

Abstract
Iron–sulfur clusters (ISCs) are known to play a major role in various protein functions. Located in the mitochondria, cytosol, endoplasmic reticulum and nucleus, they contribute to various core cellular functions. Until recently, only a few human diseases related to mitochondrial ISC biogenesis defects have been described. Such diseases include Friedreich ataxia, combined oxidative phosphorylation deficiency 19, infantile complex II/III deficiency defect, hereditary myopathy with lactic acidosis and mitochondrial muscle myopathy, lipoic acid biosynthesis defects, multiple mitochondrial dysfunctions syndromes and non ketotic hyperglycinemia due to glutaredoxin 5 gene defect. Disorders of mitochondrial import, export and translation, including sideroblastic anemia with ataxia, EVEN-PLUS syndrome and mitochondrial complex I deficiency due to nucleotide-binding protein-like protein gene defect, have also been implicated in ISC biogenesis defects. With advances in next generation sequencing technologies, more disorders related to ISC biogenesis defects are expected to be elucidated. In this article, we aim to shed the light on mitochondrial ISC biogenesis, related proteins and their function, pathophysiology, clinical phenotypes of related disorders, diagnostic approach, and future implications.
Iron sulfur clusters (ISCs) were first described in the early 1960s by Helmut Beinert.1-3 Subsequent research highlighted the crucial role of these proteins in different biological cellular processes in plants, prokaryotes and eukaryotes and their link to human diseases.4 The first human disease related to ISCs biogenesis pathway was Friedreich ataxia (FRDA) which was described in the 1860s. Since the elucidation of the molecular basis causing FRDA in 1996,5 several studies have been conducted to explore the types of mutations and the exact role of the mitochondrial membrane protein frataxin and its involvement in ISC pathway.6-8 More recently, with the application of advanced molecular diagnostic technologies along with whole exome sequencing, additional human disorders have been unveiled beyond FRDA. In this article, we aim to shed the light on mitochondrial ISC biogenesis, related proteins and their function, pathophysiology clinical phenotypes of related disorders, diagnostic approach, and future implications.
Iron-sulfur proteins: biological function and relevance
Iron sulfur clusters are known to play a major role in various protein functions. Located in the mitochondria, cytosol, endoplasmic reticulum and nucleus, they contribute to respiration, iron homeostasis, heme biosynthesis, oxidative phosphorylation, citric acid cycle, and DNA replication and repair, among regulation of other pathways.5-7
Assembly of ISCs usually begins in mitochondria, where several other proteins essential for iron-sulfur (Fe-S) components maturation are found. There, ISCs are involved in enzymatic reactions of aconitase 1 and 2 of citric acid cycle, electron transfer of complexes I, II and III, fatty acid oxidation specifically electron-transfer-flavoprotein-ubiquinone oxidoreductase, biotin and lipoic acid,9 which is a fundamental cofactor involved in many cellular pathways. Iron-sulfar (Fe-S) components continue their maturation in mitochondria till they reach their target apoproteins, a process that’s been well investigated in prokaryotes and human equivalents.10
There are approximately 20 different proteins involved in the mitochondrial ISC biogenesis. These include: The sulfur donor nitrogen fixation gene 1,11 scaffold proteins such as Fe-S cluster scaffold (ISCU),12 LYR motif-containing protein 4 (LYRM4)13 - a eukaryotic specific accessory factor- required for cysteine desulfurase activity, Frataxin (FXN)8 as cysteine desulfurase depressor, Ferredoxins (FDX1, FDX2)14 and Ferredoxin reductase (FDXR)15 which are necessary for electron transport. Moreover, Fe-S cluster assembly 1(ISCA1),16,17 iron-sulfur cluster assembly 2 (ISCA2), and iron-sulfur cluster assembly factor for biotin synthase- and aconitase-like mitochondrial proteins, with a mass of 57kDa (IBA57)17 are also implicated in [4Fe-4S] assembly. In addition, there are molecular chaperone such as Mortalin (heat shock protein family A (Hsp70) member 9 (HSPA9),18,19 HSC20 gene20 that binds target proteins containing the LYR motif, an intermediate carrier glutaredoxin 5 (GLRX5),21 iron-sulfur cluster scaffold (NFU1)22 and bolA family member 3 (BOLA3)23,24 which are dedicated targeting factors for lipoic acid synthase,25 nucleotide-binding protein-like protein (NUBPL) - essential for complex I assembly-,26 ATP-binding cassette subfamily b, member 7 (ABCB7) that exports a sulfur containing compound from mitochondria,27 and growth factor, ERV1-like.28
Pathophysiology
Mitochondrial ISCs process is crucial for the biosynthesis of mitochondrial, cytosolic and nuclear Fe-S containing proteins.29-31 In eukaryotes, ISCs are present in 2 forms: [2Fe-2S] rhombic, and [4Fe-4S] cubane.32
The process is composed of three sequential steps (Figure 1); the first step starts by sulfur donation from cysteine desulfurase (NFS), which in turn forms a complex with the stabilizing protein LYRM4, to complete the reaction and release sulfane (-SSH).33,34 Simultaneously, iron is imported into the mitochondria through the inner membrane protein, SLC25A37 (Mitochondrial Iron Transporter 1).35 Meanwhile FXN probably (as there is no clear evidence) acts as iron chaperone to deliver the imported iron to scaffold protein ISCU and regulates the above mentioned cysteine desulfurase reaction.36,37 A reduction reaction then takes place by FDX2 and FDXR to reduce sulfane to sulfide, after which Fe-S assembly is accomplished on ISCU.14,38,39 In the second step, a series of proteins, HSC20, HSPA9 and GrpE like 1, react with the Fe-S loaded ISCU leading to detachment of [2Fe-2S] cluster.20,40 Monothiol GLRX5 then binds transiently to the released [2Fe-2S], in coordination with tripeptide glutathione to form a glutathione-containing complex41,42 and finally transfer the mature [2Fe-2S] cluster to apoproteins or export it outside the mitochondria, through the inner membrane protein ABCB7,27 to take part in the cytosol iron assembly (CIA) machinery. The aforementioned proteins are essential for the synthesis of all cellular ISC proteins, hence called the core components of ISC machinery.9 In the third and final step, A-type proteins ISCA1, ISCA2, and IBA57 carry out the maturation of all cellular [4Fe-4S] clusters and carry them to their apoproteins either directly or through other specific proteins, like NFU1, NUBPL, and BOLA3 (Figure 1), which contribute to the maturation of specific [4Fe-4S] containing proteins.17 Further details of the process can be reviewed in references.9,43
Figure 1.
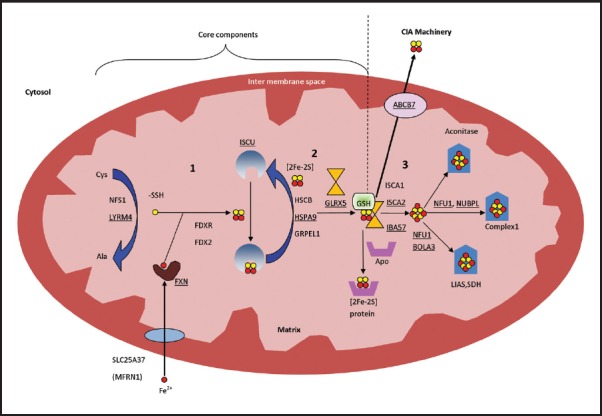
- Mitochondrial iron-sulfur cluster assembly: all involved proteins are written in their genetic names. Yellow circle presents sulfur and red circle presents iron. Numbers indicate the steps of the process; number 1 and 2 include the core components of the process. Proteins associated with known medical conditions are underlined. Ala - Alanine, BOLA3 - bolA family member 3, Cys - Cysteine, FDX2 - Ferredoxin 2, FDXR - Ferredoxin reductase, FXN - Frataxin, GLRX5 - glutaredoxin 5, GRPEL1 - GrpE like 1, GSH - tripeptide glutathione, HSCB - HscB mitochondrial iron-sulfur cluster cochaperone, HSPA9 - heat shock protein family A (Hsp70) member 9, IBA57 - iron-sulfur cluster assembly factor for biotin synthase- and aconitase-like mitochondrial proteins, with a mass of 57kDa, ISCA1 - iron-sulfur cluster assembly 1, ISCA2 - iron-sulfur cluster assembly 2, ISCU - Iron–sulfur cluster scaffold homolog, LIAS - lipoic acid synthetase, LYRM4 - LYR motif containing 4, NFS - Cysteine desulfurase, NFU1 - NFU1 iron-sulfur cluster scaffold, NUBPL - nucleotide binding protein like, SDH - succinate dehydrogenase.
Classification of mitochondrial ISC biogenesis defects
The ISCs are fundamental for many cellular pathways. Therefore, it is not unexpected that impairment of this pathway will cause a wide variety of diseases in humans (Table 1). Depending on the step of the pathway where there is a defect, disorders are classified into 3 categories:
Table 1.
Diseases caused by mitochondrial iron-sulfur cluster biogenesis.
| Variables | Frataxin (FXN) | ISD11 (LYRM4) | NFS1 | ISCU | FDX1L | GLRX5 | NFU1 | BOLA3 | IBA57 | ISCA2 | NUBPL | ABCB7 | HSPA9 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OMIM# | 22930 | 615595 | 603485 | 255125 | 614585 | 616860 and 616859 | 605711 | 614299 | 615330 | 616370 | 613621 | 301310 | 616854 |
| Disease | Friedreich ataxia (FRDA) | Combined oxidative phosphorylation deficiency 19 | Infantile complex II/III deficiency (IMC23D) | Myopathy with lactic acidosis, hereditary | mitochondrial muscle myopathy | 616860: Anemia, sideroblastic, 3, pyridoxine-616859: refractory Spasticity, childhood- | Multiple mitochondrial dysfunctions syndrome 1 | Multiple mitochondrial dysfunctions syndrome 2 | Multiple mitochondrial dysfunctions syndrome 3 | Multiple mitochondrial dysfunctions syndrome 4 | Mitochondrial complex I deficiency | Sidroblastic anemia with ataxia | Even-plus syndrome |
| Year | 1996 | 2013 | 2014 | 2008 | 2014 | 2007 and 2011 | 2011 | 2011 | 2013 | 2015 | 2010 | 1999 | 2015 |
| Pathway defect | Core [Fe-S] assembly | Core [Fe-S] assembly | Core [Fe-S] assembly | Core [Fe-S] assembly | Core [Fe-S] assembly | [Fe-S] transfer to specific recipients | [Fe-S] transfer to specific recipients | [Fe-S] transfer to specific recipients | [Fe4-S4] assembly | [Fe4-S4] assembly | Mitochondrial translation; complex I assembly | Mitochondrial export | Mitochondrial iron import |
| Number of patients/Prevalence | 1:50000 | 2 | 3 | 25 | 1 | 5 | 20 | 3 | 2 | 6 | 7 | 22 | 3 |
| Age of onset | Childhood-Adult (usually 2nd decade) | Neonatal | Infantile | Childhood | Childhood | Adult and childhood | neonatal and infantile | Infantile | Neonatal | Infantile | Infantile | Childhood | Prenatal |
| Origin | Panethnic | Lebanon and Syria | Canada | Sweden and Norway | Morocco | Italy and China | Mexico, Germany, Serbia, Romania, Pakistan | India, Australia, Africa | Morocco | Saudi | Argentina, Germany, Canada, Australia, Netherlands | USA | Chile and Korea |
| Clinical hints | Ataxia, dysarthria, muscle weakness, spasticity in the lower limbs, scoliosis, bladder dysfunction, absent lower limb reflexes, and loss of position and vibration sense, cardiomyopathy, DM | Hypotonia, respiratory distress | Hypotonia, respiratory distress, seizure, multisystem organ failure | Muscle weakness, exercise intolerance and cardiomyopathy | Severe proximal lower limb weakness and muscle cramps | 616860: Sideroblastic anemia, hepatosplenomegaly and jaundice 616859: Spastic paraplegia, spinal lesion, and optic atrophy | Hypotonia, respiratory distress, seizure, Neurologic regression pulmonary hypertension, lethargy, poor feeding, White matter lesions seen on brain imaging | Hypotonia, respiratory distress, seizure, Neurologic regression, lethargy, poor feeding, optic atrophy, white matter lesions seen on brain imaging | Severe hypotonia, generalized muscle weakness, absent primitive reflexes, microcephaly and dysmorphic features (retrognathia, high palate, widely spaced nipples, arthrogryposis, cerebral atrophy and polymicrogyria on Brain MRI | Neuroregression, developmental delay, nystagmus with optic atrophy and diffuse white matter disease of the brain and spine | Hypotonia, muscle weakness, muscle atrophy exercise intolerance Muscle biopsy shows abnormal mitochondria, developmental delay, neuroregression, seizure, white matter lesions seen on brain imaging | Sidroblastic anemia and ataxia | EVEN-PLUS syndrome is characterized by short stature, vertebral and epiphyseal changes, microtia, midface hypoplasia with flat nose and triangular nares, cardiac malformations, and other findings including anal atresia, hypodontia, and aplasia cutis. The features overlap those reported in patients with CODAS syndrome |
| Biochemical hints | None | Lactic acidosis, metabolic acidosis, high liver enzymes low complexes I-IV in the muscle | Lactic acidosis, metabolic acidosis, high CK level and high liver enzymes, DIC picture, low complexes II and III in the muscle | Lactic acidosis, Myoglobinuria Histopathology showed succinate dehydrogenase and cytochrome c oxidase (COX) deficienc | Lactic acidosis, myoglobinuria and low complexes I, II and III in the muscle | 616860: hypochromic microcytic anemia, increase ferritin level, ringed sideroblasts on bone marrow aspirate | Hyperglycinemia, metabolic acidosis, lactic acidosis, Increased urinary 2-hydroxybutyrate, Decreased activity of pyruvate dehydrogenase complex, low complexes I and II in the muscle | same as NFU1 gene defect | Hyperglycinemia, metabolic acidosis, lactic acidosis | Hyperglycinemia, metabolic acidosis, lactic acidosis | hypoglycemia, lactic acidosis, low complex I in the muscle | Increased free erythrocyte protoporphyrin, hypochromic microcytic anemia, ringed sideroblasts on bone marrow aspirate | Not specific |
| Mutation reported | 90 % have expanded GAA repeat in intron 1 of FXN gene* | Missense mutation c.203G>T, p.Arg68Lys | Missense mutation c.251G>A, p.Arg72Gln | Splicing defect IVS5 + 382G>C, heterozygosity for the splicing defect and the missense mutation c.149G>A, p.Gly50Glu | homozygous mutation c.1A>T | 616860: A> G homozygous transition 616859: Homozygous deletion c.151_153delAAG, p.K51del or compound heterozygosity for p.K51del and 8bp insertion | A homozygous missense mutation, c.545G>A(p.Arg182Gln), compound heterozygous for aforementioned mutation and a splice-site (c.545+5G>A) mutation, compound heterozygous mutation (g.69400462C>A, p.Gly208Cys); g.69592691_ 69648327del, [?]) compound heterozygous mutation (c.544C>T, [?], p.Arg182Trp);[?]), (c. 565G>A, p. Gly189Arg);[568G>A],;[Gly190 Arg]), (c.[544C>T];[?], p.[Arg182Trp];[?]), homozygous frameshift mutation c.302+3A>G (p.Val56Glyfs*9), compound heterozygous mutation (c.62G>C, p.Arg21Pro); (c.622G>T, p.Gly208Cys) | (c.136C4T, p.R46X) | (c.941A > C, p.Gln314Pro) | (c.229G>A, p.Gly77Ser) | Homozygous missense mutation (c.166G>A, p.Gly56Arg), intronic mutation: c.815-27T>C or compound heterozygous for (c.166G>A, p.Gly56Arg) and other mutation | Homozygous missense mutation(c.1200T>G(p.Ile400Met), Other several mutations near to or in transmembrane domains of the ABC transporter | Compound hgetrozygous mutation (c.383A > G (p.Y128C) and c.882_883delAG (p.V296*), homozygous missense mutation (c.376C > T;p.Arg126Trp). |
| Mortality | The average age of death was at 37.5 years (range, 5–71 years) | 1/2 | 2/3 | None | None | None | 20/20 | 3/3 | 2/2 | 4/6 | None | None | None |
1) Iron-sulfur assembly defects which include 5 disorders: Friedreich ataxia due to FXN gene defect, combined oxidative phosphorylation deficiency 19 due to LYRM4 gene defect, infantile complex II/III deficiency (IMC23D) due to NFS1 gene defect, hereditary myopathy with lactic acidosis due to ISCU gene defect and mitochondrial muscle myopathy due to Ferredoxin 1-Like protein (FDX1L) gene defect.
2) Lipoic acid biosynthesis defects which include 5 disorders: Multiple mitochondrial dysfunctions syndrome 1 due to NFU1 gene defect, multiple mitochondrial dysfunctions syndrome 2 due to BOLA3 gene defect, multiple mitochondrial dysfunctions syndrome 3 due to IBA57 gene defect, multiple mitochondrial dysfunctions syndrome 4 due to ISCA2 gene defect and non ketotic hyperglycinemia due to GLRX5 gene defect.
3) Disorders of mitochondrial import, export and translation: Sideroblastic anemia with ataxia due to ABCB7 gene defect, EVEN-PLUS syndrome due to HSPA9 gene defect and, mitochondrial complex I deficiency due to NUBPL gene defect.
Clinical phenotypes
1. Friedreich ataxia (OMIM#229300)
Inherited as an autosomal recessive disorder and considered as the most common spinocerebellar degenerative disease, with prevalence of 1:20,000-1:50,000. Friedreich ataxia is characterized by the triad: ataxia, areflexia and positive plantar response. Additional features include cardiomyopathy, diabetes mellitus, visual loss and deafness. Cognitive function and intelligence on the other hand are usually preserved.
Friedreich ataxia presents during childhood or adolescence. Clinically, ataxia is usually the first sign often followed by pyramidal signs, dysarthria and upper-limb ataxia. Areflexia and distal sensory loss are also present in most cases, and spasticity which occurs later can lead to discomfort, pain and contractures. Cardiac involvement is initially clinically asymptomatic. However, hypertrophic cardiomyopathy usually develops after the neurological symptoms. Approximately 30% of patients with FRDA develop diabetes mellitus.
Ophthalmological findings which present early on include fixation instability (square wave jerks), nystagmus, and blindness at a later age. Auditory neuropathy often leads to hearing abnormalities. Other features include mild dysphagia which progresses with disease advancement, skeletal abnormalities (scoliosis, and foot deformities such as pes cavus and talipes equinovarus).
Diagnosis of FRDA is made by combining clinical findings and molecular testing. Approximately 90% of cases of FRDA are homozygous for a disease causing GAA repeat expansion in intron 1 of FXN gene encoding frataxin, while 10% are compound heterozygotes for a disease causing GAA repeat expansion in one allele and another intragenic pathogenic variant in the other allele. The disease causing alleles exhibit expansion from 66 to 1700 GAA repeats, with most patients having between 600 and 1200 GAA repeats. Treatment is usually supportive but without a cure. Therefore prognosis has improved, however morbidity and quality of life remain a major concern. Cardiac complications including arrhythmias (especially atrial fibrillation) and congestive heart failure are the main causes of death. Early age of onset and other comorbidities, including diabetes mellitus, contribute to a survival age of around 40 years old.44-49
2. Combined oxidative phosphorylation deficiency type 19 due to LYRM4 gene defect (OMIM# 615595)
Combined oxidative phosphorylation deficiency is one of the most common enzymatic defects in patients with mitochondrial disorders, contributing up to 30% of all cases.50 To date, there are approximately 30 different types of defects, each classified according to the gene defect. Combined oxidative phosphorylation deficiency type 19 due to defect in LYRM4 gene is an autosomal recessive disease that was first described by Lim et al51 2013 in 2 double first cousins with neonatal onset of hypotonia, respiratory distress, stridor, failure to thrive, poor feeding and hepatic steatosis. Biochemical workup showed lactic acidosis, metabolic acidosis, high liver enzymes, and low complexes I-IV in muscle and liver samples. Diagnosis was confirmed by identifying homozygous missense mutation in LYRM4 gene (c.203G>T, p.Arg68Leu).51 One of these 2 relatives died at 12 weeks of age while the other was reported healthy at the age of 20 years old.51
3. Infantile mitochondrial complex II/III deficiency due to NFS1 gene defect (OMIM#603485)
Farhan et al52 2014 first described this autosomal recessive disease in 3 affected siblings to consanguineous parents from Old Order Mennonite community in Canada.52 These patients presented with hypotonia, respiratory distress, seizures, and multisystem organ failure. Biochemical workup revealed lactic acidosis, metabolic acidosis, high CK levels, high liver enzymes, disseminated intravascular coagulation features, and low complexes II and III in muscle sample. Diagnosis was confirmed through autozygosity mapping and whole exome sequencing, which identified homozygous missense mutation (c.215G>A, p.Arg72Gln) in NFS1 gene in all three affected siblings. Two of the 3 reported siblings died at 7 month of age due to cardiac failure.52
4. ISCU myopathy, also known as hereditary myopathy with lactic acidosis or as Swedish myopathy due to ISCU gene defect (OMIM#255125)
Inherited in an autosomal recessive pattern, these patients classically present with muscular manifestations including: myopathy, exercise intolerance, and premature exertional muscle weakness where muscles become hard, painful and tender during exercise. Laboratory investigations show mitochondrial respiratory chain complex I, II, and III defect, rhabdomyolysis, abnormal mitochondria in muscle biopsy, subsarcolemmal mitochondrial and lipid droplet accumulation, abnormal iron deposition, and decreased muscle succinate dehydrogenase and muscle mitochondrial aconitase.53-56
5. Mitochondrial muscle myopathy due to FDX1L gene defect (OMIM#614585)
It was reported only once, in a Jewish Moroccan female adolescent to consanguineous parents, who presented with severe proximal lower limb myopathy, muscle cramps and lactic acidosis. Biochemical workup showed lactic acidosis, myoglobinuria and low complexes I, II and III in the muscle. Notably, subsequent annual cardiac evaluation remained normal.57
6. Mitochondrial complex I deficiency due to NUBPL gene defect (OMIM# 613621)
Respiratory chain deficiency of complex I is the most common cause for mitochondrial disorders, accounting for approximately one third of all reported patients.58 As was recently demonstrated, NUBPL plays an important role in mitochondrial translation, which secondarily affects complex I assembly, of which many core subunits are mitochondrially encoded.59 Inherited in as an autosomal recessive pattern, clinical phenotype comprises the following: hypotonia, muscle weakness, muscle atrophy, exercise intolerance, developmental delay, neuroregression, and seizures. Muscle biopsy shows abnormal mitochondria, and brain imaging shows white matter lesions and pontocerebellar hypoplasia. Biochemically, affected individuals show hypoglycemia, lactic acidosis, and low complex I in the muscle.60-63
7. X-Linked sideroblastic anemia with ataxia due to ABCB7 gene defect (OMIM#301310)
The function of ABCB7 gene is to export a sulfur containing compound from mitochondria to the cytosol. Inherited in an X-linked recessive pattern, patients with defects in ABCB7 gene present clinically with ataxia and sideroblastic anemia in early childhood. Laboratory investigations show increased free erythrocyte protoporphyrin, hypochromic microcytic anemia, and ringed sideroblasts on bone marrow aspirate.64-66 Interestingly, heterozygous females have a normal neurologic examination and may have a dimorphic peripheral blood smear with both hypochromic microcytic anemia and normal red blood cells, and may additionally have ring sideroblasts on bone marrow examination.67
8. EVEN-PLUS syndrome due to HSPA9 gene defect (OMIM#616854)
EVEN-PLUS syndrome was described recently. The phenotype involves epiphyses, vertebrae, ears, and nose which are included in the EVEN part of the acronym, and PLUS associated findings include short stature, vertebral and epiphyseal changes, microtia, midface hypoplasia with flat nose and triangular nares, cardiac malformations. Other findings include anal atresia, hypodontia, and aplasia cutis. These features can overlap with findings reported in patients with Cerebral, Ocular, Dental, Auricular and Skeletal syndrome characterized by multiple congenital anomalies including Cerebral, Ocular, Dental, Auricular and Skeletal anomalies.68
Royer-Bertrand et al69 2015 reported 3 girls from Korean and Chilean origins with EVEN-PLUS syndrome and confirmed the diagnosis with mutations in HSPA9 gene, a gene that is essential for mitochondrial protein import. While homozygous mutations in HSPA9 can cause EVEN-PLUS syndrome, heterozygous mutations can cause autosomal dominant sideroblastic anemia type 4.69,70
9. Lipoic acid biosynthesis defects
One important biochemical phenotype of many mitochondrial ISCs biogenesis disorders is hyperglycinemia, which occurs due to defects in the third and final step that leads to inhibition of lipoic acid synthase which is a covalently bound cofactor essential for glycine cleavage system (GCS), an impairment of which results in hyperglycinemia (Figure 2). Lipoic acid is a vital cofactor for five redox reactions in humans: Two enzymes that are essential for energy production (a-ketoglutarate dehydrogenase, and pyruvate dehydrogenase PDH), and three that contribute to amino acid pathways (branched-chain ketoacid dehydrogenase (BCKDH), 2-oxoadipate dehydrogenase, and the GCS) (Figure 2). Lipoic acid biosynthesis defects were previously described in five syndromes: a) Multiple mitochondrial dysfunctions syndrome 1 due to NFU1 gene defect (OMIM#605711);24,71-73 b) Multiple mitochondrial dysfunctions syndrome 2 due to BOLA3 gene defect (OMIM#614299);24,74,75 c) Multiple mitochondrial dysfunctions syndrome 3 due to IBA57 gene defect (OMIM#615330);76-78 d) Multiple mitochondrial dysfunctions syndrome 4 due to ISCA2 gene defect (OMIM#616370);79 e) Non ketotic hyperglycinemia due to GLRX5 gene defect (OMIM# 616860 and 616859).75
Figure 2.
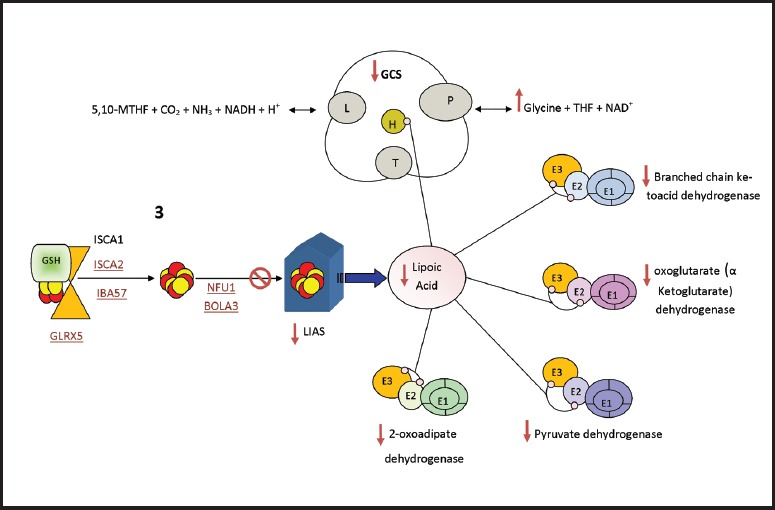
- Lipoic Acid Synthetase (LIAS) biosynthesis and function, LIAS is essential in the maturation of lipoic acid (LA) which acts as cofactor for many enzymes. lipoic acid is bound to E2 and E3 subunits of the mitochondrial a-ketoacid dehydrogenase complex family (Pyruvate dehydrogenase, oxoglutarate (a ketoglutarate) dehydrogenase and branched chain ketoacid dehydrogenase, 2-oxoadipate dehydrogenase). lipoic acid is also bound to protein H in the Glycine cleavage system (GCS). Diseases which cause inhibition of any of ISC assembly step 3 proteins (shown in red and underlined) will affect the synthesis of the target apoprotein LIAS and subsequently the end product LA. As a result, all the biological process which include lipoic acid will be affected; the activity of a-ketoacid dehydrogenase complex family will decrease and the GCS activity will decrease which result in hyperglycinemia. ISC - Iron–sulfur clusters, 5,10-MTHF - 5, 10 methylene tetrahydrofolate, GCS - Glycine Cleavage System, THF - tetrahydrofolate
These 5 disorders share one biochemical abnormality which is hyperglycinemia. The 4 multiple mitochondrial dysfunction syndromes have similar clinical and biochemical phenotypes which include: hypotonia, respiratory distress, seizures, encephalopathy, myopathy, neurologic regression, lethargy, poor feeding, optic atrophy, and diffuse white matter lesions seen on brain imaging. Furthermore, metabolic acidosis, lactic acidosis, increased urinary 2-hydroxybutyrate, and decreased activity of lipoic acid dependent enzymes including PDH, a-KGDH, BCKDH, and GCS. Additionally, muscle biopsy shows low complexes I and II.24,68,71-75,78,79
The last disorder due to GLRX5 gene defect has two clinical phenotypes: one is sideroblastic anemia, and the other constitutes childhood spasticity. However, biochemically, both phenotypes present with hyperglycinemia in the plasma of affected individuals.75
Diagnostic approach
Mitochondrial ISC biogenesis disorders should be considered in the differential diagnosis when there is sideroblastic anemia, neurological manifestations or myopathic phenotypes, and follow up biochemical workup should be pursued. Hyperglycinemia, for instance, should point to lipoic acids biosynthesis defect disorders after excluding classical non ketotic hyperglycinemia caused by glycine decarboxylase deficiency, aminomethyltransferase gene, and glycine cleavage system H gene. Sideroblastic anemia with neurological manifestations could suggest GLRX5, ABCB7 or HSPA9 gene defects. Short stature, skeletal abnormalities and dysmorphic features could suggest EVEN-PLUS syndrome. One diagnostic approach for patients of consanguineous parents presenting with neurological or myopathic findings, is to consider homozygosity mapping and whole exome sequencing (WES) or whole genome sequencing.
Future implications and conclusion
Prior to 2010, only a few human diseases related to mitochondrial ISC biogenesis defects were elucidated. With the introduction of next generation sequencing technologies, it has become more possible to expand these disorders and better characterize their clinical phenotypes, consequently more disorders are expected to be identified in the near future. The complexity and mixture of genetic and clinical heterogeneity (as illustrated by previous disorders) make ISC biogenesis defects difficult to recognize, and it is plausible that their prevalence is under estimated.
In conclusion, the combined clinical and advanced molecular diagnostic efforts will likely identify more disorders linked to the mitochondrial ISC biogenesis pathway, and potentially discover new genes in association with such diseases. Continued studies will help further elucidate mitochondrial iron homeostasis regulation, as understanding of mitochondrial iron overload could potentially yield better therapeutic approaches for such defects.
Footnotes
Disclosure.
References
- 1.Beinert H, Holm RH, Munck E. Iron-sulfur clusters:nature’s modular, multipurpose structures. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 2.Beinert H, Lee W. Evidence for a new type of iron containing electron carrier in mitochondria. Biochem Biophys Res Commun. 1961;5:40–45. doi: 10.1016/0006-291x(61)90077-8. [DOI] [PubMed] [Google Scholar]
- 3.Beinert H, Sands RH. Studies on succinic and DPNH dehydrogenase preparations by paramagnetic resonance (EPR) spectroscopy. Biochemical and Biophysical Research Communications. 1960;3:47–52. [Google Scholar]
- 4.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, et al. Friedreich’s ataxia:autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 6.Santos R, Lefevre S, Sliwa D, Seguin A, Camadro JM, Lesuisse E. Friedreich ataxia:molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid Redox Signal. 2010;13:651–690. doi: 10.1089/ars.2009.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol. 2009;256:9–17. doi: 10.1007/s00415-009-1003-2. [DOI] [PubMed] [Google Scholar]
- 8.Huynen MA, Snel B, Bork P, Gibson TJ. The phylogenetic distribution of frataxin indicates a role in iron-sulfur cluster protein assembly. Hum Mol Genet. 2001;10:2463–2468. doi: 10.1093/hmg/10.21.2463. [DOI] [PubMed] [Google Scholar]
- 9.Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta. 2012;1823:1491–1508. doi: 10.1016/j.bbamcr.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Roche B, Aussel L, Ezraty B, Mandin P, Py B, Barras F. Reprint of:Iron/sulfur proteins biogenesis in prokaryotes:formation, regulation and diversity. Biochim Biophys Acta. 2013;1827:923–937. doi: 10.1016/j.bbabio.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Zheng L, White RH, Cash VL, Jack RF, Dean DR. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc Natl Acad Sci U S A. 1993;90:2754–2758. doi: 10.1073/pnas.90.7.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong WH, Rouault T. Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 2000;19:5692–5700. doi: 10.1093/emboj/19.21.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y, Ghosh MC, Tong WH, Rouault TA. Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum Mol Genet. 2009;18:3014–3025. doi: 10.1093/hmg/ddp239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheftel AD, Stehling O, Pierik AJ, Elsasser HP, Muhlenhoff U, Webert H, et al. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A. 2010;107:11775–11780. doi: 10.1073/pnas.1004250107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Y, Ghosh M, Kovtunovych G, Crooks DR, Rouault TA. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim Biophys Acta. 2012;1823:484–492. doi: 10.1016/j.bbamcr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cozar-Castellano I, del Valle Machargo M, Trujillo E, Arteaga MF, Gonzalez T, Martin-Vasallo P, et al. hIscA:a protein implicated in the biogenesis of iron-sulfur clusters. Biochim Biophys Acta. 2004;1700:179–188. doi: 10.1016/j.bbapap.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsasser HP, Muhlenhoff U, et al. The human mitochondrial ISCA1, ISCA2 and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol Biol Cell. 2012;23:1157–1166. doi: 10.1091/mbc.E11-09-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan Y, Napoli E, Cortopassi G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum Mol Genet. 2007;16:929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- 19.Dores-Silva PR, Minari K, Ramos CH, Barbosa LR, Borges JC. Structural and stability studies of the human mtHsp70-escort protein 1:an essential mortalin co-chaperone. Int J Biol Macromol. 2013;56:140–148. doi: 10.1016/j.ijbiomac.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Uhrigshardt H, Singh A, Kovtunovych G, Ghosh M, Rouault TA. Characterization of the human HSC20, an unusual DnaJ type III protein, involved in iron-sulfur cluster biogenesis. Hum Mol Genet. 2010;19:3816–3834. doi: 10.1093/hmg/ddq301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest. 2010;120:1749–1761. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong WH, Jameson GN, Huynh BH, Rouault TA. Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc Natl Acad Sci U S A. 2003;100:9762–9767. doi: 10.1073/pnas.1732541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou YB, Cao JB, Wan BB, Wang XR, Ding GH, Zhu H, et al. hBolA, novel non-classical secreted proteins, belonging to different BolA family with functional divergence. Mol Cell Biochem. 2008;317:61–68. doi: 10.1007/s11010-008-9809-2. [DOI] [PubMed] [Google Scholar]
- 24.Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, et al. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–495. doi: 10.1016/j.ajhg.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayr JA, Zimmermann FA, Fauth C, Bergheim C, Meierhofer D, Radmayr D, et al. Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am J Hum Genet. 2011;89:792–797. doi: 10.1016/j.ajhg.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheftel AD, Stehling O, Pierik AJ, Netz DJ, Kerscher S, Elsasser HP, et al. Human ind1, an iron-sulfur cluster assembly factor for respiratory complex I. Mol Cell Biol. 2009;29:6059–6073. doi: 10.1128/MCB.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kispal G, Csere P, Guiard B, Lill R. The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 1997;418:346–350. doi: 10.1016/s0014-5793(97)01414-2. [DOI] [PubMed] [Google Scholar]
- 28.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, et al. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 29.Lill R, Diekert K, Kaut A, Lange H, Pelzer W, Prohl C, et al. The essential role of mitochondria in the biogenesis of cellular iron-sulfur proteins. Biol Chem. 1999;380:1157–1166. doi: 10.1515/BC.1999.147. [DOI] [PubMed] [Google Scholar]
- 30.Kispal G, Csere P, Prohl C, Lill R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Netz DJ, Mascarenhas J, Stehling O, Pierik AJ, Lill R. Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol. 2014;24:303–312. doi: 10.1016/j.tcb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 33.Zheng L, White RH, Cash VL, Dean DR. Mechanism for the desulfurization of L-cysteine catalyzed by the nifS gene product. Biochemistry. 1994;33:4714–4720. doi: 10.1021/bi00181a031. [DOI] [PubMed] [Google Scholar]
- 34.Kaiser JT, Clausen T, Bourenkow GP, Bartunik HD, Steinbacher S, Huber R. Crystal structure of a NifS-like protein from Thermotoga maritima:implications for iron sulphur cluster assembly. J Mol Biol. 2000;297:451–464. doi: 10.1006/jmbi.2000.3581. [DOI] [PubMed] [Google Scholar]
- 35.Chen W, Paradkar PN, Li L, Pierce EL, Langer NB, Takahashi-Makise N, et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc Natl Acad Sci U S A. 2009;106:16263–16268. doi: 10.1073/pnas.0904519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerber J, Muhlenhoff U, Lill R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003;4:906–911. doi: 10.1038/sj.embor.embor918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- 38.Muhlenhoff U, Gerber J, Richhardt N, Lill R. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003;22:4815–4825. doi: 10.1093/emboj/cdg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lange H, Kaut A, Kispal G, Lill R. A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. Proc Natl Acad Sci U S A. 2000;97:1050–1055. doi: 10.1073/pnas.97.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonomi F, Iametti S, Morleo A, Ta D, Vickery LE. Studies on the mechanism of catalysis of iron-sulfur cluster transfer from IscU[2F.e2S] by HscA/HscB chaperones. Biochemistry. 2008;47:12795–12801. doi: 10.1021/bi801565j. [DOI] [PubMed] [Google Scholar]
- 41.Herrero E, de la Torre-Ruiz MA. Monothiol glutaredoxins:a common domain for multiple functions. Cell Mol Life Sci. 2007;64:1518–1530. doi: 10.1007/s00018-007-6554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bandyopadhyay S, Gama F, Molina-Navarro MM, Gualberto JM, Claxton R, Naik SG, et al. Chloroplast monothiol glutaredoxins as scaffold proteins for the assembly and delivery of [2Fe-2S] clusters. EMBO J. 2008;27:1122–1133. doi: 10.1038/emboj.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maio N, Rouault TA. Iron-sulfur cluster biogenesis in mammalian cells:New insights into the molecular mechanisms of cluster delivery. Biochim Biophys Acta. 2015;1853:1493–1512. doi: 10.1016/j.bbamcr.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Payne RM, Wagner GR. Cardiomyopathy in Friedreich ataxia:clinical findings and research. J Child Neurol. 2012;27:1179–1186. doi: 10.1177/0883073812448535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Regner SR, Wilcox NS, Friedman LS, Seyer LA, Schadt KA, Brigatti KW, et al. Friedreich ataxia clinical outcome measures:natural history evaluation in 410 participants. J Child Neurol. 2012;27:1152–1158. doi: 10.1177/0883073812448462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delatycki MB, Corben LA. Clinical features of Friedreich ataxia. J Child Neurol. 2012;27:1133–1137. doi: 10.1177/0883073812448230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lynch DR, Farmer JM, Balcer LJ, Wilson RB. Friedreich ataxia:effects of genetic understanding on clinical evaluation and therapy. Arch Neurol. 2002;59:743–747. doi: 10.1001/archneur.59.5.743. [DOI] [PubMed] [Google Scholar]
- 48.Alikasifoglu M, Topaloglu H, Tuncbilek E, Ceviz N, Anar B, Demir E, et al. Clinical and genetic correlate in childhood onset Friedreich ataxia. Neuropediatrics. 1999;30:72–76. doi: 10.1055/s-2007-973463. [DOI] [PubMed] [Google Scholar]
- 49.De Michele G, Di Maio L, Filla A, Majello M, Cocozza S, Cavalcanti F, et al. Childhood onset of Friedreich ataxia:a clinical and genetic study of 36 cases. Neuropediatrics. 1996;27:3–7. doi: 10.1055/s-2007-973740. [DOI] [PubMed] [Google Scholar]
- 50.Thorburn DR, Chow CW, Kirby DM. Respiratory chain enzyme analysis in muscle and liver. Mitochondrion. 2004;4:363–375. doi: 10.1016/j.mito.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Lim SC, Friemel M, Marum JE, Tucker EJ, Bruno DL, Riley LG, et al. Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum Mol Genet. 2013;22:4460–4473. doi: 10.1093/hmg/ddt295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farhan SM, Wang J, Robinson JF, Lahiry P, Siu VM, Prasad C, et al. Exome sequencing identifies NFS1 deficiency in a novel Fe-S cluster disease, infantile mitochondrial complex II/III deficiency. Mol Genet Genomic Med. 2014;2:73–80. doi: 10.1002/mgg3.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mochel F, Haller RG. Myopathy with Deficiency of ISCU. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. Gene Reviews(R) Seattle (WA): University of Washington; 1993. [Google Scholar]
- 54.Sanaker PS, Toompuu M, Hogan VE, He L, Tzoulis C, Chrzanowska-Lightowlers ZM, et al. Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy. Biochim Biophys Acta. 2010;1802:539–244. doi: 10.1016/j.bbadis.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 55.Kollberg G, Tulinius M, Melberg A, Darin N, Andersen O, Holmgren D, et al. Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain. 2009;132:2170–2179. doi: 10.1093/brain/awp152. [DOI] [PubMed] [Google Scholar]
- 56.Olsson A, Lind L, Thornell LE, Holmberg M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum Mol Genet. 2008;17:1666–1672. doi: 10.1093/hmg/ddn057. [DOI] [PubMed] [Google Scholar]
- 57.Spiegel R, Saada A, Halvardson J, Soiferman D, Shaag A, Edvardson S, et al. Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur J Hum Genet. 2014;22:902–906. doi: 10.1038/ejhg.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fassone E, Rahman S. Complex I deficiency:clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159. [DOI] [PubMed] [Google Scholar]
- 59.Wydro MM, Sharma P, Foster JM, Bych K, Meyer EH, Balk J. The evolutionarily conserved iron-sulfur protein INDH is required for complex I assembly and mitochondrial translation in Arabidopsis [corrected] Plant Cell. 2013;25:4014–4027. doi: 10.1105/tpc.113.117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kevelam SH, Rodenburg RJ, Wolf NI, Ferreira P, Lunsing RJ, Nijtmans LG, et al. NUBPL mutations in patients with complex I deficiency and a distinct MRI pattern. Neurology. 2013;80:1577–1583. doi: 10.1212/WNL.0b013e31828f1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tenisch EV, Lebre AS, Grevent D, de Lonlay P, Rio M, Zilbovicius M, et al. Massive and exclusive pontocerebellar damage in mitochondrial disease and NUBPL mutations. Neurology. 2012;79:391. doi: 10.1212/WNL.0b013e3182611232. [DOI] [PubMed] [Google Scholar]
- 62.Tucker EJ, Mimaki M, Compton AG, McKenzie M, Ryan MT, Thorburn DR. Next-generation sequencing in molecular diagnosis:NUBPL mutations highlight the challenges of variant detection and interpretation. Hum Mutat. 2012;33:411–418. doi: 10.1002/humu.21654. [DOI] [PubMed] [Google Scholar]
- 63.Calvo SE, Tucker EJ, Compton AG, Kirby DM, Crawford G, Burtt NP, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dolatshad H, Pellagatti A, Liberante FG, Llorian M, Repapi E, Steeples V, et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia. 2016;30:2322–2331. doi: 10.1038/leu.2016.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Protasova MS, Grigorenko AP, Tyazhelova TV, Andreeva TV, Reshetov DA, Gusev FE, et al. Whole-genome sequencing identifies a novel ABCB7 gene mutation for X-linked congenital cerebellar ataxia in a large family of Mongolian ancestry. Eur J Hum Genet. 2016;24:550–555. doi: 10.1038/ejhg.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.D’Hooghe M, Selleslag D, Mortier G, Van Coster R, Vermeersch P, Billiet J, et al. X-linked sideroblastic anemia and ataxia:a new family with identification of a fourth ABCB7 gene mutation. Eur J Paediatr Neurol. 2012;16:730–735. doi: 10.1016/j.ejpn.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 67.Bekri S, D’Hooghe M, Vermeersch P. X-Linked Sideroblastic Anemia and Ataxia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): University of Washington; 1993. [Google Scholar]
- 68.Shebib SM, Reed MH, Shuckett EP, Cross HG, Perry JB, Chudley AE. Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies:CODAS syndrome--a case report. Am J Med Genet. 1991;40:88–93. doi: 10.1002/ajmg.1320400118. [DOI] [PubMed] [Google Scholar]
- 69.Royer-Bertrand B, Castillo-Taucher S, Moreno-Salinas R, Cho TJ, Chae JH, Choi M, et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci Rep. 2015;5:17154. doi: 10.1038/srep17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmitz-Abe K, Ciesielski SJ, Schmidt PJ, Campagna DR, Rahimov F, Schilke BA, et al. Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood. 2015;126:2734–2738. doi: 10.1182/blood-2015-09-659854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahting U, Mayr JA, Vanlander AV, Hardy SA, Santra S, Makowski C, et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front Genet. 2015;6:123. doi: 10.3389/fgene.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrer-Cortes X, Font A, Bujan N, Navarro-Sastre A, Matalonga L, Arranz JA, et al. Protein expression profiles in patients carrying NFU1 mutations. Contribution to the pathophysiology of the disease. J Inherit Metab Dis. 2013;36:841–847. doi: 10.1007/s10545-012-9565-z. [DOI] [PubMed] [Google Scholar]
- 73.Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–667. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haack TB, Rolinski B, Haberberger B, Zimmermann F, Schum J, Strecker V, et al. Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis. 2013;36:55–62. doi: 10.1007/s10545-012-9489-7. [DOI] [PubMed] [Google Scholar]
- 75.Baker PR, 2nd, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2014;137:366–379. doi: 10.1093/brain/awt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Debray FG, Stumpfig C, Vanlander AV, Dideberg V, Josse C, Caberg JH, et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes fatal infantile leukodystrophy. J Inherit Metab Dis. 2015;38:1147–1153. doi: 10.1007/s10545-015-9857-1. [DOI] [PubMed] [Google Scholar]
- 77.Lossos A, Stumpfig C, Stevanin G, Gaussen M, Zimmerman BE, Mundwiller E, et al. Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology. 2015;84:659–667. doi: 10.1212/WNL.0000000000001270. [DOI] [PubMed] [Google Scholar]
- 78.Ajit Bolar N, Vanlander AV, Wilbrecht C, Van der Aa N, Smet J, De Paepe B, et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet. 2013;22:2590–2602. doi: 10.1093/hmg/ddt107. [DOI] [PubMed] [Google Scholar]
- 79.Al-Hassnan ZN, Al-Dosary M, Alfadhel M, Faqeih EA, Alsagob M, Kenana R, et al. ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet. 2015;52:186–194. doi: 10.1136/jmedgenet-2014-102592. [DOI] [PubMed] [Google Scholar]