

Abstract
In this review, we discuss the intricate roles of the Wnt signalling network in the development and progression of mature B‐cell‐derived haematological malignancies, with a focus on chronic lymphocytic leukaemia (CLL) and related B‐cell lymphomas. We review the current literature and highlight the differences between the β‐catenin‐dependent and ‐independent branches of Wnt signalling. Special attention is paid to the role of the non‐canonical Wnt/planar cell polarity (PCP) pathway, mediated by the Wnt‐5–receptor tyrosine kinase‐like orphan receptor (ROR1)–Dishevelled signalling axis in CLL. This is mainly because the Wnt/PCP co‐receptor ROR1 was found to be overexpressed in CLL and the Wnt/PCP pathway contributes to numerous aspects of CLL pathogenesis. We also discuss the possibilities of therapeutically targeting the Wnt signalling pathways as an approach to disrupt the crucial interaction between malignant cells and their micro‐environment. We also advocate the need for research in this direction for other lymphomas, namely, diffuse large B‐cell lymphoma, Hodgkin lymphoma, mantle cell lymphoma, Burkitt lymphoma and follicular lymphoma where the Wnt signalling pathway probably plays a similar role.
Linked Articles
This article is part of a themed section on WNT Signalling: Mechanisms and Therapeutic Opportunities. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.24/issuetoc
Abbreviations
- APC
adenomatous polyposis coli
- BL
Burkitt lymphoma
- BM
bone marrow
- CAR
chimeric antigen receptor
- CCND1
cyclin D1 gene
- CELSR
cadherin EGF laminin G seven‐pass G‐type receptor
- CK
casein kinase
- CLL
chronic lymphocytic leukaemia
- CSNK1E
casein kinase ε gene
- CTNNB1
β‐catenin gene
- CYLD
cylindromatosis protein
- DKK
Dickkopf protein
- DLBCL
diffuse large B‐cell lymphoma
- DVL
Dishevelled
- EA
ethacrynic acid
- FL
follicular lymphoma
- FZD
Frizzled
- GC
germinal centre
- GSK‐3β
glycogen synthase kinase‐3 β
- HL
Hodgkin lymphoma
- HSC
haematopoietic stem cell
- IGHV
immunoglobulin heavy chain
- LEF
lymphoid enhancer‐binding factor
- LRP
LDL receptor‐related protein
- MCL
mantle cell lymphoma
- M‐CLL/U‐CLL
mutated/unmutated immunoglobulin heavy chain status of chronic lymphocytic leukaemia patients
- OS
overall survival
- PB
peripheral blood
- PCP
planar cell polarity
- PI3K
phosphoinositide 3‐kinase
- PORCN
porcupine protein
- PRICKLE
prickle‐like protein
- RAC1
Ras‐related C3 botulinum toxin substrate 1
- ROR
receptor tyrosine kinase‐like orphan receptor
- RS
Richter syndrome
- RYK
tyrosine‐protein kinase
- sFRP
secreted frizzled‐related protein
- TFS
therapy‐free survival
- VANGL
Vang‐like protein
Introduction
Wnt signalling activity is tightly regulated in time and space and has been considered a cornerstone of mammalian embryonic development as well as adult tissue homeostasis. Wnt signalling is now seen more as a network of interacting pathways instead of a linear signal transduction (Kestler and Kühl, 2008). This can be illustrated by the number and heterogeneity of the processes controlled by the Wnt pathway. These include not only a balance between stemness and cell differentiation, cell‐cycle regulation, proliferation and apoptosis but also cytoskeletal rearrangement, cell and tissue polarity, cell adhesion and motility, directed migration and invasion and overall interaction with the micro‐environment (Clevers, 2006; Seifert and Mlodzik, 2007). These key developmental processes are important for the normal physiological function of adult tissues and, therefore, commonly impaired in various diseases, including cancer (Clevers and Nusse, 2012).
The Wnt signalling network has been linked to the haematopoiesis mainly via its role in the biology of haematopoietic stem cells (HSCs) (Staal et al., 2008; Malhotra and Kincade, 2009; Lento et al., 2013; Staal et al., 2016b). Consequently, the Wnt signalling pathway was shown to be crucial for the leukaemogenesis of malignancies originating from HSCs – namely, acute myeloid leukaemia, acute lymphoid leukaemia and chronic myeloid leukaemia – and these connections have been reviewed extensively (Luis et al., 2012; Laranjeira and Yang, 2016; Staal et al., 2016a). However, not all the principles described in the context of HSCs can be transferred to mature B‐cells and their transformed counterparts. Thus, in this review we focus on the role of the Wnt signalling cascade in the mature B‐cell‐derived haematological malignancies – mainly chronic lymphocytic leukaemia (CLL). In these cells, β‐catenin‐independent signalling, predominantly the Wnt/planar cell polarity (PCP) pathway, has a prominent role – due to the important function of the Wnt/PCP co‐receptor receptor tyrosine kinase like orphan receptor 1 (ROR1)‐driven signalling in CLL (Baskar et al., 2008; Daneshmanesh et al., 2008; Fukuda et al., 2008). Wherever possible, we will extend the findings from CLL to other malignancies originating from the various stages of mature B‐cell development, to provide a perspective on the so far poorly characterized links between Wnt signalling and other lymphomas, such as diffuse large B‐cell lymphoma (DLBCL), Hodgkin lymphoma (HL), mantle cell lymphoma (MCL), Burkitt lymphoma (BL) and follicular lymphoma (FL) (Ott and Rosenwald, 2008; Frick et al., 2012; Kuppers et al., 2012; Vogt et al., 2017). These diseases are connected by having similar molecular mechanisms involved in their pathogenesis, are clearly dependent on their micro‐environment and cell–cell interactions, which suggest analogous roles of the Wnt signalling pathways.
Chronic lymphocytic leukaemia and related lymphomas
Chronic lymphocytic leukaemia – aetiology and treatment
CLL is a lymphoproliferative disease characterized by a progressive accumulation of mature non‐functional CD5+ B cells in the peripheral blood (PB), lymphoid tissue and bone marrow (BM). CLL is the most common adult leukaemia in Western countries; with a median age of diagnosis of 67–72 years and an overall incidence of 4–5/100 000 per year in the USA and Europe, which rapidly rises to >30/100 000 above the age of 80 years (Sant et al., 2010; Hallek, 2015). The course of the disease is highly heterogeneous – while some CLL patients remain asymptomatic, others develop an active disease with one or more symptoms requiring therapy, that is, massive lymphadenopathy, BM failure manifested by anaemia and/or thrombocytopaenia and constitutional symptoms (Hallek, 2015). Among the biological markers used for clinical evaluation of patient prognosis, the mutational status of the immunoglobulin heavy chain (IGHV) variable region and recurrent cytogenetic aberrations [del(13q), trisomy 12, del(11q) or del(17p)] were shown to reliably predict the survival of patients with CLL (Delgado et al., 2017). The presence of mutations in TP53, MYD88, SF3B1, BIRC3 or NOTCH1 and other genes further help not only to assess the prognosis of patients, but also to understand the biology of the disease and its dependence on different cell‐signalling pathways (Lazarian et al., 2017).
CLL patients are typically not treated unless/until they suffer from an aggressive form of the disease. Treatment options involve immunochemotherapy (typically fludarabine/cyclophosphamide/rituximab – regimen) and, more recently, also novel inhibitors that target pro‐survival B‐cell receptor or anti‐apoptotic B‐cell lymphoma 2 (BCL2) signalling (Jamroziak et al., 2017). Despite the fact that new treatment options have significantly improved patient response, this therapy needs to be mostly infinite to prevent relapse (Burger et al., 2016; Jain et al., 2017). CLL is thus still considered incurable. This creates a real need for new therapeutic agents that could target the disease on a different signalling pathway to decrease the chance of emergence of a more aggressive and resistant clone.
Richter syndrome and similarities between CLL and lymphomas
CLL patients can develop so‐called Richter syndrome (RS), which is when the disease transforms into a high‐grade lymphoma (Rossi and Gaidano, 2016), most commonly to DLBCL (2–7% of CLL patients in clinical trials) and HL (0.4–0.7% of CLL patients) (Mauro et al., 2017). An analysis of IGHV‐D‐J genes revealed that 80% of the DLBCL‐RS are clonally related to the preceding CLL phase, while this is true for only 40–50% of HL‐RS. These findings indicate that RS is largely an actual transformation of the disease, while the others represent the de novo development of lymphoma alongside the CLL clone. The RS prognosis is also highly unfavourable due to the presence of genetic lesions in TP53, NOTCH1, MYC or CDKN2A, connected to chemoresistance and rapid disease progression, which are present in 90% of these patients (Rossi and Gaidano, 2016). Transformation of CLL to RS supports the hypothesis that CLL is functionally related to mature B‐cell lymphomas on a molecular level and findings from the field of lymphomas should also be considered in CLL pathogenesis and vice versa. This standpoint is also applied in this review. The main features of lymphomas and CLL are summarized in Table 1.
Table 1.
The main features of lymphomas in comparison with CLL
| Leukaemia/lymphoma type | Main features | Origin | Disease‐initiating genetic aberration | Reference |
|---|---|---|---|---|
| CLL | Infiltration of BM and lymphoid organs, high degree of cell migration – recirculation and proliferation in typical pseudofolicles in the lymph nodes | Small mature B cells | — | Hallek et al. (2015) |
| HL | Disseminated lymphoma cells: peripheral lymph nodes, liver, lung and BM | Transformed GC B cells | — | Kuppers et al. (2012) |
| MCL | Systemic dissemination | Small mature B cells | t(11;14), CCND1 translocation | Vogt et al. (2017) |
| DLBCL | Large B cells with a high proliferation index resembling that of germinal centroblasts | B cells in various stage of GC reaction | t(14;18) ectopic expression of BCL2 (45% GC B cell‐DLBCL, but not inactivated B cell‐DLBCL) | Frick et al. (2012) |
| BL | Actively proliferating differentiating lymphocytes, often detected at site of origin | Transformed GC B cells | MYC locus translocation | Frick et al. (2012) |
| FL | Proliferation of neoplastic GC B cells, with at least a partial follicular pattern | Transformed GC B cells | t(14;18), ectopic expression of BCL2 | Ott and Rosenwald (2008) |
Wnt signalling pathways
Wnt proteins can drive several distinct pathways that are typically divided into β‐catenin‐dependent (Wnt/β‐catenin) and independent branches. Often, when the Wnt signalling pathway is discussed in the literature in association with leukaemia and lymphoma, the authors refer to the Wnt/β‐catenin pathway (Lu et al., 2004; Peiffer et al., 2014; Wang et al., 2014). However, activation of β‐catenin‐dependent and independent signalling cascades have different functional consequences even though they share several signalling proteins (Figure 1). In this review, we thus distinguish, wherever possible, between the two to avoid confusion and provide a clearer implication.
Figure 1.
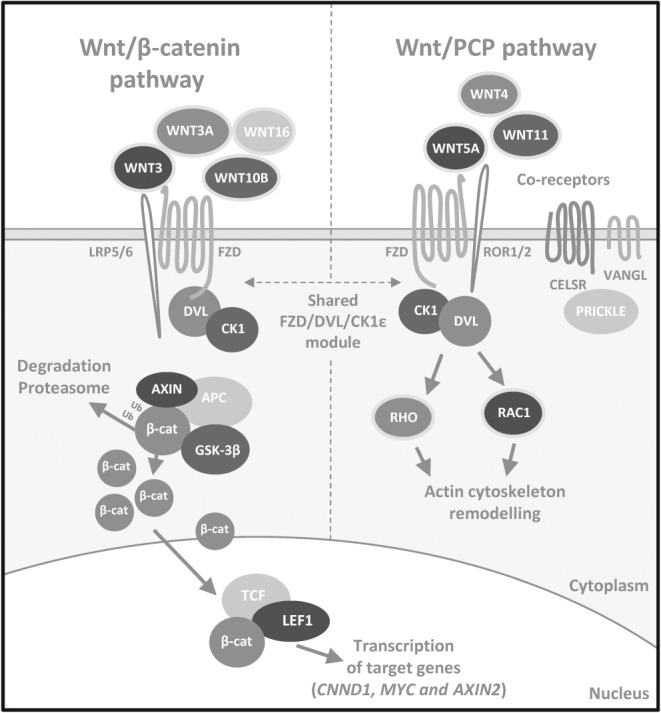
Simplified overview of mammalian Wnt/β‐catenin and Wnt/PCP pathways. The Wnt/β‐catenin signalling pathway (on the left) is activated upon binding of Wnt proteins (typical ligands Wnt‐3, Wnt‐3a, Wnt‐10b and Wnt‐16 are indicated) to dedicated receptors and co‐receptors FZD and LRP5/6. This leads to activation of cytoplasmic effector proteins from the DVL family that are phosphorylated by CK1ε, destabilizing the AXIN/GSK‐3β/APC destruction complex, normally responsible for β‐catenin degradation by proteasome. Cytoplasmic β‐catenin accumulates and transports to the nucleus, where it binds TCF/LEF transcription factors and activates transcription of target genes (such as CCND1, MYC or AXIN2). The Wnt/PCP pathway (on the right) is activated by a different set of ligands – typically Wnt‐4, Wnt‐5a, Wnt‐5b and Wnt‐11. The Wnt/PCP pathway shares a module composed of FZD, DVL and CK1ε with the Wnt/β‐catenin pathway but also contains numerous other transmembrane proteins – ROR1/2, VANGL1/2 and CELSR1–3 (and others not shown on this scheme) and cytoplasmic effectors from the PRICKLE family dedicated to the Wnt/PCP pathway only. This receptor complex conveys the signal downstream via a poorly known mechanism involving small G proteins Rho and/or Rac1 and their effectors that remodel the actin cytoskeleton.
Wnt/β‐catenin pathway
The Wnt/β‐catenin pathway has been closely connected to cell proliferation, cell‐cycle regulation and stem‐cell homeostasis, and therefore, its malfunction is a hallmark of many cancers (Clevers and Nusse, 2012). The pathway (Figure 1, on the left) is activated upon the binding of ligands – Wnt proteins (typical ligands: Wnt‐1, Wnt‐3, Wnt‐3a, Wnt‐8b, Wnt‐10b and Wnt‐16) – to the dedicated receptors and co‐receptors – Class Frizzled (FZD) and LDL receptor‐related protein (LRP) 5/6 (MacDonald et al., 2009). Ligand binding triggers the formation of a complex on the cell surface, which activates cytoplasmic effector proteins Dishevelled (DVL) and casein kinase 1 (CK1). This leads to destabilization of the AXIN/glycogen synthase kinase‐3 β (GSK‐3β)/adenomatous polyposis coli (APC) destruction complex that is in the absence of the ligand responsible for β‐catenin degradation by proteasome. Consequently, cytoplasmic β‐catenin accumulates and is transported to the nucleus, where it can bind T‐cell‐specific /lymphoid enhancer‐binding factor (LEF) transcription factors and activate the transcription of target genes (CCND1, encoding cyclin D1; MYC; DKK1, encoding Dickkopf 1; AXIN2; and many more).
β‐Catenin‐independent Wnt signalling – Wnt/PCP pathway
This signalling branch itself contains several pathways that do not require β‐catenin stabilization. For the purpose of the review, the Wnt/PCP pathway (Butler and Wallingford, 2017) is the most important. Other β‐catenin‐independent pathways will not be discussed here, and we refer the interested readers to one of the recent reviews (Semenov et al., 2007; Kestler and Kühl, 2008; van Amerongen, 2012). Compared with the β‐catenin‐dependent pathway, this signalling network is significantly less explored, and the current understanding of the function of this pathway's components and regulation mechanisms is limited.
The Wnt/PCP pathway (Figure 1, on the right) is implicated in the regulation of cell polarity, migration and invasion (Seifert and Mlodzik, 2007; Butler and Wallingford, 2017). Wnt/PCP is crucial mainly in embryonic development, directing the processes of convergent extension, neurulation or axon guidance. In mammals, the Wnt factors able to activate this signalling branch, typically Wnt‐4, Wnt‐5a/b or Wnt‐11, bind to FZD receptors and co‐receptors [family of receptor tyrosine pseudokinases – ROR1, ROR2, tyrosine‐protein kinase (RYK) and protein tyrosine kinase 7(CCK4)], which transduce the signal. The other core Wnt/PCP components include transmembrane proteins Vang‐like protein (VANGL)1/2 and cadherin EGF laminin G seven‐pass G‐type receptor (CELSR)1–3, cytoplasmic effectors prickle‐like protein (PRICKLE)1–4, CK1ε/δ or DVL1–3 and small G proteins RHO/Ras‐related C3 botulinum toxin substrate 1 (Rac1). Activation of Rho and/or Rac1 via their effectors ROCK (Rho‐associated protein kinase) and JNK leads to the actin cytoskeleton remodelling (Schlessinger et al., 2009).
The role of Wnt signalling pathways in CLL pathogenesis
The Wnt signalling pathway was suggested to be involved in the pathogenesis of CLL (Lu et al., 2004; Gutierrez et al., 2010; Kaucka et al., 2013; Wang et al., 2014; Yu et al., 2016). However, so far, no consensus has been reached about the importance of individual Wnt branches in different physiological processes and cell–cell interactions controlling CLL pathogenesis and CLL response to treatment. The expression of Wnt signalling molecules from both the Wnt/β‐catenin and Wnt/PCP pathways (summarized in Table 2 including references) is defective in CLL, but their role in CLL often remains elusive. Below, we describe in detail the reported connections between CLL and the Wnt signalling pathways.
Table 2.
Changes in the expression of Wnt pathway components in CLL
| Wnt components | Note | References | |
|---|---|---|---|
| Upregulated | ROR1 |
Wnt/PCP co‐receptor Surface marker expressed on CLL B cells Not present on normal mature B cells Described role in CLL cell migration and proliferation |
Baskar et al. (2008), Daneshmanesh et al. (2008), Fukuda et al. (2008), Broome et al. (2011), Kaucka et al. (2013), Cui et al. (2016) and Yu et al. (2016) |
| Wnt‐5a and Wnt‐5b |
High Wnt‐5a/b expression associates with adverse prognosis in CLL ROR1 ligands Regulation of CLL cell chemotaxis and proliferation Expression varies in PB/tonsilar B‐cell subsets |
Kaucka et al. (2013), Janovska et al. (2016) and Yu et al. (2016) | |
| Wnt‐3, Wnt‐10a and Wnt‐16 |
Wnt‐3 is among the most overexpressed genes in CLL compared with normal B cells Significantly lower Wnt‐3 expression in U‐CLL Low expression Wnt‐3 = independent marker of short TFS in M‐CLL Expression varies in PB/tonsilar B‐cell subsets |
Rosenwald et al. (2001), Lu et al. (2004), Memarian et al. (2009) and Poppova et al. (2016) | |
| LEF1 |
Absent in normal mature PB/GC B cells LEF1 silencing in primary CLL cells induces cell death Up‐regulated in monoclonal B‐cell lymphocytosis and CLL High expression associated with adverse prognosis |
Lu et al. (2004), Memarian et al. (2009), Rosenwald et al. (2001) and Gutierrez et al. (2010) | |
| DVL1/2/3 | Cytosolic components of both Wnt/β‐catenin and Wnt/PCP pathways | Kaucka et al. (2013) and Khan et al. (2016) | |
| CSNK1E, FZD3/7, PRICKLE1, CELSR1 and VANGL2 |
Wnt/PCP pathway components up‐regulated in CLL compared with normal PB B cells FZD3/7 and PRICKLE1 high expression associates with adverse prognosis in CLL |
Kaucka et al. (2013) | |
| Downregulated | DKKs and SFRPs |
Soluble inhibitors of Wnt signalling Often epigenetically silenced in CLL |
Seeliger et al. (2009), Moskalev et al. (2012) and Pei et al. (2012) |
ROR1 – a key player in CLL
ROR1 is a co‐receptor acting in the Wnt/PCP pathway. It is a Wnt‐5‐dedicated receptor that was found to be expressed on the surface of CLL cells and not on mature healthy B‐cells (Baskar et al., 2008; Daneshmanesh et al., 2008; Fukuda et al., 2008), with the exception of a subset of non‐neoplastic B‐cell precursors in the BM, so‐called hematogones (Broome et al., 2011). ROR1 can be used as a sensitive marker to analyse residual disease in CLL patients in remission (Kotaskova et al., 2016). Until recently, its mRNA expression and presence on the cell surface of CLL cells was described as uniform; however, a study performed on a larger patient dataset showed that high ROR1 surface levels might distinguish patients with a more aggressive course of the disease (Cui et al., 2016). Due to its unique expression pattern, ROR1 represents an important therapeutic feature for targeting CLL cells, even though the novel data show that some CLL patients may be ROR1‐negative (5% of analysed samples) (Cui et al., 2016). Hence, these ROR1‐directed therapies may not be applicable for all cases of CLL.
Wnt/PCP pathway governs polarity and migration in CLL cells
Earlier studies proposed that ROR1 might not be the only Wnt/PCP component up‐regulated in CLL (Mahadevan et al., 2009; Kotaskova et al., 2010), and currently, this has been ahown to be the case for many other Wnt/PCP pathway components. These include the typical ligands Wnt‐5a and Wnt‐5b (Lu et al., 2004; Memarian et al., 2009; Janovska et al., 2016), receptors (FZD3 and FZD7, VANGL2 and CELSR1) as well as cytoplasmic effectors (DVL2/3, CK1ε and PRICKLE1) (Kaucka et al., 2013; Khan et al., 2016). High expression of FZD3/7, PRICKLE1 and Wnt‐5a/b is also associated with a shorter therapy‐free survival (TFS) and poor prognosis in CLL patients (Kaucka et al., 2013; Janovska et al., 2016). Functional experiments confirmed the importance of the Wnt/PCP signalling in the pathogenesis of CLL: genetic experiments in mice showed that FZD6, a receptor dedicated to β‐catenin‐independent Wnt pathway (Golan et al., 2004; Wang et al., 2010), is crucial for the leukaemogenesis in the Eμ‐TCL1 CLL mouse model (Bichi et al., 2002; Wu et al., 2009), and the overexpression of ROR1 in the same model leads to earlier disease development (Widhopf et al., 2014).
The Wnt/PCP pathway controls cell polarity (Butler and Wallingford, 2017), and there is evidence that the pathway components (VANGL, ROR and DVL) can also be expressed in a polarized manner in the migrating CLL cells, namely, in the CLL‐derived MEC‐1 cell line (Kaucka et al., 2015) (schematized in Figure 2). It was demonstrated that the Wnt/PCP proteins control chemotactic responses and cell homing in CLL (Kaucka et al., 2013), which are processes that require B‐cell polarization (Parameswaran et al., 2011). The migration capacity of primary CLL cells in the CXCL12 gradient is increased after activation of the pathway by recombinant Wnt‐5a, an effect that can be blocked by anti‐ROR1 treatment (Kaucka et al., 2013; Yu et al., 2016) and is dependent on the activity of Rho activity, not Rac1 (Kaucka et al., 2013; Hofbauer et al., 2014; Yu et al., 2016). This is in line with the importance of Rho‐dependent signalling in CLL cell migration that was recognized earlier (Sanchez‐Aguilera et al., 2010; Troeger et al., 2012). In a cohort of patients characterized by the autocrine production of Wnt‐5a, cell migration and motility of CLL cells was affected in vitro (Janovska et al., 2016). These patients showed an aggressive disease course and were mostly from the unmutated IGHV (U‐CLL) subgroup.
Figure 2.
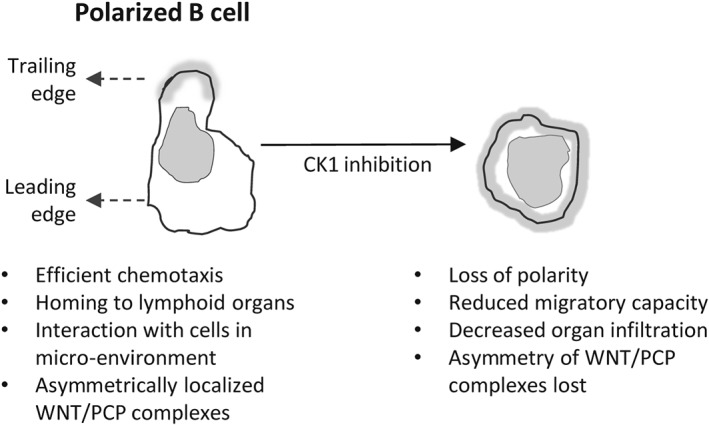
Impact of CK1 inhibition on CLL lymphocytes. Normal morphology of a polarized migrating lymphocyte with clearly distinguishable leading and trailing edge is shown. Wnt/PCP proteins were shown to localize asymmetrically, which can be best demonstrated by trailing edge‐specific distribution of VANGL2 (shown in grey). CK1 inhibitor treatment results in B‐cell polarity loss. As a consequence, CLL cells are unable to migrate, respond to micro‐environmental stimuli or home into distant organs.
Another line of evidence comes from in vivo studies in mice. The homing of CLL cells in vivo can be blocked by inhibition at the level of the Wnt/PCP receptors – ROR1 (Kaucka et al., 2013; Widhopf et al., 2014; Yu et al., 2016) and FZD7 (Kaucka et al., 2013) – as well as downstream at the level of the cytoplasmic effector CK1 (Kaucka et al., 2013). This was shown in numerous experimental models – xenograft of primary CLL cells to immunodeficient NOD/SCID IL2Rγ‐null (NSG) mice (Kaucka et al., 2013), adoptive transfer of leukemic splenocytes to ROR1 Tg Eμ‐TCL1 mice (Widhopf et al., 2014) or engraftment of MEC‐1–ROR1 cell line to Rag2−/−γc −/− mice (Yu et al., 2016). In addition to the well‐defined effects on migration, signalling through Wnt‐5a/ROR1/Rac1 (Yu et al., 2016; 2017) and Wnt‐5a/ROR1/ PI3K/Akt (Daneshmanesh et al., 2015; Cui et al., 2016) pathways has been proposed to have pro‐survival and pro‐proliferative effects in CLL, which is similar to the ROR1 function in other cancers such as melanoma or breast cancer (Zhang et al., 2012; Fernandez et al., 2016). Future work will determine what the dominant mode of action of ROR1 is in CLL.
Overexpression of Wnts associated with the Wnt/β‐catenin pathway in CLL cells
The involvement of the Wnt/β‐catenin pathway in CLL was postulated based on the high expression of genes encoding ligands able to activate Wnt/β‐catenin signalling in other systems – mainly WNT3 but also WNT10A and WNT16 (Rosenwald et al., 2001; Lu et al., 2004; Memarian et al., 2009). WNT3 is among the most up‐regulated genes in CLL, and this fact has long been considered one of the strongest arguments supporting an active role of the Wnt/β‐catenin pathway in CLL. A recent study performed a detailed analysis of the expression of its ligands in a cohort of 137 patients and correlated the results with the clinical information available (Poppova et al., 2016). This work suggested that in spite of the very high levels of expression of WNT3 in CLL cells, this was not associated with an aggressive form of this disease. The expression of WNT3 was significantly lower in U‐CLL patients, and moreover, low WNT3 expression could be used as an independent marker to identify patients with short TFS in the generally indolent subgroup with mutated IGHV (M‐CLL). In addition, this study showed that a reduced expression of WNT3 accompanies the onset of disease activity within U‐CLL (Poppova et al., 2016). Importantly, it is currently not clear whether or not Wnt/β‐catenin‐associated ligands can efficiently activate downstream signalling in CLL – so far, attempts to trigger Wnt/β‐catenin pathway by Wnt‐3 or Wnt‐3a in CLL cells have been unsuccessful (Kaucka et al., 2013; Poppova et al., 2016). This contrasts with the finding that direct inhibition of the β‐catenin destruction complex by a GSK‐3β inhibitor acting downstream was effective at activating the Wnt/β‐catenin pathway (Lu et al., 2004; Poppova et al., 2016). In conclusion, these findings suggest that the ligands produced by CLL cells do not act in an autocrine manner but rather serve to communicate with the other cell types present in the CLL micro‐environment.
Somatic mutations in genes encoding Wnt pathway components
Next‐generation sequencing techniques, namely, whole‐exome sequencing, have recently enabled the discovery of novel genetic aberrations in components of key signalling pathways. Among them, somatic mutations in several Wnt‐related genes were demonstrated in CLL and suggested to affect activation of the Wnt pathway and, therefore, disease pathogenesis (Wang et al., 2011; 2014). The authors sequenced 91 samples and discovered 15 mutations in 12 components of the Wnt pathway, totalling 14% of samples (Wang et al., 2014). Interestingly, the mutations were found in genes encoding extracellular factors (WNT1, WNT10, DKK2 and RSPO4), transmembrane receptors (FZD5 and RYK), cytoplasmic [casein kinase ε gene (CSNK1E) and PRICKLE1] as well as nuclear factors (CHD8, BRD7, CREBBP and BCL9). The mutations did not associate with any commonly used CLL prognostic factors (IGHV status, ZAP70 expression, age at diagnosis, clinical stage, presence of cytogenetic abnormalities, mutation rate or time to first therapy), and six of the patients were chemotherapy naïve, which ruled out their occurrence as a result of chemotherapy exposure. Functional experiments with overexpression of WT and mutant alleles in HEK293 cells showed that these mutations result in the activation (BCL9, DKK2 and RYK), inactivation (CSNK1E – encoding for CK1ε, Wnt1 and FZD5) or no (FZD5 – second mutation) functional change in the Wnt/β‐catenin pathway, an effect which was validated in primary CLL carrying the WT or mutated alleles of BCL9, DKK2, RYK and CSNK1E. Silencing of these genes showed that these cells were highly dependent on the mutated gene expression for cell survival. Because the study of Wang et al. focused on the effect of the mutations on the level of β‐catenin‐dependent activity in HEK293 cells and cell survival in primary CLL and normal B‐cells, no data were obtained regarding the activity of the Wnt/PCP pathway, even though some of the mutated genes encode for proteins that function in either both Wnt pathways (CSNK1E and DVL1) or in the β‐catenin‐independent branch only (RYK and PRICKLE1).
Lymphoid enhancer‐binding factor 1 (LEF1)
LEF1 is a critical transcription factor that is activated by the Wnt/β‐catenin pathway and drives the expression of its target genes (Behrens et al., 1996; Huber et al., 1996). LEF1 is required in the early phases of B‐cell development (Reya et al., 2000; Gutierrez et al., 2010), but there is virtually no expression of LEF1 in PB B cells or germinal centre (GC) mature B cells (Reya et al., 2000; Lu et al., 2004; Gutierrez et al., 2010; Kaucka et al., 2013). In contrast, LEF1 expression is very high in CLL and also in the pre‐leukaemic state of CLL called monoclonal B‐cell lymphocytosis (Gutierrez et al., 2010). Of note, no such difference was detected in the case of β‐catenin (Gutierrez et al., 2010; Kaucka et al., 2013). Experimental silencing of LEF1 reduced CLL cell survival (Gutierrez et al., 2010; Wang et al., 2014), but not the survival of normal CD19+ B cells (Wang et al., 2014), in contrast to the silencing of CTNNB1 (encoding β‐catenin) or DVL1 that caused cell death in both cell types. Higher LEF1 expression was also associated with adverse prognosis in CLL patients (Erdfelder et al., 2010; Wu et al., 2016). LEF1 expression levels, among other CLL‐pathogenesis‐related factors including ROR1 or PI3K, were shown to decrease when the CLL cells were forced towards differentiation to plasma cells in vitro using phorbol myristate acetate or CpG oligodeoxynucleotide, in combination with a CD40 ligand and cytokines (Gutierrez et al., 2011; Ghamlouch et al., 2015).
One of the candidate gene targets regulated by LEF1 in the CLL context is the cylindromatosis gene (CYLD) (Liu et al., 2012). Low CYLD expression was associated with U‐CLL status, and shorter overall survival (OS) in all major CLL cohorts, including the M‐CLL subgroup. In this context, LEF1 acts as a transcriptional repressor of CYLD – Wu et al. (2014) showed that interference with LEF1 binding to DNA restored CYLD expression. CYLD acts as a deubiquitinase and a defect in its activity has been implied in several cancers, including CLL (Mathis et al., 2015). Interestingly, the CYLD −/− mice exhibited abnormalities in B‐cell development, marked by spontaneous B‐cell activation and hyperplasia in the periphery, with enlarged lymphoid organs and with cells being hyperproliferative upon stimulation in vitro (Jin et al., 2007). Overall, the data support the role of CYLD as a tumour suppressor that is directly controlled by LEF1, which acts to repress its activity.
The ambiguous role of Dishevelled proteins
Other components of the Wnt pathway up‐regulated in CLL, but practically undetectable in normal B‐cells, are DVL proteins (1–3) (Kaucka et al., 2013; Khan et al., 2016). This finding is interesting, because DVL proteins have been shown to play a key role in both β‐catenin‐dependent and ‐independent Wnt pathways (Gao and Chen, 2010; Bryja and Bernatík, 2014). The siRNA knockdown of various DVL isoforms lead to different outcomes; Wang et al. (2014) showed that DVL1 knockdown in primary CLL cells leads to increased CLL cell death, similar to LEF1 or CTNNB1 silencing; however, we did not observe such effects with the DVL2 isoform in the CLL‐derived cell line MEC‐1 (Kaucka et al., 2013). However, DVL2 silencing caused a decrease in chemotaxis in MEC‐1 cells, suggesting it has a role in the Wnt/PCP pathway. Similarly, Khan et al. (2016) did not observe any effects on viability in the EHEB cell line (also CLL derived) after the silencing of all three DVL isoforms. DVL acts as a switch between the Wnt/β‐catenin and Wnt/PCP pathway. Therefore, the recently reported alternative spliced variants of DVL in CLL cells, produced as a result of a mutated SF3B1 (common recurrent mutation in CLL, associated with poor prognosis) (Wang et al., 2016), is of considerable interest. The region missing in the SF3B1 mutation‐associated spliced variant was shown to regulate the DVL tertiary structure and its role in the individual Wnt signalling branches (Lee et al., 2015; Qi et al., 2017).
Epigenetic silencing of Wnt inhibitory factors
Another verification for the involvement of Wnt/β‐catenin activation in CLL comes from studies using the epigenetic silencing of genes encoding soluble Wnt inhibitors in CLL cells, mainly from the family of secreted frizzled‐related proteins (sFRPs) or Dickkopf proteins (DKKs) (Cruciat and Niehrs, 2013), with sFRP‐1 being one of the most hypermethylated genes in CLL (Seeliger et al., 2009; Moskalev et al., 2012; Pei et al., 2012). However, the role of the soluble Wnt inhibitors is not straightforward: recent studies showed that both sFRP and DKK proteins can act as activators and supressors of Wnt/β‐catenin or Wnt/PCP pathways (Mii and Taira, 2009; Esteve et al., 2011; Kagey and He, 2017). Better insight into the role of epigenetic mechanisms controlling the Wnt pathway in CLL can be obtained by detailed analysis of two recent reports that describe epigenome on large datasets of CLL primary samples and several mature B‐cell populations (Kulis et al., 2012; Oakes et al., 2016). Kulis et al. (2012) showed that an epigenetic signature can distinguish the main CLL prognostic subgroups – U‐CLL samples resembled the profiles of normal naive B cells and M‐CLL related to memory B cells. The data were confirmed and expanded by Oakes et al. (2016), who by comparing the epigenome of mature B cells and CLL proposed that CLL cells probably derive from a continuum of B‐cell maturation stages. This is of interest because the expression of Wnt ligands (and probably also of Wnt pathway inhibitors) changes during the B‐cell maturation process (see Future directions section).
Similarities between CLL and lymphomas – Wnt‐biased view
Similar to CLL, the Wnt pathway was shown to be malfunctioning in HL and non‐HL. These malignancies derive from various stages of B‐cell maturation, and not surprisingly, their gene expression patterns differ significantly. However, several recent studies suggested a role for Wnt pathways in their disease pathophysiology, mainly via effects on the interaction of the lymphoma cells with their micro‐environment. The complexity of the cellular composition of lymphomas makes in vitro studies more complicated in comparison with CLL – where typically large numbers of homogenous primary cells are available for functional analysis. In most lymphomas, the findings are limited (in contrast to CLL) to immunohistochemical staining and analysis of lymphoma‐derived cell lines. For a list of Wnt components that show altered levels in lymphomas, see Table 3. The most important observations are discussed further below.
Table 3.
Changes in the expression of Wnt pathway components in B‐cell lymphomas
| Lymphoma type | Wnt components | Note | Source of information |
|---|---|---|---|
| HL | LEF1 |
Up‐regulation compared with normal B‐cell subtypes Regulation of chemotaxis towards endothelial cells (ECs), adhesion to EC layers and cell invasion LEF1 and β‐catenin‐regulate cHL secretome – effects on ECs: promoted migration, sprouting and vascular tube formation |
Linke et al. (2017b) |
| β‐catenin | Present only rarely and highly expressed in cell lines | Morrison et al. (2004) and Sohlbach et al. (2012) | |
| Wnt‐5a |
Increased Wnt‐5a expression compared with normal B cells cHL cell migration, invasion and adhesion depend on autocrine Wnt‐5a signalling (Wnt‐5a‐FZD5‐DVL3‐RhoA axis) |
Linke et al. (2017a) | |
| MCL | CCND1 |
Up‐regulation induced by t(11;14) Initial oncogenic event |
Vogt et al. (2017) |
| ROR1 | 13–93% positive cells, median 56% | Daneshmanesh et al. (2013) | |
| Nuclear β‐catenin |
Primary MCL staining Marker of Wnt/β‐catenin activity |
Gelebart et al. (2008) | |
| LEF1 | Expression reported rarely (4–9% of MCL cases) | O'Malley et al. (2017) | |
| DLBCL | LEF1 | Up‐regulated in GC‐DLBCL subtype | Cubedo et al. (2012) |
| ROR1 | 30–81% positive cells, median 50% | Daneshmanesh et al. (2013) | |
| BL | MYC |
Locus translocated to Ig enhancer elements 80% patients harbours t(8;14) |
Frick et al. (2012) |
| LEF1 |
Up‐regulated in ‘molecular BL’ subtype Nuclear localization Transcriptionally active |
Walther et al. (2013) | |
| FL | ROR1 | 1–89% positive cells, median 28% | Daneshmanesh et al. (2013) |
Hodgkin lymphoma
HL originates from transformed germinal centre (GC) B‐cells (Hodgkin–Rees–Sternberg cells) that are rare in the lymphoma tissue (0.1–2%) and depend heavily on the interactions with their micro‐environment (Kuppers et al., 2012). This lymphoid malignancy involves peripheral lymph nodes and can also affect liver, lung and bone marrow. Previously, it was reported that only a small subset of primary HL samples express cytoplasmic and nuclear β‐catenin and inactivated GSK‐3β (Ser9 phosphorylation) (Morrison et al., 2004), even though HL‐derived cell lines show high levels of β‐catenin, as well as other components of the Wnt pathway (Sohlbach et al., 2012). It was shown that the majority of classical HL (cHL) samples are positive for the activated form of GSK‐3β (Y216 phosphorylation), normally responsible for the inhibition of Wnt/β‐catenin signalling (Agostinelli et al., 2017). Recently, a new insight was provided by results of studies that have functionally analysed local Wnt signalling in HL cells’ interactions with the micro‐environment (Linke et al., 2017a,b). These highlighted the importance of the autocrine Wnt‐5a/FZD5/DVL3/RHOA signalling axis in HL cell motility, chemotaxis and adhesion to endothelial cells (Linke et al., 2017b) and suggested that LEF1 and β‐catenin are required for chemotaxis in HL cells and their signalling to endothelial cells (Linke et al., 2017a). The HL cells were shown to produce extracellular factors in a LEF1‐ and β‐catenin‐dependent manner, which affected the migration, sprouting and tube formation of the endothelial cells normally present in their micro‐environment. Secretome analysis revealed that one of these factors is VEGF, whose high expression is associated with a shorter OS in cHL patients, emphasizing the clinical importance of these findings.
Non‐Hodgkin lymphomas
Non‐HLs represent a heterogeneous group with diverse pathology and several well‐defined types of lymphoma. MCL is a rare but aggressive type of lymphoma, where chromosomal translocation t(11;14) is considered an initial tumourigenic event, leading to an up‐regulated expression of CCND1 and impairments in the cell‐cycle (Perez‐Galan et al., 2011; Vogt et al., 2017). BL is characterized by translocation of the MYC gene to one of the three immunoglobulin gene loci, leading to aberrant expression of this proto‐oncogene (Frick et al., 2012). FL is a common lymphoma type (Ott and Rosenwald, 2008) marked by the proliferation of the neoplastic cells with the phenotypic features of GC cells – including both centrocyte‐like and centroblast‐like phenotypes. DLBCL is the most common subtype of malignant lymphoma (Frick et al., 2012). It is characterized by high heterogeneity with respect to clinical presentation, morphology, molecular pathogenesis and patient survival.
In general, compared with CLL, much less is known about the role of Wnt pathways in lymphoma. However, several close similarities exist suggesting the activation of common tumourigenic programmes. This fact can be illustrated by two examples: high expression levels of ROR1 and LEF1. Expression levels of both these genes are virtually negligible in mature B cells – in PB and GC (Gutierrez et al., 2010; Broome et al., 2011), but they are highly expressed in both CLL and lymphomas. Increased ROR1 gene expression, a hallmark of CLL, was observed in non‐HLs – MCL (13–93% positive cells in analysed samples), DLBCL (30–81%) and FL (1–89%) (Barna et al., 2011; Daneshmanesh et al., 2013). LEF1 was recently detected at different levels in a large portion of DLBCL samples (Cubedo et al., 2012). Furthermore, LEF1 gene expression in DLBCLs was comparable with that in CLL. High LEF1 expression was also identified as a signature gene and possible therapeutic target in the so‐called ‘molecular BL’ subtype (Walther et al., 2013). In MCL, the role of the Wnt/β‐catenin pathway is still debatable due to the fact that nuclear β‐catenin was detected in the samples (Gelebart et al., 2008). In this case, DVL2 siRNA silencing and sFRP1 treatment lead to a significant decrease in the proliferation of the MCL cell line and increased apoptosis in the case of knock down. LEF1 expression was reported only in 4–9% of MCL cases (O'Malley et al., 2017).
Therapeutic possibilities
The maladjusted Wnt signalling pathways in CLL and lymphomas may represent an interesting therapeutic possibility. The candidate targets in the Wnt signalling pathway are summarized in Figure 3 and described in detail below.
Figure 3.
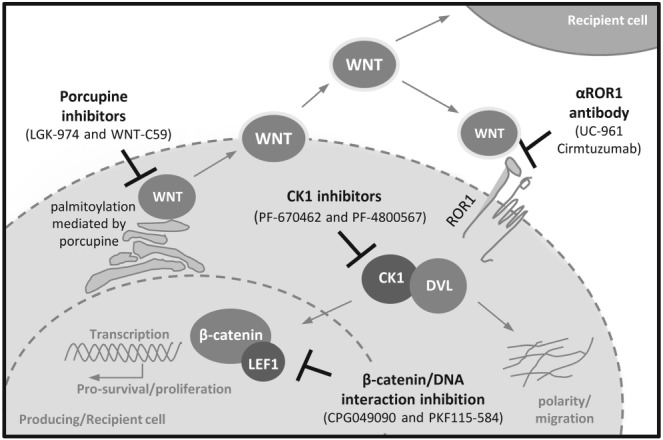
Possible Wnt pathway‐related therapeutic targets in CLL. Scheme summarizes the candidate therapeutic targets in CLL and their position in the Wnt pathway. Examples of compounds tested in the preclinical or early phase clinical trials are shown. See text for details.
ROR1 – targeting migration and pro‐survival signalling in B‐cell malignancies
As mentioned earlier, ROR1 is uniquely expressed in CLL cells and also in other lymphomas. In CLL, higher cell‐surface ROR1 is associated with earlier disease progression (Cui et al., 2016) and ROR1 thus represents an attractive target for monoclonal antibodies (mABs)‐based therapeutic interventions in aggressive CLL; ROR1 signalling can be blocked by anti‐ROR1 mABs (Yang et al., 2011; Kaucka et al., 2013; Janovska et al., 2016). Conflicting data exist as to whether antibodies against ROR1 can (Daneshmanesh et al., 2012; 2015) or cannot (Yang et al., 2011) induce apoptosis in CLL; a discrepancy that can probably be explained by differences in the epitopes recognized by these antibodies. The best characterized anti‐ROR1 antibody (UC‐961, cirmtuzumab) has undergone preclinical specificity and safety testing (Choi et al., 2015) and has entered phase I clinical trials (ID: NCT02222688). It efficiently blocked Wnt‐5a‐induced effects such as migration and proliferation and reduced ROR1‐triggered CLL development in mice (Yu et al., 2016), which raises hope for its success in the clinical trials. An alternative approach that takes advantage of specific ROR1 expression in CLL, explored the possibility of targeting CLL via chimeric antigen receptor (CAR) T‐cells that recognize ROR1 (Hudecek et al., 2010). ROR1–CARs were reported to have in vivo activity against ROR1+ B‐cell lymphoma in a xenograft mouse model (Hudecek et al., 2013) and were well tolerated in preclinical safety studies performed in primates (Berger et al., 2015).
Wnt proteins
Wnt proteins are modified by glycosylation and palmitoylation in order to be secreted from cells in an active form (Willert et al., 2003). This lipid modification is dependent on the activity of porcupine protein (PORCN) (Kadowaki et al., 1996), an enzyme that is present in the membrane of endoplasmic reticulum, and its chemical inhibition leads to disruption of Wnt protein secretion. Wnt modification via PORCN can be targeted by small molecules such as Wnt‐C59 and LGK‐974 (Liu et al., 2013; Proffitt et al., 2013; Madan et al., 2016), which have already shown their potential in preclinical in vitro and in vivo investigations. LGK‐974 has entered phase I clinical trials to treat Wnt‐dependent solid tumours (ID: NCT01351103). The inhibition of PORCN represents an approach that blocks both β‐catenin‐dependent and ‐independent Wnt pathways activated by the respective Wnt proteins produced in an autocrine/paracrine manner by tumour cells or released by other cell types into the micro‐environment. This feature may be very important in diseases like CLL and B‐cell lymphoma, or any metastatic cancers where the tumour progression depends on the interaction of several cell types in the BM stromal niche or lymph nodes.
LEF1
Targeting LEF1 transcription activity was suggested as a therapeutic possibility due to the fact that its inhibition by ethacrynic acid (EA) led to an increase in both primary CLL cell apoptosis and necroptosis (Wu et al., 2016). EA disrupted LEF1 binding to DNA and decreased the expression of its target genes, such as CCND1 or MYC. In another study, the transactivation properties of the LEF1/β‐catenin complex were targeted by two small molecule inhibitors (CGP049090 and PKF115‐584) that disrupt the interaction between the two proteins (Gandhirajan et al., 2010). Treatment induced apoptosis of primary CLL cells, while healthy B cells were not affected, and these effects were also confirmed in vivo in a xenograft mouse model.
Casein kinase 1
Another candidate that could act as a therapeutic target is CK1, an enzyme that phosphorylates various targets in the Wnt pathway – mainly DVLs, VANGL2 and LEF1 (Hammerlein et al., 2005; Bryja et al., 2007; Gao et al., 2011). An increased expression of CK1, together with other Wnt/PCP components, was demonstrated in primary CLL compared with normal PB B‐cells (Kaucka et al., 2013). Evidence was presented that CLL cells are sensitive to CK1 inhibition, which leads to CLL cell polarity disruption, blocked chemotaxis in vitro both in primary CLL cells and CLL‐derived cell lines and diminished primary CLL cell homing in vivo in a xenograft mouse model (Kaucka et al., 2013; Kaucka et al., 2015). Because the cell polarity, established as a consequence of directed chemokine signalling or a polarized interaction with T‐cells (Troeger et al., 2012; Yuseff and Lennon‐Dumenil, 2015), is crucial for the physiological function of B‐cells, interference with the process via CK1 inhibitors could be explored as a treatment option in future. Selective CK1 inhibitors have not yet reached the stage of clinical trials; however, some compounds (PF‐670462 and PF‐4800567) were tested and found to be well‐tolerated in vivo in preclinical studies (Meng et al., 2010; Arey and McClung, 2012) and could be used in a wide variety of Wnt‐driven cancers (Cheong and Virshup, 2016). Interestingly, a PI3Kδ inhibitor, TGR‐1202, currently in the phases II and III of ongoing CLL and DLBCL clinical trials (ID: NCT02793583, NCT02612311) was shown to affect the activity of CK1ε in vitro (Deng et al., 2017), and the upcoming results of these trials will provide important information as to whether targeting CK1 in these patients is beneficial when compared with other PI3K inhibitors.
Future directions
Studies examining the role of Wnt signalling in CLL and B‐cell lymphomas reviewed here often present divergent or even contradictory findings. One of the reasons may be that the complex Wnt signalling network often tends to be oversimplified and generalized. Individual Wnt pathways might have a completely different outcome – for example, proliferation versus migration – even in the same cell type. In addition, during the complex process of B‐cell maturation/pathogenesis, cells differentiate and gradually change their receptor composition and also their response to Wnt activators/inhibitors from the micro‐environment.
A thorough depiction of the expression and role of the components of the Wnt pathway in B‐cell differentiation and maturation provides important insights. As an example of this approach, we have summarized in Figure 4 the known expression patterns of ROR1, LEF1, WNT3 and WNT5A/5B during the B‐cell maturation and in the various CLL subgroups. Even this descriptive correlation between the physiological and pathological situation provides a helpful insight and indicates the importance of the dynamic regulation of Wnt signalling activity throughout the B‐cell maturation process and the context‐dependent switch between the Wnt/β‐catenin and Wnt/PCP pathways. It also pinpoints the important, though still unknown, position of ROR1 and LEF1 – the two genes that are very highly expressed in CLL but missing in mature B cells, which either reflects their driving role in CLL or, alternatively, their high expression in the CLL cell of origin.
Figure 4.
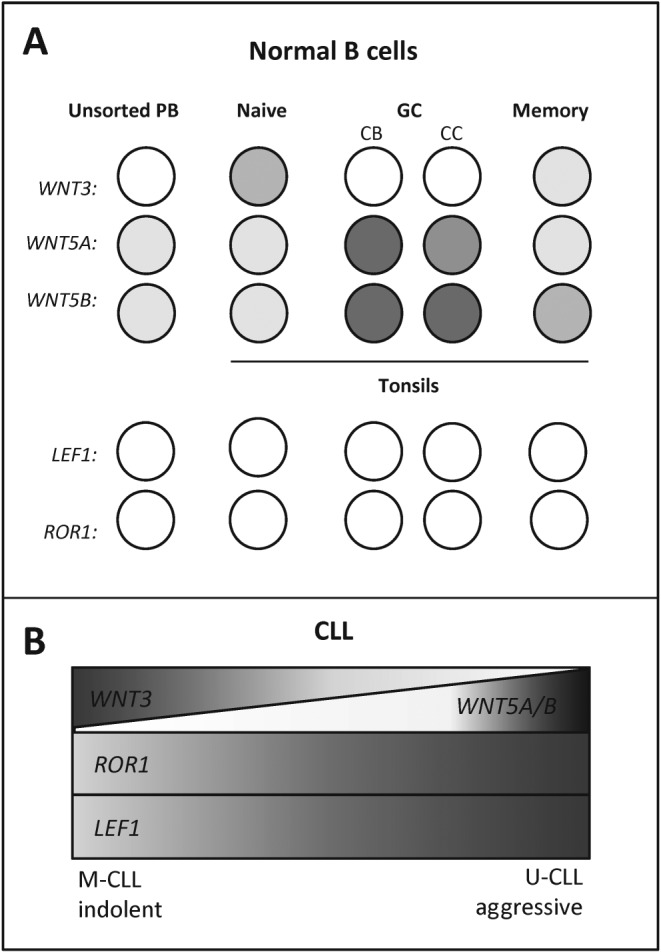
Similarities between CLL and normal B‐cell development. (A) Expression of selected Wnt pathway components (ROR1, LEF1, Wnt‐3 and Wnt‐5a/b) in major mature B‐cell subtypes. Wnt proteins associated with β‐catenin‐dependent or independent Wnt pathways are expressed differentially in mature B‐cell development. CB, centroblasts, CC, centrocytes. (B) Expression of the same genes in CLL subtypes. Generally a high expression of LEF1 and ROR1 is a hallmark of CLL; however, additional level of Wnt signalling regulation may be represented by the expression of distinct Wnt proteins, which associate with different CLL subgroups. Based on (WNT3 – Poppova et al., 2016; WNT5A/B – Janovska et al., 2016; ROR1 – Cui et al., 2016; Kaucka et al., 2013; Broome et al., 2011; LEF1 – Gutierrez et al., 2010; Wu et al., 2016).
Another important question related to the function of Wnt signalling in CLL and lymphomas is: are malignant cells receiving or sending the Wnt signals? Are Wnts used by CLL cells to orchestrate their environment or vice versa? For example, currently, there is no convincing evidence that CLL cells can respond to Wnt proteins by activation of the Wnt β‐catenin‐dependent pathway (Kaucka et al., 2013; Poppova et al., 2016). CLL cells can be activated downstream by a GSK‐3β inhibitor that inhibits the β‐catenin destruction complex, which demonstrates the presence of functional molecular machinery (Lu et al., 2004; Poppova et al., 2016). This suggests that Wnt‐3 produced in large quantities by CLL cells can be used to communicate with other cell types, for example, bone marrow stromal cells, which respond strongly to Wnt‐3 stimulation (Poppova et al., 2016). This hypothesis is in line with findings from previous studies, which showed that the effects of the CLL–stroma interaction are bidirectional (Ding et al., 2009) and that the expression pattern of both Wnt‐associated ligands and receptors in CLL cells changes dynamically – as illustrated in the study by Mittal et al. (2014) who performed gene expression profiling in a set of primary CLL samples and compared cells isolated from the PB, BM and lymph nodes (LN). The intraclonal expression changes of Wnt components in primary CLL cells, which emerged from the LN to the PB, also support the idea that the Wnt signalling is modulated in mature B cells in response to different micro‐environmental stimuli (Calissano et al., 2011). To determine the functional importance of these Wnt pathway changes and its relevance to the pathogenesis of CLL/lymphoma and response to therapy remains an open question for further investigation.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
Research of V.B. and P.J. on Wnt signalling in relation to leukaemia pathogenesis was funded by the Ministry of Health of the Czech Republic (15‐29793A). We thank Sarka Pavlova and Karla Plevova for consultation and Matthew Smith and Karol Kaiser for language corrections.
Janovská, P. , and Bryja, V. (2017) Wnt signalling pathways in chronic lymphocytic leukaemia and B‐cell lymphomas. British Journal of Pharmacology, 174: 4701–4715. doi: 10.1111/bph.13949.
References
- Agostinelli C, Carloni S, Limarzi F, Righi S, Laginestra MA, Musuraca G, et al (2017). The emerging role of GSK‐3beta in the pathobiology of classical Hodgkin lymphoma. Histopathology 71: 72–80. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arey R, McClung CA (2012). An inhibitor of casein kinase 1 epsilon/delta partially normalizes the manic‐like behaviors of the ClockDelta19 mouse. Behav Pharmacol 23: 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna G, Mihalik R, Timar B, Tombol J, Csende Z, Sebestyen A et al (2011). ROR1 expression is not a unique marker of CLL. Hematol Oncol 29: 17–21. [DOI] [PubMed] [Google Scholar]
- Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E et al (2008). Unique cell surface expression of receptor tyrosine kinase ROR1 in human B‐cell chronic lymphocytic leukemia. Clin Cancer Res 14: 396–404. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R et al (1996). Functional interaction of beta‐catenin with the transcription factor LEF‐1. Nature 382: 638–642. [DOI] [PubMed] [Google Scholar]
- Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ et al (2015). Safety of targeting ROR1 in primates with chimeric antigen receptor‐modified T cells. Cancer Immunol Res 3: 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R et al (2002). Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A 99: 6955–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome HE, Rassenti LZ, Wang HY, Meyer LM, Kipps TJ (2011). ROR1 is expressed on hematogones (non‐neoplastic human B‐lymphocyte precursors) and a minority of precursor‐B acute lymphoblastic leukemia. Leuk Res 35: 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryja V, Bernatík O (2014). Dishevelled at the crossroads of pathways In: Wnt Signaling in Development and Disease: Molecular Mechanisms and Biological Functions (eds Hoppler S. and Moon R. T.), John Wiley & Sons, Inc, Hoboken, NJ, USA: https://doi.org/10.1002/9781118444122.ch15 [Google Scholar]
- Bryja V, Schulte G, Rawal N, Grahn A, Arenas E (2007). Wnt‐5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1‐dependent mechanism. J Cell Sci 120 (Pt 4): 586–595. [DOI] [PubMed] [Google Scholar]
- Burger JA, Landau DA, Taylor‐Weiner A, Bozic I, Zhang H, Sarosiek K et al (2016). Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun 7: 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MT, Wallingford JB (2017). Planar cell polarity in development and disease. Nat Rev Mol Cell Biol 18: 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calissano C, Damle RN, Marsilio S, Yan XJ, Yancopoulos S, Hayes G et al (2011). Intraclonal complexity in chronic lymphocytic leukemia: fractions enriched in recently born/divided and older/quiescent cells. Mol Med 17: 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta‐catenin signaling in development and disease. Cell 127: 469–480. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R (2012). Wnt/beta‐catenin signaling and disease. Cell 149: 1192–1205. [DOI] [PubMed] [Google Scholar]
- Cruciat CM, Niehrs C (2013). Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol 5: a015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubedo E, Gentles AJ, Huang C, Natkunam Y, Bhatt S, Lu X et al (2012). Identification of LMO2 transcriptome and interactome in diffuse large B‐cell lymphoma. Blood 119: 5478–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B, Ghia EM, Chen L, Rassenti LZ, DeBoever C, Widhopf GF 2nd et al (2016). High‐level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood 128: 2931–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshmanesh AH, Hojjat‐Farsangi M, Khan AS, Jeddi‐Tehrani M, Akhondi MM, Bayat AA et al (2012). Monoclonal antibodies against ROR1 induce apoptosis of chronic lymphocytic leukemia (CLL) cells. Leukemia 26: 1348–1355. [DOI] [PubMed] [Google Scholar]
- Daneshmanesh AH, Hojjat‐Farsangi M, Moshfegh A, Khan AS, Mikaelsson E, Osterborg A et al (2015). The PI3K/AKT/mTOR pathway is involved in direct apoptosis of CLL cells induced by ROR1 monoclonal antibodies. Br J Haematol 169: 455–458. [DOI] [PubMed] [Google Scholar]
- Daneshmanesh AH, Mikaelsson E, Jeddi‐Tehrani M, Bayat AA, Ghods R, Ostadkarampour M et al (2008). Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int J Cancer 123: 1190–1195. [DOI] [PubMed] [Google Scholar]
- Daneshmanesh AH, Porwit A, Hojjat‐Farsangi M, Jeddi‐Tehrani M, Tamm KP, Grander D et al (2013). Orphan receptor tyrosine kinases ROR1 and ROR2 in hematological malignancies. Leuk Lymphoma 54: 843–850. [DOI] [PubMed] [Google Scholar]
- Delgado J, Doubek M, Baumann T, Kotaskova J, Molica S, Mozas P et al (2017). Chronic lymphocytic leukemia: a prognostic model comprising only two biomarkers (IGHV mutational status and FISH cytogenetics) separates patients with different outcome and simplifies the CLL‐IPI. Am J Hematol 92: 375–380. [DOI] [PubMed] [Google Scholar]
- Deng C, Lipstein MR, Scotto L, Jirau Serrano XO, Mangone MA, Li S et al (2017). Silencing c‐Myc translation as a therapeutic strategy through targeting PI3Kdelta and CK1epsilon in hematological malignancies. Blood 129: 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Nowakowski GS, Knox TR, Boysen JC, Maas ML, Schwager SM et al (2009). Bi‐directional activation between mesenchymal stem cells and CLL B‐cells: implication for CLL disease progression. Br J Haematol 147: 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdfelder F, Hertweck M, Filipovich A, Uhrmacher S, Kreuzer KA (2010). High lymphoid enhancer‐binding factor‐1 expression is associated with disease progression and poor prognosis in chronic lymphocytic leukemia. Hematol Rep 2 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve P, Sandonis A, Ibanez C, Shimono A, Guerrero I, Bovolenta P (2011). Secreted frizzled‐related proteins are required for Wnt/beta‐catenin signalling activation in the vertebrate optic cup. Development 138: 4179–4184. [DOI] [PubMed] [Google Scholar]
- Fernandez NB, Lorenzo D, Picco ME, Barbero G, Dergan‐Dylon LS, Marks MP et al (2016). ROR1 contributes to melanoma cell growth and migration by regulating N‐cadherin expression via the PI3K/Akt pathway. Mol Carcinog 55: 1772–1785. [DOI] [PubMed] [Google Scholar]
- Frick M, Dorken B, Lenz G (2012). New insights into the biology of molecular subtypes of diffuse large B‐cell lymphoma and Burkitt lymphoma. Best Pract Res Clin Haematol 25: 3–12. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Chen L, Endo T, Tang L, Lu D, Castro JE et al (2008). Antisera induced by infusions of autologous Ad‐CD154‐leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc Natl Acad Sci U S A 105: 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhirajan RK, Staib PA, Minke K, Gehrke I, Plickert G, Schlosser A et al (2010). Small molecule inhibitors of Wnt/beta‐catenin/lef‐1 signaling induces apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Neoplasia 12: 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Song H, Bishop K, Elliot G, Garrett L, English MA et al (2011). Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev Cell 20: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Chen YG (2010). Dishevelled: the hub of Wnt signaling. Cell Signal 22: 717–727. [DOI] [PubMed] [Google Scholar]
- Gelebart P, Anand M, Armanious H, Peters AC, Dien Bard J, Amin HM et al (2008). Constitutive activation of the Wnt canonical pathway in mantle cell lymphoma. Blood 112: 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamlouch H, Darwiche W, Hodroge A, Ouled‐Haddou H, Dupont S, Singh AR et al (2015). Factors involved in CLL pathogenesis and cell survival are disrupted by differentiation of CLL B‐cells into antibody‐secreting cells. Oncotarget 6: 18484–18503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan T, Yaniv A, Bafico A, Liu G, Gazit A (2004). The human Frizzled 6 (HFz6) acts as a negative regulator of the canonical Wnt. beta‐catenin signaling cascade. J Biol Chem 279: 14879–14888. [DOI] [PubMed] [Google Scholar]
- Gutierrez A Jr, Arendt BK, Tschumper RC, Kay NE, Zent CS, Jelinek DF (2011). Differentiation of chronic lymphocytic leukemia B cells into immunoglobulin secreting cells decreases LEF‐1 expression. PLoS One 6 e26056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A Jr, Tschumper RC, Wu X, Shanafelt TD, Eckel‐Passow J, Huddleston PM 3rd et al (2010). LEF‐1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B‐cell lymphocytosis. Blood 116: 2975–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek M (2015). Chronic lymphocytic leukemia: 2015 update on diagnosis, risk stratification, and treatment. Am J Hematol 90: 446–460. [DOI] [PubMed] [Google Scholar]
- Hammerlein A, Weiske J, Huber O (2005). A second protein kinase CK1‐mediated step negatively regulates Wnt signalling by disrupting the lymphocyte enhancer factor‐1/beta‐catenin complex. Cell Mol Life Sci 62: 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer SW, Krenn PW, Ganghammer S, Asslaber D, Pichler U, Oberascher K et al (2014). Tiam1/Rac1 signals contribute to the proliferation and chemoresistance, but not motility, of chronic lymphocytic leukemia cells. Blood 123: 2181–2188. [DOI] [PubMed] [Google Scholar]
- Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R (1996). Nuclear localization of beta‐catenin by interaction with transcription factor LEF‐1. Mech Dev 59: 3–10. [DOI] [PubMed] [Google Scholar]
- Hudecek M, Lupo‐Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C et al (2013). Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1‐specific chimeric antigen receptor T cells. Clin Cancer Res 19: 3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M, Schmitt TM, Baskar S, Lupo‐Stanghellini MT, Nishida T, Yamamoto TN et al (2010). The B‐cell tumor‐associated antigen ROR1 can be targeted with T cells modified to express a ROR1‐specific chimeric antigen receptor. Blood 116: 4532–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JK, Virshup DM (2016). CK1delta: a pharmacologically tractable Achilles' heel of Wnt‐driven cancers? Ann Transl Med 4: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MY, Widhopf GF 2nd, Wu CC, Cui B, Lao F, Sadarangani A et al (2015). Pre‐clinical specificity and safety of UC‐961, a first‐in‐class monoclonal antibody targeting ROR1. Clin Lymphoma Myeloma Leuk 15 (Suppl): S167–S169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Thompson PA, Keating M, Estrov Z, Ferrajoli A, Jain N et al (2017). Long‐term outcomes for patients with chronic lymphocytic leukemia who discontinue ibrutinib. Cancer 7: 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroziak K, Pula B, Walewski J (2017). Current treatment of chronic lymphocytic leukemia. Curr Treat Options Oncol 18: 5. [DOI] [PubMed] [Google Scholar]
- Janovska P, Poppova L, Plevova K, Plesingerova H, Behal M, Kaucka M et al (2016). Autocrine signaling by Wnt‐5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin Cancer Res 22: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Reiley WR, Lee AJ, Wright A, Wu X, Zhang M et al (2007). Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem 282: 15884–15893. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Wilder E, Klingensmith J, Zachary K, Perrimon N (1996). The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev 10: 3116–3128. [DOI] [PubMed] [Google Scholar]
- Kagey MH, He X (2017). Rationale for targeting the Wnt signaling modulator Dickkopf‐1 for oncology. Br J Pharmacol 2: 13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaucka M, Petersen J, Janovska P, Radaszkiewicz T, Smyckova L, Daulat AM et al (2015). Asymmetry of VANGL2 in migrating lymphocytes as a tool to monitor activity of the mammalian Wnt/planar cell polarity pathway. Cell communication and signaling : CCS 13: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaucka M, Plevova K, Pavlova S, Janovska P, Mishra A, Verner J et al (2013). The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B‐lymphocyte migration. Cancer Res 73: 1491–1501. [DOI] [PubMed] [Google Scholar]
- Kestler HA, Kühl M (2008). From individual Wnt pathways towards a Wnt signalling network. Philosophical Transactions of the Royal Society B: Biological Sciences 363: 1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AS, Hojjat‐Farsangi M, Daneshmanesh AH, Hansson L, Kokhaei P, Osterborg A et al (2016). Dishevelled proteins are significantly upregulated in chronic lymphocytic leukaemia. Tumour Biol 37: 11947–11957. [DOI] [PubMed] [Google Scholar]
- Kotaskova J, Pavlova S, Greif I, Stehlikova O, Plevova K, Janovska P et al (2016). ROR1‐based immunomagnetic protocol allows efficient separation of CLL and healthy B cells. Br J Haematol 175: 339–342. [DOI] [PubMed] [Google Scholar]
- Kotaskova J, Tichy B, Trbusek M, Francova HS, Kabathova J, Malcikova J et al (2010). High expression of lymphocyte‐activation gene 3 (LAG3) in chronic lymphocytic leukemia cells is associated with unmutated immunoglobulin variable heavy chain region (IGHV) gene and reduced treatment‐free survival. J Mol Diagn 12: 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G et al (2012). Epigenomic analysis detects widespread gene‐body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 44: 1236–1242. [DOI] [PubMed] [Google Scholar]
- Kuppers R, Engert A, Hansmann ML (2012). Hodgkin lymphoma. J Clin Invest 122: 3439–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laranjeira AB, Yang SX (2016). Therapeutic target discovery and drug development in cancer stem cells for leukemia and lymphoma: from bench to the clinic. Expert Opin Drug Discov 11: 1071–1080. [DOI] [PubMed] [Google Scholar]
- Lazarian G, Guieze R, Wu CJ (2017). Clinical implications of novel genomic discoveries in chronic lymphocytic leukemia. J Clin Oncol 35: 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H‐J, Shi D‐L, Zheng JJ (2015). Conformational change of Dishevelled plays a key regulatory role in the Wnt signaling pathways. Elife 4 e08142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lento W, Congdon K, Voermans C, Kritzik M, Reya T (2013). Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb Perspect Biol 5 pii: a008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke F, Harenberg M, Nietert MM, Zaunig S, von Bonin F, Arlt A et al (2017a). Microenvironmental interactions between endothelial and lymphoma cells: a role for the canonical Wnt pathway in Hodgkin lymphoma. Leukemia 31: 361–372. [DOI] [PubMed] [Google Scholar]
- Linke F, Zaunig S, Nietert MM, von Bonin F, Lutz S, Dullin C et al (2017b). Wnt5A: a motility‐promoting factor in Hodgkin lymphoma. Oncogene 36: 13–23. [DOI] [PubMed] [Google Scholar]
- Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T et al (2013). Targeting Wnt‐driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A 110: 20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y et al (2012). Dysregulation of TNFalpha‐induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia 26: 1293–1300. [DOI] [PubMed] [Google Scholar]
- Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM et al (2004). Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 101: 3118–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJ (2012). Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 26: 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X (2009). Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J et al (2016). Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 35: 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan D, Choi J, Cooke L, Simons B, Riley C, Klinkhammer T et al (2009). Gene expression and serum cytokine profiling of low stage CLL identify Wnt/PCP, Flt‐3L/Flt‐3 and CXCL9/CXCR3 as regulators of cell proliferation, survival and migration. Hum Genomics Proteomics 2009: 453634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Kincade PW (2009). Wnt‐related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell 4: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis BJ, Lai Y, Qu C, Janicki JS, Cui T (2015). CYLD‐mediated signaling and diseases. Curr Drug Targets 16: 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro FR, Galieni P, Tedeschi A, Laurenti L, Del Poeta G, Reda G et al (2017). Factors predicting survival in chronic lymphocytic leukemia patients developing Richter syndrome transformation into Hodgkin lymphoma. Am J Hematol 92: 529–535. [DOI] [PubMed] [Google Scholar]
- Memarian A, Hojjat‐Farsangi M, Asgarian‐Omran H, Younesi V, Jeddi‐Tehrani M, Sharifian RA et al (2009). Variation in Wnt genes expression in different subtypes of chronic lymphocytic leukemia. Leuk Lymphoma 50: 2061–2070. [DOI] [PubMed] [Google Scholar]
- Meng QJ, Maywood ES, Bechtold DA, Lu WQ, Li J, Gibbs JE et al (2010). Entrainment of disrupted circadian behavior through inhibition of casein kinase 1 (CK1) enzymes. Proc Natl Acad Sci U S A 107: 15240–15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mii Y, Taira M (2009). Secreted Frizzled‐related proteins enhance the diffusion of Wnt ligands and expand their signalling range. Development 136: 4083–4088. [DOI] [PubMed] [Google Scholar]
- Mittal AK, Chaturvedi NK, Rai KJ, Gilling‐Cutucache CE, Nordgren TM, Moragues M et al (2014). Chronic lymphocytic leukemia cells in a lymph node microenvironment depict molecular signature associated with an aggressive disease. Mol Med 20: 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JA, Gulley ML, Pathmanathan R, Raab‐Traub N (2004). Differential signaling pathways are activated in the Epstein‐Barr virus‐associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer Res 64: 5251–5260. [DOI] [PubMed] [Google Scholar]
- Moskalev EA, Luckert K, Vorobjev IA, Mastitsky SE, Gladkikh AA, Stephan A et al (2012). Concurrent epigenetic silencing of wnt/beta‐catenin pathway inhibitor genes in B cell chronic lymphocytic leukaemia. BMC Cancer 12: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley DP, Lee JP, Bellizzi AM (2017). Expression of LEF1 in mantle cell lymphoma. Ann Diagn Pathol 26: 57–59. [DOI] [PubMed] [Google Scholar]
- Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS et al (2016). DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet 48: 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott G, Rosenwald A (2008). Molecular pathogenesis of follicular lymphoma. Haematologica 93: 1773–1776. [DOI] [PubMed] [Google Scholar]
- Parameswaran N, Matsui K, Gupta N (2011). Conformational switching in ezrin regulates morphological and cytoskeletal changes required for B cell chemotaxis. J Immunol 186: 4088–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM et al (2012). Genome‐wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics 7: 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer L, Poll‐Wolbeck SJ, Flamme H, Gehrke I, Hallek M, Kreuzer KA (2014). Trichostatin A effectively induces apoptosis in chronic lymphocytic leukemia cells via inhibition of Wnt signaling and histone deacetylation. J Cancer Res Clin Oncol 140: 1283–1293. [DOI] [PubMed] [Google Scholar]
- Perez‐Galan P, Dreyling M, Wiestner A (2011). Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood 117: 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppova L, Janovska P, Plevova K, Radova L, Plesingerova H, Borsky M et al (2016). Decreased Wnt3 expression in chronic lymphocytic leukaemia is a hallmark of disease progression and identifies patients with worse prognosis in the subgroup with mutated IGHV. Br J Haematol 175: 851–859. [DOI] [PubMed] [Google Scholar]
- Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA et al (2013). Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of Wnt‐driven mammary cancer. Cancer Res 73: 502–507. [DOI] [PubMed] [Google Scholar]
- Qi J, Lee HJ, Saquet A, Cheng XN, Shao M, Zheng JJ et al (2017). Autoinhibition of Dishevelled protein regulated by its extreme C terminus plays a distinct role in Wnt/beta‐catenin and Wnt/planar cell polarity (PCP) signaling pathways. J Biol Chem 292: 5898–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, O'Riordan M, Okamura R, Devaney E, Willert K, Nusse R et al (2000). Wnt signaling regulates B lymphocyte proliferation through a LEF‐1 dependent mechanism. Immunity 13: 15–24. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X et al (2001). Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 194: 1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Gaidano G (2016). Richter syndrome: pathogenesis and management. Semin Oncol 43: 311–319. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Aguilera A, Rattmann I, Drew DZ, Muller LU, Summey V, Lucas DM et al (2010). Involvement of RhoH GTPase in the development of B‐cell chronic lymphocytic leukemia. Leukemia 24: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sant M, Allemani C, Tereanu C, De Angelis R, Capocaccia R, Visser O et al (2010). Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 116: 3724–3734. [DOI] [PubMed] [Google Scholar]
- Schlessinger K, Hall A, Tolwinski N (2009). Wnt signaling pathways meet Rho GTPases. Genes Dev 23: 265–277. [DOI] [PubMed] [Google Scholar]
- Seeliger B, Wilop S, Osieka R, Galm O, Jost E (2009). CpG island methylation patterns in chronic lymphocytic leukemia. Leuk Lymphoma 50: 419–426. [DOI] [PubMed] [Google Scholar]
- Seifert JR, Mlodzik M (2007). Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat Rev Genet 8: 126–138. [DOI] [PubMed] [Google Scholar]
- Semenov MV, Habas R, Macdonald BT, He X (2007). SnapShot: noncanonical Wnt signaling pathways. Cell 131: 1378. [DOI] [PubMed] [Google Scholar]
- Sohlbach K, Moll R, Gossmann J, Nowak O, Barth P, Neubauer A et al (2012). β‐Catenin signaling: no relevance in Hodgkin lymphoma? Leuk Lymphoma 53: 996–998. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal FJ, Famili F, Garcia Perez L, Pike‐Overzet K (2016a). Aberrant Wnt signaling in leukemia. Cancers (Basel) 8: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal FJ, Chhatta A, Mikkers H (2016b). Caught in a Wnt storm: complexities of Wnt signaling in hematopoiesis. Exp Hematol 44: 451–457. [DOI] [PubMed] [Google Scholar]
- Staal FJ, Luis TC, Tiemessen MM (2008). Wnt signalling in the immune system: Wnt is spreading its wings. Nat Rev Immunol 8: 581–593. [DOI] [PubMed] [Google Scholar]
- Troeger A, Johnson AJ, Wood J, Blum WG, Andritsos LA, Byrd JC et al (2012). RhoH is critical for cell‐microenvironment interactions in chronic lymphocytic leukemia in mice and humans. Blood 119: 4708–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R (2012). Alternative Wnt pathways and receptors. Cold Spring Harb Perspect Biol 4 pii: a007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt N, Dai B, Erdmann T, Berdel WE, Lenz G (2017). The molecular pathogenesis of mantle cell lymphoma. Leuk Lymphoma 58: 1530–1537. [DOI] [PubMed] [Google Scholar]
- Walther N, Ulrich A, Vockerodt M, von Bonin F, Klapper W, Meyer K et al (2013). Aberrant lymphocyte enhancer‐binding factor 1 expression is characteristic for sporadic Burkitt's lymphoma. Am J Pathol 182: 1092–1098. [DOI] [PubMed] [Google Scholar]
- Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S et al (2016). Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell 30: 750–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K et al (2011). SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Shalek AK, Lawrence M, Ding R, Gaublomme JT, Pochet N et al (2014). Somatic mutation as a mechanism of Wnt/beta‐catenin pathway activation in CLL. Blood 124: 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chang H, Nathans J (2010). When whorls collide: the development of hair patterns in frizzled 6 mutant mice. Development 137: 4091–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widhopf GF 2nd, Cui B, Ghia EM, Chen L, Messer K, Shen Z et al (2014). ROR1 can interact with TCL1 and enhance leukemogenesis in Emu‐TCL1 transgenic mice. Proc Natl Acad Sci U S A 111: 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T et al (2003). Wnt proteins are lipid‐modified and can act as stem cell growth factors. Nature 423: 448–452. [DOI] [PubMed] [Google Scholar]
- Wu QL, Zierold C, Ranheim EA (2009). Dysregulation of Frizzled 6 is a critical component of B‐cell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood 113: 3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zhu H, Fu Y, Shen W, Miao K, Hong M et al (2016). High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. Oncotarget 7: 21631–21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zhu H, Fu Y, Shen W, Xu J, Miao K et al (2014). Clinical significance of down‐regulated cylindromatosis gene in chronic lymphocytic leukemia. Leuk Lymphoma 55: 588–594. [DOI] [PubMed] [Google Scholar]
- Yang J, Baskar S, Kwong KY, Kennedy MG, Wiestner A, Rader C (2011). Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B‐cell malignancies. PLoS One 6 e21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Chen L, Cui B, Widhopf GF 2nd, Shen Z, Wu R et al (2016). Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J Clin Invest 126: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Chen L, Cui B, Wu C, Choi MY, Chen Y et al (2017). Cirmtuzumab inhibits Wnt5a‐induced Rac1 activation in chronic lymphocytic leukemia treated with ibrutinib. Leukemia 31: 1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuseff MI, Lennon‐Dumenil AM (2015). B cells use conserved polarity cues to regulate their antigen processing and presentation functions. Front Immunol 6: 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chen L, Cui B, Chuang HY, Yu J, Wang‐Rodriguez J et al (2012). ROR1 is expressed in human breast cancer and associated with enhanced tumor‐cell growth. PLoS One 7 (3) e31127. [DOI] [PMC free article] [PubMed] [Google Scholar]