates of duplicated genes. However, an explanation that provides a general and inherent mechanism that strongly determines the duplicability of genes has been poorly investigated. Here, we present strong evidence that the levels of gene expression determine the likelihood of a gene to persist duplicated in the genome. We find that genes that are highly expressed in species pre-dating the whole-genome duplication event in Saccharomyces are more likely to be preserved in duplicate in species originated after duplication. This observation is in agreement with the dosage sub-functionalization hypothesis (DSH), as high levels of gene expression would ensure purifying selection on the gene copies despite the stochastic variation in their expression levels.28 We, nevertheless, show that this is not a link exclusively seen in WGDs because the preservation of SSDs is also dependent on the expression levels of genes, however to a lesser extent than WGDs.
We also show that highly expressed genes exhibit greater phylogenetic stability (i.e. they are preserved in greater number of post-duplication species) than lowly expressed genes, perhaps due to the higher likelihood of functional divergence between gene copies of highly expressed duplicates, and thus emergence of purifying selection to maintain the functions of gene copies. Therefore, we conclude that the levels of gene expression determine the survival of duplicates across long evolutionary times. Our observations are not confounded by other factors such as dosage balance, mainly an issue in the case of genes encoding proteins that are part of protein complexes,28,39,43,45,46 as neither WGDs nor SSDs are enriched for complex-encoding genes when compared to singletons in S. cerevisiae.
A pivotal conclusion derived from the link between gene expression and duplicability is that gene copies with higher expression levels can be noisier in terms of expression without undergoing an imbalance in their selective pressures. This noise could eventually lead to higher divergence between the gene copies of these duplicates when such divergence becomes adaptive. Our results are in agreement with this prediction and show that the magnitude of expression divergence, and perhaps functional divergence, is dependent on the levels of gene expression. Therefore, duplicates with higher expression are more likely to lead to higher divergence between the gene copies. This higher divergence between gene copies has been likely important for the origin of novel adaptations to stressful environments in the yeast Saccharomyces cerevisiae.26,27,51–53 In agreement with this hypothesis, when S. cerevisiae was faced with a number of stress conditions, the levels of expression divergence between gene copies was positively correlated with their expression plasticity. Importantly, we show that the levels of gene expression also influence the patterns of transcriptional divergence between the gene copies of duplicates. Those duplicates with copies exhibiting discordant expression patterns or with only one copy altered under stress are likely those in which each copy encodes a function under different conditions compared to the other copy.
Expression noise is generally coupled with plasticity if noise and plasticity do not present a cost-benefit conflict (i.e. a conflict emerges when plasticity is important for adaptation but noisy expression can be detrimental).50,54,55 We show that duplicates are both more enriched for transcriptionally plastic genes and for genes with TATA motifs in their promoters when compared with singletons. TATA-motifs have been shown to be associated with higher noise and plasticity in TATA-containing genes.49,56,57 The expression properties of genes that have been preserved as duplicates are, therefore, remarkably different from those that returned to single copy genes. Noticeably, non-plastic duplicates that showed higher expression levels than transcriptionally plastic singletons also exhibited greater enrichment for TATA-containing genes than plastic singletons, linking TATA-containing genes to higher levels of gene expression. The question that remains is then whether it is the expression properties or duplication itself that have determined the fate of duplicates and the origin of adaptations. Our results lead to the conclusion that duplicated genes exhibit significantly different expression properties than singleton genes but also that gene duplication is mainly responsible for the origin of plasticity, as the transcriptional plasticity observed in S. cerevisiae originated after the duplication of genes which were not transcriptionally more plastic before duplication than other non-duplicable genes. Therefore, gene duplication provides the appropriate genetic and selective opportunity for the evolution of transcriptional plasticity. A remarkable result is that the percentage of singletons transcriptionally altered in S. cerevisiae was significantly lower than that of their orthologs altered in L. kluyveri. However, the absolute percentages of altered genes cannot be compared between L. kluyveri and S. cerevisiae as both species represent completely different metabolisms and the conditions of stress under which they were subjected were different. It is, therefore, likely that duplication itself may relax the cost–benefit conflict between noise and plasticity, as previously suggested,50 allowing for the emergence of plasticity and adaptation to environmental perturbations. In conclusion, our result point to a strong correlation between the expression properties of genes, their duplicability, transcriptional plasticity, and ability to give rise to novel adaptations.
Supplementary Material
Acknowledgements
We would like to thank members of Fares’ Lab for a careful reading and discussion of the results in the manuscript. We are also grateful to colleagues at Trinity College for helpful discussions. This work was supported by a grant from the Spanish Ministerio de Economía y Competitividad (MINECO-FEDER; BFU2015-66073-P) to M.A.F. F.M. is supported by a PhD grant from the Spanish Ministerio de Economía y Competitividad (reference: BES-2016-076677). C.T. was supported by a grant Juan de la Cierva from the Spanish Ministerio de Economía y Competitividad (reference: JCA-2012-14056).
Conflict of interest
None declared.
Accession numbers
All newly sequenced RNA sequences are available from the Sequence Read Archive with the following accession number (SRP074821).
Supplementary data
Supplementary data are available at DNARES online.
References
- 1. Ohno S. 1970, Evolution by Gene Duplication. Springer Verlag, New York. [Google Scholar]
- 2. Ohno S. 1999, Gene duplication and the uniqueness of vertebrate genomes circa 1970–1999, Semin. Cell Dev. Biol., 10, 517–22. [DOI] [PubMed] [Google Scholar]
- 3. Lynch M., Conery J.S.. 2000, The evolutionary fate and consequences of duplicate genes, Science, 290, 1151–5. [DOI] [PubMed] [Google Scholar]
- 4. Otto S.P., Whitton J.. 2000, Polyploid incidence and evolution, Annu. Rev. Genet., 34, 401–37. [DOI] [PubMed] [Google Scholar]
- 5. Carretero-Paulet L., Fares M.A.. 2012, Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications, Mol. Biol. Evol., 29, 3541–51. [DOI] [PubMed] [Google Scholar]
- 6. Cui L., Wall P.K., Leebens-Mack J.H., et al. 2006, Widespread genome duplications throughout the history of flowering plants, Genome Res., 16, 738–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holub E.B. 2001, The arms race is ancient history in Arabidopsis, the wildflower, Nat. Rev. Genet., 2, 516–27. [DOI] [PubMed] [Google Scholar]
- 8. Lespinet O., Wolf Y.I., Koonin E.V., Aravind L.. 2002, The role of lineage-specific gene family expansion in the evolution of eukaryotes, Genome Res., 12, 1048–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wendel J.F. 2000, Genome evolution in polyploids, Plant Mol. Biol., 42, 225–49. [PubMed] [Google Scholar]
- 10. Soltis D.E., Albert V.A., Leebens-Mack J., et al. 2009, Polyploidy and angiosperm diversification, Am. J. Bot., 96, 336–48. [DOI] [PubMed] [Google Scholar]
- 11. Van de Peer Y. 2004, Computational approaches to unveiling ancient genome duplications, Nat. Rev. Genet., 5, 752–63. [DOI] [PubMed] [Google Scholar]
- 12. Hoegg S., Brinkmann H., Taylor J.S., Meyer A.. 2004, Phylogenetic timing of the fish-specific genome duplication correlates with the diversification of teleost fish, J. Mol. Evol., 59, 190–203. [DOI] [PubMed] [Google Scholar]
- 13. Grant S.G. 2016, The molecular evolution of the vertebrate behavioural repertoire, Philos. Trans. R. Soc. Lond. B: Biol. Sci., 371, 20150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Green S.A., Bronner M.E.. 2013, Gene duplications and the early evolution of neural crest development, Semin. Cell Dev. Biol., 24, 95–100. [DOI] [PubMed] [Google Scholar]
- 15. Huminiecki L., Conant G.C.. 2012, Polyploidy and the evolution of complex traits, Int. J. Evol. Biol., 2012, 292068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Conant G.C., Wolfe K.H.. 2007, Increased glycolytic flux as an outcome of whole-genome duplication in yeast, Mol. Syst. Biol., 3, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wolfe K.H., Shields D.C.. 1997, Molecular evidence for an ancient duplication of the entire yeast genome, Nature, 387, 708–13. [DOI] [PubMed] [Google Scholar]
- 18. Fares M.A., Keane O.M., Toft C., Carretero-Paulet L., Jones G.W.. 2013, The roles of whole-genome and small-scale duplications in the functional specialization of Saccharomyces cerevisiae genes, PLoS Genet., 9, e1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blanc G., Wolfe K.H.. 2004, Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes, Plant Cell, 16, 1667–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blanc G., Wolfe K.H.. 2004, Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution, Plant Cell, 16, 1679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conant G.C., Wolfe K.H.. 2008, Turning a hobby into a job: how duplicated genes find new functions, Nat. Rev. Genet., 9, 938–50. [DOI] [PubMed] [Google Scholar]
- 22. Fares M.A., Byrne K.P., Wolfe K.H.. 2006, Rate asymmetry after genome duplication causes substantial long-branch attraction artifacts in the phylogeny of Saccharomyces species, Mol. Biol. Evol., 23, 245–53. [DOI] [PubMed] [Google Scholar]
- 23. Scannell D.R., Wolfe K.H.. 2008, A burst of protein sequence evolution and a prolonged period of asymmetric evolution follow gene duplication in yeast, Genome Res., 18, 137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Freeling M. 2009, Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition, Annu. Rev. Plant Biol., 60, 433–53. [DOI] [PubMed] [Google Scholar]
- 25. Conant G.C., Birchler J.A., Pires J.C.. 2014, Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time, Curr. Opin. Plant Biol., 19, 91–8. [DOI] [PubMed] [Google Scholar]
- 26. Keane O.M., Toft C., Carretero-Paulet L., Jones G.W., Fares M.A.. 2014, Preservation of genetic and regulatory robustness in ancient gene duplicates of Saccharomyces cerevisiae, Genome Res., 24, 1830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fares M.A. 2015, The origins of mutational robustness, Trends Genet., 31, 373–81. [DOI] [PubMed] [Google Scholar]
- 28. Gout J.F., Lynch M.. 2015, Maintenance and loss of duplicated genes by dosage subfunctionalization, Mol. Biol. Evol., 32, 2141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altschul S.F., Madden T.L., Schaffer A.A., et al. 1997, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25, 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Byrne K.P., Wolfe K.H.. 2005, The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species, Genome Res., 15, 1456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lohse M., Bolger A.M., Nagel A., et al. 2012, RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics, Nucleic Acids Res., 40, W622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson M.D., McCarthy D.J., Smyth G.K.. 2010, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, 26, 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anders S., Huber W.. 2010, Differential expression analysis for sequence count data, Genome Biol., 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benjamini Y., Yekutieli D.. 2001, The control of the false discovery rate in multiple testing under dependency, Ann. Stat., 29, 24. [Google Scholar]
- 35. Brion C., Pflieger D., Souali-Crespo S., Friedrich A., Schacherer J.. 2016, Differences in environmental stress response among yeasts is consistent with species-specific lifestyles, Mol. Biol. Cell, 27, 1694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Linde J., Duggan S., Weber M., et al. 2015, Defining the transcriptomic landscape of Candida glabrata by RNA-Seq, Nucleic Acids Res., 43, 1392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seoighe C., Wolfe K.H.. 1999, Yeast genome evolution in the post-genome era, Curr. Opin. Microbiol., 2, 548–54. [DOI] [PubMed] [Google Scholar]
- 38. Aury J.M., Jaillon O., Duret L., et al. 2006, Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia, Nature, 444, 171–8. [DOI] [PubMed] [Google Scholar]
- 39. Gout J.F., Kahn D., Duret L., Paramecium Post-Genomics C.. 2010, The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution, PLoS Genet., 6, e1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGrath C.L., Gout J.F., Doak T.G., Yanagi A., Lynch M.. 2014, Insights into three whole-genome duplications gleaned from the Paramecium caudatum genome sequence, Genetics, 197, 1417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McGrath C.L., Gout J.F., Johri P., Doak T.G., Lynch M.. 2014, Differential retention and divergent resolution of duplicate genes following whole-genome duplication, Genome Res., 24, 1665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Albert F.W., Muzzey D., Weissman J.S., Kruglyak L.. 2014, Genetic influences on translation in yeast, PLoS Genet., 10, e1004692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gout J.F., Duret L., Kahn D.. 2009, Differential retention of metabolic genes following whole-genome duplication, Mol. Biol. Evol., 26, 1067–72. [DOI] [PubMed] [Google Scholar]
- 44. Papp B., Pal C., Hurst L.D.. 2003, Dosage sensitivity and the evolution of gene families in yeast, Nature, 424, 194–7. [DOI] [PubMed] [Google Scholar]
- 45. Qian W., Liao B.Y., Chang A.Y., Zhang J.. 2010, Maintenance of duplicate genes and their functional redundancy by reduced expression, Trends Genet., 26, 425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Birchler J.A., Veitia R.A.. 2012, Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines, Proc. Natl. Acad. Sci. USA., 109, 14746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pu S., Wong J., Turner B., Cho E., Wodak S.J.. 2009, Up-to-date catalogues of yeast protein complexes, Nucleic Acids Res., 37, 825–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scannell D.R., Byrne K.P., Gordon J.L., Wong S., Wolfe K.H.. 2006, Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts, Nature, 440, 341–5. [DOI] [PubMed] [Google Scholar]
- 49. Landry C.R., Lemos B., Rifkin S.A., Dickinson W.J., Hartl D.L.. 2007, Genetic properties influencing the evolvability of gene expression, Science, 317, 118–21. [DOI] [PubMed] [Google Scholar]
- 50. Lehner B. 2010, Conflict between noise and plasticity in yeast, PLoS Genet., 6, e1001185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mattenberger F., Sabater-Munoz B., Hallsworth J.E., Fares M.A.. 2017, Glycerol stress in Saccharomyces cerevisiae: cellular responses and evolved adaptations, Environ. Microbiol, 19(3), 990–1007. [DOI] [PubMed] [Google Scholar]
- 52. Mattenberger F., Sabater-Munoz B., Toft C., Fares M.A.. 2017, The phenotypic plasticity of duplicated genes in Saccharomyces cerevisiae and the origin of adaptations, G3 (Bethesda), 7, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mattenberger F., Sabater-Munoz B., Hallsworth J.E., Fares M.A.. 2017, Glycerol stress in Saccharomyces cerevisiae: cellular responses and evolved adaptations, Environ. Microbiol., 19, 990–1007. [DOI] [PubMed] [Google Scholar]
- 54. Blake W.J., Balazsi G., Kohanski M.A., et al. 2006, Phenotypic consequences of promoter-mediated transcriptional noise, Mol. Cell, 24, 853–65. [DOI] [PubMed] [Google Scholar]
- 55. Raser J.M., O'Shea E.K.. 2005, Noise in gene expression: origins, consequences, and control, Science, 309, 2010–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Newman J.R., Ghaemmaghami S., Ihmels J., et al. 2006, Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise, Nature, 441, 840–6. [DOI] [PubMed] [Google Scholar]
- 57. Tirosh I., Weinberger A., Carmi M., Barkai N.. 2006, A genetic signature of interspecies variations in gene expression, Nat. Genet., 38, 830–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.