

Abstract
Background and Purpose
We recently proposed the existence of mGlu3‐preferring autoreceptors in spinal cord terminals and of mGlu2‐preferring autoreceptors in cortical terminals. This study aims to verify our previous conclusions and to extend their pharmacological characterization.
Experimental Approach
We studied the effect of LY566332, an mGlu2 receptor positive allosteric modulator (PAM), and of LY2389575, a selective mGlu3 receptor negative allosteric (NAM) modulator, on the mGlu2/3 agonist LY379268‐mediated inhibition of glutamate exocytosis [measured as KCl‐evoked release of preloaded [3H]‐D‐aspartate]. The mGlu2 PAM BINA and the mGlu3 NAM ML337, as well as selective antibodies recognizing the N‐terminal of the receptor proteins, were used to confirm the pharmacological characterization of the native receptors.
Key Results
Cortical synaptosomes possess LY566332‐sensitive autoreceptors that are slightly, although significantly, susceptible to LY2389575. In contrast, LY566332‐insensitive and LY2389575‐sensitive autoreceptors are present in spinal cord terminals. BINA and ML337 mimicked LY566332 and LY2389575, respectively, in controlling LY379268‐mediated inhibition of glutamate exocytosis from both cortical and spinal cord synaptosomes. Incubation of cortical synaptosomes with anti‐mGlu2 antibody prevented the LY379268‐induced inhibition of glutamate exocytosis, and this response was partially reduced by the anti‐mGlu3 antibody. Incubation of spinal cord synaptosomes with the anti‐mGlu3 antibody abolished LY379268‐mediated reduction of glutamate exocytosis from these terminals, while the anti‐mGlu2 antibody was inactive. Western blot analysis and confocal microscopy data were largely consistent with these functional observations.
Conclusions and Implications
We confirmed that mGlu3‐preferring autoreceptors exist in spinal cord terminals. Differently, cortical glutamatergic terminals possess mGlu2/mGlu3 heterodimers, whose inhibitory effect is largely mediated by mGlu2 receptors.
Abbreviations
- [3H]‐D‐Asp
[3H]‐D‐aspartate
- BINA
3′‐[[(2‐Cyclopentyl‐2,3‐dihydro‐6,7‐dimethyl‐1‐oxo‐1H‐inden‐5‐yl)oxy]methyl]‐[1,1′‐biphenyl]‐4‐carboxylic acid
- LY2389575
(3S)‐1‐(5‐bromopyrimidin‐2‐yl)‐N‐(2,4‐dichlorobenzyl)pyrrolidin‐3‐amine methanesulfonate hydrate
- LY379268
(1R,4R,5S,6R)‐4‐Amino‐2‐oxabicyclo[3.1.0]‐hexane‐4,6‐dicarboxylic acid
- LY541850
(1S,2S,4R,5R,6S)‐2‐amino‐4‐methylbicyclo[3.1.0]‐hexane2,6‐dicarboxylic acid
- LY566332
N‐(4′‐cyano‐biphenyl‐3‐yl)‐N‐(3‐pyridinylmethyl)‐ethanesulfonamide hydrochloride
- mGlu
metabotropic glutamate
- ML337
[2‐Fluoro‐4‐[2‐(4‐methoxyphenyl)ethynyl]phenyl][(3R)‐3‐hydroxy‐1‐piperidinyl]methanone
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- TRIS
Tris‐(hydroxymethyl)‐amino methane
- t‐TBS
Tris‐buffered saline‐Tween
- VGLUT1
vesicular glutamate transporters type 1
Introduction
The group II metabotropic glutamate (mGlu) receptors comprise mGlu2 and mGlu3 receptors. The high sequence homology of the two receptors for a long time limited the possibility of distinguishing the respective contribution of mGlu2 and mGlu3 units to the expression of native group II mGlu receptors in the CNS (Niswender and Conn, 2010; Nicoletti et al., 2011; 2015). However, there is now evidence demonstrating that mGlu2 and mGlu3 receptors are differently expressed in particular CNS regions (Gu et al., 2008; Wright et al., 2013) and in cells (neurons and astrocytes; Ohishi et al., 1993; Tamaru et al., 2001; Copeland et al., 2017) and could have different roles in controlling synaptic transmission (Kew et al., 2002; Corti et al., 2007; Hanna et al., 2013; Sanger et al., 2013). Altogether, these observations support the existence of group II mGlu receptor subtypes.
The recent discovery of new ligands able to separate the role of the two receptors has allowed the pharmacological characterization of the native mGlu2/3 receptors. Consistently, we recently proposed the existence of presynaptic mGlu3‐preferring autoreceptors in spinal cord glutamate terminals and presynaptic mGlu2‐preferring autoreceptors in the cortex of adult mice (Di Prisco et al., 2016a). These conclusions originated from the results obtained in release studies with N‐acetyl‐aspartyl‐glutamate (NAAG), which has been shown to selectively activate the mGlu3 receptor subtype (Neale et al., 2011; Romei et al., 2012; Di Prisco et al., 2016a) and LY541850, a selective mGlu2 receptor agonist with mGlu3 receptor antagonist activity (Hanna et al., 2013; Sanger et al., 2013).
In this study we aimed to extend our previous work on the pharmacological characterization of the presynaptic mGlu2/3 autoreceptors in cortical and spinal cord synaptosomes. We investigated the effects of LY566332 and BINA, two positive allosteric modulators (PAMs) of the mGlu2 receptor subtype (Rorick‐Kehn et al., 2005; Benneyworth et al., 2007; Molinaro et al., 2009; Caraci et al., 2011; Sanger et al., 2013), and of LY2389575 and ML337, which are selective mGlu3 receptor negative allosteric modulators (NAMs; Caraci et al., 2011; Wenthur et al., 2013), on the LY379268‐induced reduction of glutamate exocytosis from both synaptosomal preparations. To further characterize the respective contribution of mGlu2 and mGlu3 receptor proteins to the assembly of the mGlu2/3 autoreceptors, we applied an unconventional immunological approach. Briefly, cortical and spinal cord synaptosomes were incubated with selective antibodies raised against the outer sequences of the mGlu2 and the mGlu3 receptor proteins in order to determine whether and to what extent these antibodies interfere with the LY379268‐mediated inhibition of glutamate exocytosis. These antibodies were also used in Western blot and confocal microscopy analyses to demonstrate the expression of mGlu3 and mGlu2 receptors in spinal cord and cortical synaptosomes and to analyse their reciprocal localization. The results from functional and immunochemical studies confirm that the mGlu2/3 autoreceptors in spinal cord and cortical terminals differ one each other in terms of mGlu receptor composition and pharmacological profile.
Methods
Animal experiments
Mice (male, strain C57BL/6J) were obtained from Charles River (Calco, Italy) and were housed in the animal facility of DIFAR, Section of Pharmacology and Toxicology. mGlu2 receptor knockout (mGlu2−/−) mice, mGlu3 receptor knockout (mGlu3−/−) mice, double mGlu2/3 receptor knockout (mGlu2/3−/−) mice on a CD1 genetic background and their CD1 wild‐type (WT) counterparts were kindly provided by Eli Lilly & Company (Indianapolis, IN, USA) (Linden et al., 2005) and housed at IRCSS Neuromed. These mice were individually genotyped for the mGlu2 and mGlu3 receptor gene by PCR. All animals were kept under environmentally controlled conditions (ambient temperature = 22°C, humidity = 40%) on a 12 h light/dark cycle with food and water available ad libitum.
Mice were killed by cervical dislocation followed by decapitation and the cortex and/or the spinal cord rapidly removed. The experimental procedures were in accordance with the European legislation (European Communities Council Directive of 24 November 1986, 86/609/EEC) and were approved by the Italian Ministry of Health (DDL 26/2014 and previous legislation; protocol number no. 50/2011‐B). Experiments were performed following the Guidelines for Animal Care and Use of the National Institutes of Health. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Sixty‐two C57BL/6 mice were used in the release experiments with selective positive and negative allosteric modulators (39 mice) and with the anti‐mGlu2 and anti‐mGlu3 selective antibodies (23 mice). In line with the 3Rs rules (replacement, refinement and reduction), both cortical and spinal cord synaptosomes were prepared from each animal, and one preparation was used for release experiments, while the other one, for Western blot analyses or confocal microscopy. Nine genetically modified mice (mGlu2−/− mice, mGlu3−/− mice and mGlu2/3−/− mice, three mice for each group) and three WT CD1 mice were used to confirm the specificity of anti‐mGlu2 and anti‐mGlu3 antibodies.
Preparation of synaptosomes
Mouse cortical and spinal cord purified synaptosomes were prepared as previously described (Musante et al., 2011). Briefly, the tissue was homogenized in 10 volumes of 0.32 M sucrose, buffered to pH 7.4 with Tris‐(hydroxymethyl)‐amino methane (TRIS; final concentration 0.01 M) using a glass/Teflon tissue grinder (clearance 0.25 mm); the homogenate was centrifuged at 1000× g for 5 min to remove nuclei and debris, and the supernatant was gently stratified on a discontinuous Percoll gradient (6, 10 and 20% v.v‐1 in Tris‐buffered sucrose) and centrifuged at 33 500× g for 5 min. The layer between 10 and 20% Percoll (synaptosomal fraction) was subsequently collected and washed by centrifugation. Synaptosomes were resuspended in a physiological solution with the following composition (mM): NaCl, 140; KCl, 3; MgSO4, 1.2; CaCl2, 1.2; NaH2PO4, 1.2; NaHCO3, 5; HEPES, 10; glucose, 10; pH 7.2–7.4.
Release experiments
Synaptosomes were incubated for 15 min a 37°C in a rotary water bath in the presence of [3H]‐D‐aspartate ([3H]‐D‐Asp; f.c.: 50 nM). In order to study the impact of anti‐mGlu antibodies on the LY379268‐mediated inhibition of glutamate exocytosis, synaptosomes were incubated for 30 min in the presence of the following antibodies: polyclonal rabbit anti‐mGlu3 (1:1000) or monoclonal mouse anti‐mGlu2 (1:1000); in these experiments, the radioactive tracer was added at t = 15 min of incubation. Identical portions of the synaptosomal suspensions were layered on microporous filters at the bottom of parallel thermostated chambers in a Superfusion System (Raiteri et al., 1974, Pittaluga, 2016; Ugo Basile, Comerio, Varese, Italy).
Synaptosomes were transiently (90 s) exposed, at t = 39 min, to high KCl‐containing medium (12 mM KCl for cortical synaptosomes or 15 mM KCl for spinal cord synaptosomes, NaCl substituting for an equimolar concentration of KCl; Zucchini et al., 2013) in the absence or in the presence of the agonist LY379268. Allosteric modulators were always added concomitantly with the agonist. Fractions were collected as follows: two 3 min fractions (basal release), one before (t = 36–39 min) and one after (t = 45–48 min) a 6 min fraction (t = 39–45 min; evoked release).The collected fractions and the superfused synaptosomes were measured for radioactivity.
The amount of radioactivity released into each superfusate fraction was expressed as a percentage of the total radioactivity. The K+‐evoked overflow was estimated by subtracting the neurotransmitter content in the first and the third fractions collected (basal release, b1 and b3) from that in the 6 min fraction collected during and after the depolarization pulse (evoked release, b2). The effect of agonists/antagonists is expressed as percentage of the KCl‐evoked overflow of tritium observed in the absence of receptor agonists and antagonists (% of control). In the figures demonstrating the impact of agonists and antagonists on [3H]‐D‐Asp release, data are always presented as the mean ± SEM of n (number within brackets under the bar) independent determinations obtained in separate experiments run in triplicate (three superfusion chambers for each experimental conditions) on different days.
Immunoblotting analysis
The cortical and spinal cord purified synaptosomes were lysed in ice‐cold lysis buffer (150 mM NaCl, 50 mM Tris, 1% Triton X‐100, protease inhibitors, pH 8.0) and quantified for protein content. Samples were boiled for 5 min at 95°C in SDS‐PAGE loading buffer. Proteins were then separated by SDS 7.5% PAGE and transferred onto PVDF membranes. Membranes were incubated for 1 h at room temperature in Tris‐buffered saline‐Tween (t‐TBS: 0.02 M Tris, 0.150 M NaCl and 0.05% Tween 20), containing 5% (w.v‐1) non‐fat dried milk and then probed with mouse monoclonal anti‐mGlu2 receptor antibody (1:1000 for cortical synaptosomes and 1:500 for spinal cord synaptosomes) or rabbit polyclonal anti‐mGlu3 receptor antibody (1:1000) and mouse anti‐β‐tubulin antibody (1:800) overnight at 4°C. After extensive washes in t‐TBS, membranes were incubated for 1 h at room temperature with appropriate horseradish peroxidise‐linked secondary antibodies (1:20 000).
In the experiments carried out to verify the specificity of the anti‐mGlu2 and anti‐mGlu3 antibodies, mouse cerebral cortices (from WT and knockout mice) were dissected out and homogenized at 4°C in Tris–HCl pH 7.5, 10 mM; NaCl, 150 mM; SDS 0.1%, EDTA, 5 mM and complete TM protease cocktail tablets. Proteins (20 μg) from supernatants were separated by 8% SDS‐PAGE and transferred on immuno‐blot PVDF membranes. Membranes were incubated with mouse anti‐mGlu2 (1:1000, 1 h at room temperature, in t‐TBS), rabbit anti‐mGlu3 receptor (1:1000, 1 h at room temperature, in t‐TBS) and mouse anti‐β‐tubulin (1:800, overnight at 4°C) antibodies and then for 1 h with the appropriate peroxidise‐coupled secondary antibody. Immunoblots were visualized with an enhanced chemiluminescence plus Western blotting detection system. β‐tubulin was used as an internal control.
Confocal microscopy and co‐localization
Mouse cortical and spinal cord synaptosomes were fixed with 2% paraformaldehyde for 15 min, permeabilized with 0.05% Triton X‐100 PBS for 5 min and incubated with the following primary antibodies: mouse anti‐mGlu2 receptor (1:1000), rabbit anti‐mGlu3 receptor (1:1000) and guinea pig anti‐vesicular glutamate transporters type 1 (VGLUT1; 1:500). After extensive washes, synaptosomes were incubated for 1 h at room temperature with the following antibodies: donkey anti‐rabbit AlexaFluor‐488 and goat anti‐guinea pig AlexaFluor‐633 (1:1000 both, confocal analysis aimed at identifying co‐localization of mGlu3 receptor and VGLUT1 proteins), with donkey anti‐mouse AlexaFluor‐488 and goat anti guinea pig AlexaFluor‐633 (1:1000 both, confocal analysis aimed at identifying co‐localization of mGlu2 receptor and VGLUT1 proteins), with donkey anti‐rabbit AlexaFluor‐488 with goat anti‐mouse AlexaFluor‐633 (1:1000 both, confocal analysis aimed at identifying co‐localization of mGlu2 and mGlu3 receptor proteins). Synaptosomes were then applied to coverslips (Musante et al., 2008a). Fluorescence images (512 × 512 × 8 bit) were then visualized by use of a six‐channel Leica TCS SP5 laser‐scanning confocal microscope, equipped with 458, 476, 488, 514, 543 and 633 nm excitation lines, through a plan‐apochromatic oil immersion objective 63X/1.4NA. Light collection configuration was optimized according to the combination of chosen fluorochromes. Sequential channel acquisition was performed to avoid crosstalk. A Leica ‘LAS AF’ software package was used for image acquisition, storage and visualization. The quantitative estimation of co‐localized proteins was performed as described previously (Musante et al., 2008a; Summa et al., 2013), by calculating the ‘co‐localization coefficients’ (Manders et al., 1993). They express the fraction of co‐localizing molecular species in each component of a dual‐colour image and are based on the Pearson's correlation coefficient, a standard procedure for matching one image with another in pattern recognition. If two molecular species are co‐localized, the overlay of their spatial distributions has a correlation value higher than what would be expected by chance alone. Costes et al. (2004) developed an automated procedure to evaluate the correlation between the green and red channels with a significance level >95%. The same procedure automatically determines an intensity threshold for each colour channel based on a linear least‐square fit of the green and red intensities in the image's 2D correlation cytofluorogramme. Costes's approach was carried out by macro routines integrated as plugins (WCIF Colocalization Plugins, Wright Cell Imaging Facility, Toronto Western Research Institute, Canada) in the ImageJ 1.51p software (Wayne Rasband, NIH, USA).
Calculations and statistical analysis
Sigma plot 10 data analysis and graphing software package were used for data handling/statistics and for graph drawing. ANOVA was performed followed by Dunnett's test or Newman–Keuls multiple comparisons test, as appropriate; direct comparisons were performed by Student's t‐test. Post hoc tests were done only if F value was significant. Data were considered significant if P < 0.05. Experiments were carried out to quantify the effects of antagonists alone and of pretreatment of the synaptosomes with antibodies on the KCl‐evoked release of [3H]‐D‐Asp. In accord with our previous results (Di Prisco et al., 2016a) and consistent with Curtis et al. (2015), at least n = 5 replicates were carried out for each experimental condition. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Chemicals
[2,3‐3H]‐D‐Asp (specific activity 11.3 Ci·mmol−1) was from Perkin Elmer (Boston, MA, USA). Mouse anti‐β‐tubulin, horseradish peroxidase‐conjugated anti‐mouse and anti‐rabbit secondary antibodies were from Sigma (Milan, Italy). LY379268, LY566332, ML337 and BINA were purchased from Tocris Bioscience (Bristol, UK). The Western blotting detection system was purchased from GeHealthcare (Italy). Mouse anti‐mGlu2 receptor antibody was from Abcam (Cambridge, UK), while rabbit anti‐mGlu3 receptor antibody was from Alomone Labs (Jerusalem, Israel). Guinea pig anti‐VGLUT1 was from Millipore Corporation, (Billerica, MA, USA). Donkey anti‐rabbit AlexaFluor‐488, goat anti guinea pig AlexaFluor‐633, donkey anti‐mouse AlexaFluor‐488 and goat anti‐mouse AlexaFluor‐633 were from Life Technologies Corporation, (Carlsbad, CA, USA). LY2389575 was kindly provided by Dr Monn and Dr McKinzie (Ely Lilly, Indianapolis, IN, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Effects of LY566332 and of LY2389575 on the LY379268‐induced inhibition of [3H]‐D‐Asp exocytosis from cortical glutamatergic synaptosomes
Purified synaptosomes isolated from the cortex of adult male mice were preloaded with [3H]‐D‐Asp and exposed to a mild (12 mM KCl) depolarizing stimulus (Grilli et al., 2004; Di Prisco et al., 2012) in the absence or in the presence of the mGlu2/3 receptor agonist LY379268. Previous studies showed that 3 nM LY379268 causes a significant, submaximal, inhibition of the [3H]‐D‐Asp exocytosis (Figure 1, see also Di Prisco et al., 2016a). LY566332 (1 μM), unable on its own to modify the 12 mM KCl‐evoked [3H]‐D‐Asp overflow, significantly enhanced the inhibitory effect of LY379268 on tritium exocytosis. A lower concentration of the mGlu2 receptor PAM (0.1 μM) slightly, although not significantly, modified the LY379268‐mediated inhibition of the 12 mM KCl‐evoked [3H]‐D‐Asp overflow (Figure 1).
Figure 1.
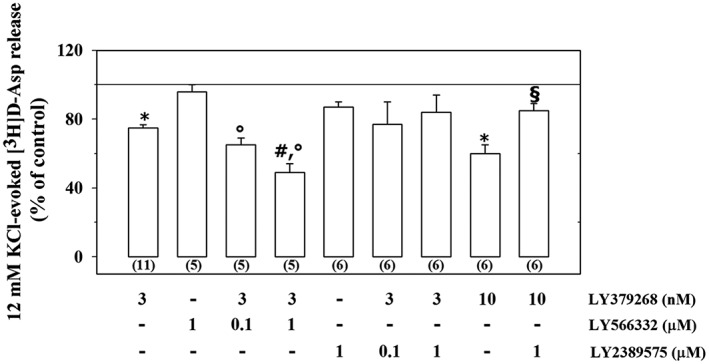
Effects of the mGlu2 PAM LY566332 and of the mGlu3 NAM LY2389575 on the inhibition of the 12 mM KCl‐evoked [3H]‐D‐Asp overflow elicited by the mGlu2/3 agonist LY379268 in mouse cortical synaptosomes. The 12 mM KCl‐evoked release of tritium over the basal release amounted to 1.39 ± 0.36 nCi. Results are expressed as a percentage of the 12 mM KCl‐evoked tritium overflow (% of control). Data are the mean ± SEM of n (number within brackets) experiments. * P < 0.05 versus the 12 mM KCl‐evoked tritium overflow; °P < 0.05 versus the 12 mM KCl/1 μM LY566332‐evoked tritium overflow; # P < 0.05 versus the 12 mM KCl/3 nM LY379268‐evoked tritium overflow; § P < 0.05 versus the 12 mM KCl/10 nM LY379268‐evoked tritium overflow.
The impact of LY2389575 on the 12 mM KCl‐evoked [3H]‐D‐Asp overflow from cortical synaptosomes was also quantified. The inhibition of the 12 mM KCl‐evoked release of tritium elicited by LY379268 (3 nM) in the presence of LY2389575 (0.1–1 μM) did not significantly differ from that caused by LY379268 alone, nor was it significantly modified when compared to control. The mGlu3 receptor NAM (1 μM), however, significantly modified the inhibitory effect elicited by a higher concentration (10 nM) of LY379268 on the 12 mM KCl‐evoked release of tritium from cortical synaptosomes. LY2389575 failed to modify significantly, on its own, the 12 mM KCl‐evoked release of [3H]‐D‐Asp (Figure 1).
Effects of LY566332 and of LY2389575 on the LY379268‐induced inhibition of [3H]‐D‐Asp exocytosis from spinal cord glutamatergic synaptosomes
Exposure of spinal cord synaptosomes to 15 mM KCl‐enriched solution causes the Ca2+‐dependent release of preloaded [3H]‐D‐Asp from these terminals (Di Prisco et al., 2013; 2016a). The concomitant presence of LY379268 (30 pM) significantly reduced this release (Figure 2; see also Di Prisco et al., 2013; 2016a). LY566332 (0.1–1 μM) failed to significantly modify the 30 pM LY379268‐induced inhibition of tritium exocytosis (Figure 2). Experiments were performed to investigate the impact of this compound on the tritium release evoked by 15 mM KCl in the presence of a lower (1 pM) concentration of the mGlu2/3 agonist. Again, the mGlu2 PAM was devoid of activity (Figure 2). However, the inhibitory effect elicited by LY379268 (30 pM) was totally abolished when LY2389575 (1 μM) was added concomitantly (Figure 2). A lower concentration (0.1 μM) of the mGlu3 NAM slightly, albeit not significantly, affected the 15 mM KCl/30 pM LY379268‐evoked release of [3H]‐D‐Asp. The mGlu3 receptor NAM, on its own, did not modify the 15 mM KCl‐evoked release of tritium from spinal cord synaptosomes (Figure 2).
Figure 2.
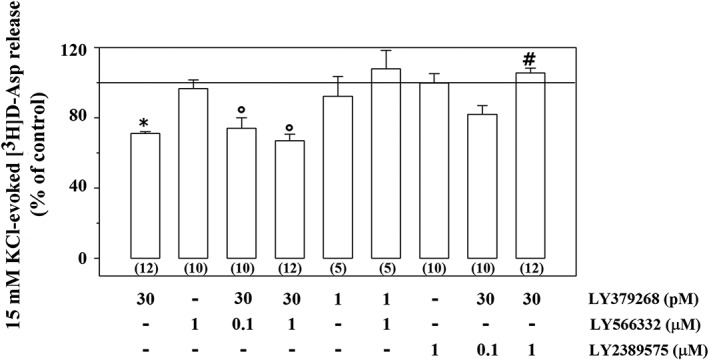
Effects of the mGlu2 PAM LY566332 and of the mGlu3 NAM LY2389575 on the inhibition of the 15 mM KCl‐evoked [3H]‐D‐Asp overflow elicited by the mGlu2/3 agonist LY379268 in mouse spinal cord synaptosomes. The 15 mM KCl‐evoked release of tritium over the basal release amounted to 4.23 ± 0.79 nCi. Results are expressed as a percentage of the 15 mM KCl‐evoked tritium overflow (% of control). Data are the mean ± SEM of n (number within brackets) experiments.* P < 0.05 versus the 15 mM KCl‐evoked tritium overflow; °P < 0.05 versus the 15 mM KCl/1 μM LY566332‐evoked tritium overflow; # P < 0.05 versus the 15 mM KCl/30 pM LY379268‐evoked tritium overflow.
Effects of BINA and of ML337 on the LY379268‐induced inhibition of [3H]‐D‐Asp exocytosis from cortical and spinal cord glutamatergic synaptosomes
To confirm the pharmacological profile of the mGlu2/3 autoreceptors under study, experiments were carried out to evaluate the impact of the mGlu2 PAM BINA and mGlu3 NAM ML337 on glutamate exocytosis in the absence and in the presence of LY379268 from both synaptosomal preparations. The concomitant addition of the mGlu2 PAM BINA (1 μM) to the depolarizing stimulus (12 mM KCl) did not significantly modify the [3H]‐D‐Asp exocytosis from cortical synaptosomes. However, the release of tritium elicited by 12 mM KCl/3 nM LY379268/1 μM BINA was significantly lower than that elicited by 12 mM KCl/3 nM LY379268 and by 12 mM KCl/1 μM BINA. The mGlu3 NAM ML337 also significantly modified the 12 mM KCl/10 nM LY379268‐evoked release of [3H]‐D‐Asp, leaving unchanged the 12 mM KCl‐induced [3H]‐D‐Asp exocytosis (Figure 3A). In contrast, in spinal cord synaptosomes, BINA (1 μM) failed to affect the 15 mM KCl‐evoked release of [3H]‐D‐Asp in the absence and in the presence of 3 pM LY379268. Moreover, ML337 did not significantly changed the 15 mM KCl‐evoked release of tritium but almost totally reversed the inhibitory effect elicited by 10 pM LY379268 on the 15 mM KCl‐evoked exocytosis of the radioactive tracer (Figure 3B).
Figure 3.
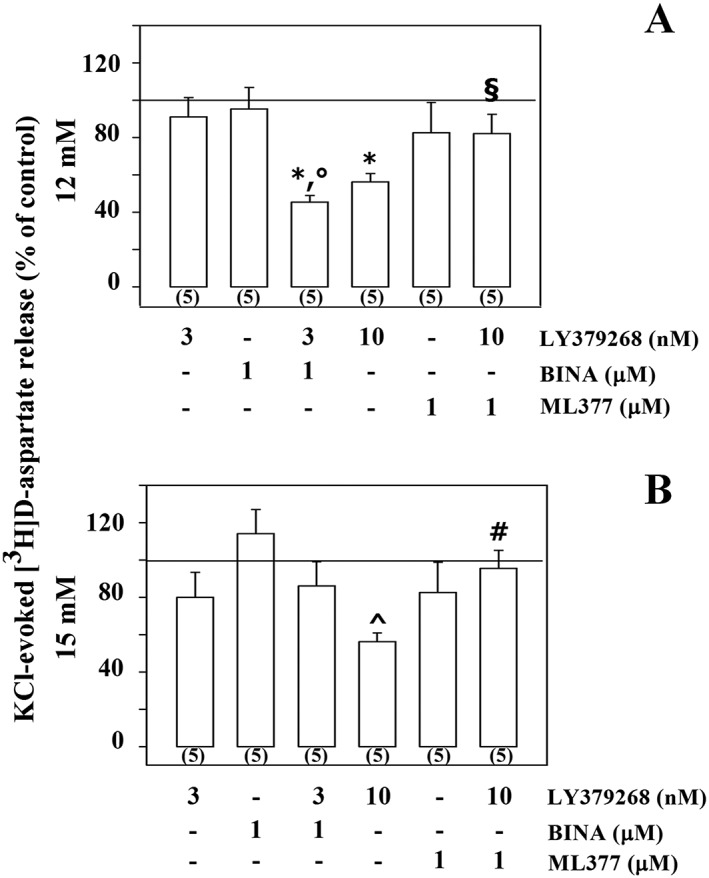
Effects of the mGlu2 PAM BINA and of the mGlu3 NAM ML337 on the inhibition of the KCl‐evoked [3H]‐D‐Asp overflow elicited by the mGlu2/3 agonist LY379268 in mouse cortical (A) and spinal cord (B) synaptosomes. (A) The 12 mM KCl‐evoked release of tritium from cortical synaptosomes amounted to 1.75 ± 0.42 nCi. (B) The 15 mM KCl‐evoked release of tritium from cortical synaptosomes amounted to 4.76 ± 0.63 nCi. Results are expressed as a percentage of the KCl‐evoked tritium overflow (% of control). Data are the mean ± SEM of n (number within brackets) experiments. * P < 0.05 versus the 12 mM KCl‐evoked tritium overflow; °P < 0.05 versus the 12 mM KCl/1 μM BINA‐evoked tritium overflow; § P < 0.05 versus the 12 mM KCl/10 nM LY379268‐evoked tritium overflow; ^ P < 0.05 versus the 15 mM KCl‐evoked tritium overflow; # P < 0.05 versus the 15 mM KCl/10 pM LY379268‐evoked tritium overflow.
Expression of mGlu2 and mGlu3 receptor proteins in mouse cortical and spinal cord synaptosomes
To assess the expression of mGlu2 and mGlu3 receptor proteins in mouse cortical and spinal cord synaptosomes, selective antibodies recognizing amino acid sequences within the outer NH2 terminus of these receptor proteins were used. Their specificity was confirmed in Western blot analysis on cortical homogenates from WT, mGlu2−/−, mGlu3−/− and mGlu2/3−/− mice. Both receptor antibodies recognized proteins with a mass of about 200 kDa in the brain homogenate from the WT mice, which is consistent with the presence of the mGlu2 and the mGlu3 receptor dimers. The anti‐mGlu2 antibody failed to recognize the band corresponding to 200 kDa in lysates from mGlu2−/− and from mGlu2/3−/− mice. Moreover, the anti‐mGlu3 antibody did not recognize the protein with comparable mass in lysates from mGlu3−/− and from mGlu2/3−/− mice (Figure 4). Both receptor antibodies recognized proteins with a mass of about 100 kDa in the brain homogenates from the mice belonging to the four different strains. In particular, the anti‐mGlu3 antibody recognized a band at 100 kDa largely conserved in the four lysates (Figure 4B), consistent with the presence of non‐specific immunoreactivity. In contrast, the band at about 100 kDa recognized by the anti‐mGlu2 antibody has a higher intensity in the WT and in the mGlu3−/− lysates when compared to mGlu2−/− and mGlu2/3−/− lysates, suggesting the presence of a monomeric form of the mGlu2 receptor protein (Figure 4A).
Figure 4.
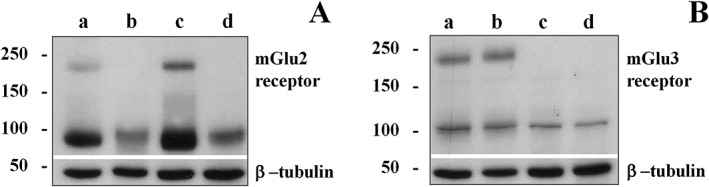
Western blot analysis of mGlu2 (A) and mGlu3 (B) receptors in mouse cortical homogenates of wild‐type mice (panel a), mGlu2 receptor knockout mice (mGlu2−/−, panel b), mGlu3 receptor knockout mice (mGlu3−/−, panel c) and double mGlu2 and mGlu3 receptor knockout mice (mGlu2/3−/−, panel d). The figure shows a representative Western blot.
Immunochemical studies showed that the anti‐mGlu2 receptor antibody recognized a clear mGlu2 immunoreactivity in the lysates of the synaptosomes from the cortex of WT mice at about 200 kDa, which is consistent with the presence of the dimeric form of the receptor (Figure 5A). Also mGlu2 immunopositivity was detectable, although to a lesser extent, in spinal cord synaptosomal lysates (Figure 5B). The anti‐mGlu3 antibody recognized a band with an apparent mass of about 200 kDa in both cortical and spinal cord synaptosomal lysates (Figure 5A,B).
Figure 5.
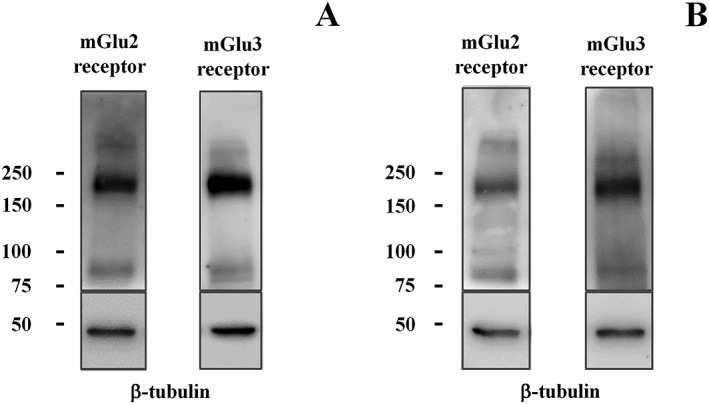
Western blot analysis reveals the presence of mGlu2 receptor and mGlu3 receptor protein dimers in mouse cortical (A) and spinal cord (B) synaptosomes (10 μg protein for cortical lysates and 20 μg protein for spinal cord lysates). The figure shows a representative blot of five (cortex) to seven (spinal cord) analyses carried out on different days.
mGlu2 and mGlu3 receptor proteins co‐localize with VGLUT1 protein in mouse cortical and spinal cord synaptosomes
Confocal microscopy was carried out to investigate whether mGlu2 and mGlu3 receptors co‐localize with VGLUT1, here used as a selective marker of glutamate terminals. A diffuse mGlu2‐immunoreactivity (Figure 6A, green) was observed in VGLUT1‐positive (Figure 6B, red) cortical synaptosomes (Figure 6C, yellow, merge, 91 ± 4%, n = 12). The mouse cortical synaptosomal preparation also showed a significant co‐localization of mGlu3 receptors and VGLUT1 (Figure 6D, green; E, red; F, yellow, merge, 74 ± 4%, n = 12). Finally, a significant percentage of mGlu2 positive particles (Figure 6G, green) was also reactive for mGlu3 receptors (Figure 6H, red; I, yellow, merge, 83 ± 4%, n = 12), while the expression of mGlu2 immunoreactivity in mGlu3 receptor‐positive particles was less pronounced (Figure 6I, yellow, merge 71 ± 3%, n = 12).
Figure 6.
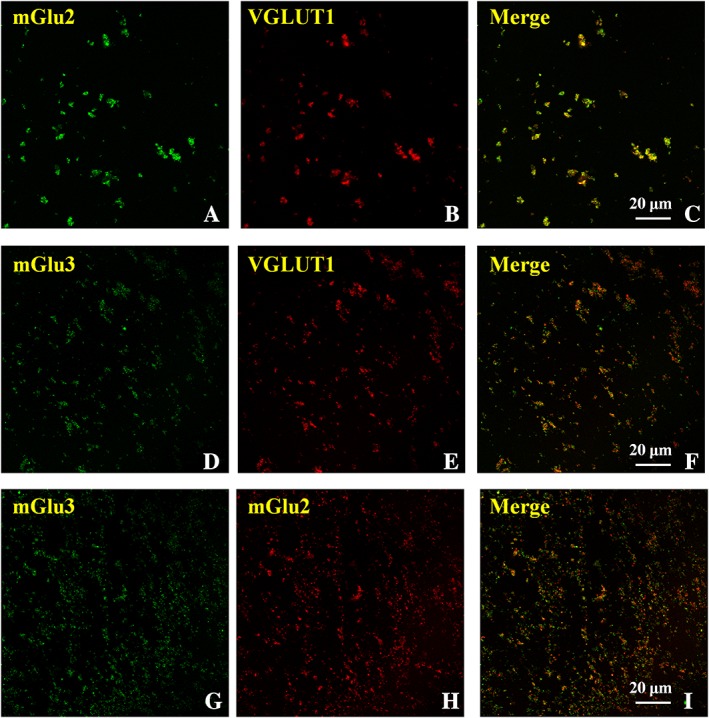
Confocal analysis of mGlu2 and mGlu3 receptor immunoreactivities in glutamatergic VGLUT1‐positive cortical nerve terminals: co‐localization of the two receptor proteins. The figure shows representative images of six independent experiments carried out on different days.
In spinal cord synaptosomes, mGlu2 receptor immunoreactivity (Figure 7A, green) and mGlu3 receptor immunoreactivity (Figure 7D, green) were observed in VGLUT1‐immunopositive (Figure 7B,E, red) particles (Figure 7C, yellow, merge, 55.7 ± 3.1 of mGlu2 receptor localization in VGLUT1‐positive cells; Figure 7F, yellow, merge, 46.5 ± 7.2 of mGlu3 receptor localization in VGLUT1‐positive cells). Finally, mGlu3 receptor immunopositive particles showed a low immunoreactivity for mGlu2 receptor signalling (Figure 7I, merge, yellow, 33.1 ± 6.2 of mGlu2 receptor immunoreactivity in mGlu3 receptor immunoreactive particles), while a larger co‐localization of mGlu3 receptor immunosignal with mGlu2 receptor‐positive particles was detected (Figure 7I, yellow, merge, 52.1 ± 9.2 mGlu3 receptor immunoreactivity in mGlu2 receptor‐immunopositive particles).
Figure 7.
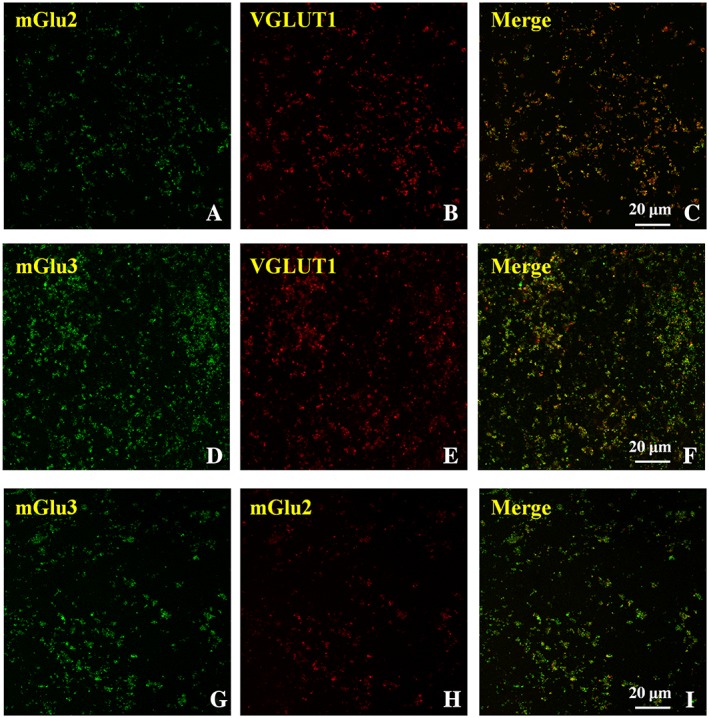
Confocal analysis highlighting the presence of mGlu2 and mGlu3 receptor immunoreactivities in glutamatergic spinal cord VGLUT1‐positive nerve terminals: co‐localization of the two receptor proteins. The figure shows representative images of eight independent experiments carried out on different days.
mGlu2 and mGlu3 receptor antibodies prevent the LY379268‐induced inhibition of the 12 mM KCl‐evoked [3H]‐D‐Asp exocytosis from cortical glutamatergic synaptosomes
Synaptosomes were incubated with anti‐mGlu2 or anti‐mGlu3 receptor antibodies and the release of preloaded [3H]‐D‐Asp elicited by the KCl stimulus was quantified in the absence or presence of LY379268. Incubation of cortical synaptosomes with the anti‐mGlu2 receptor antibody did not cause a significant changes to the 12 mM KCl‐evoked release of [3H]‐D‐Asp (control synaptosomes: 1.04 ± 0.8; synaptosomes incubated with the anti‐mGlu2 receptor antibody: 1.10 ± 0.6, n = 10, results expressed as induced overflow; mean ± SEM) but totally abolished the LY379268‐induced inhibition of the radioactive overflow (Figure 8). Similarly, incubation of cortical synaptosomes with the anti‐mGlu3 receptor antibody did not significantly modify the 12 mM KCl‐evoked tritium exocytosis (synaptosomes incubated with anti‐mGlu3 receptor antibody: 1.11 ± 0.7, n = 8, results expressed as induced overflow; mean ± SEM). In contrast, LY379268 (3 nM) still significantly inhibited the 12 mM KCl‐evoked release of tritium from cortical synaptosomes pre‐exposed to the anti‐mGlu3 receptor antibody, although the inhibitory effect was significantly less pronounced that that observed in control synaptosomes (Figure 8).
Figure 8.
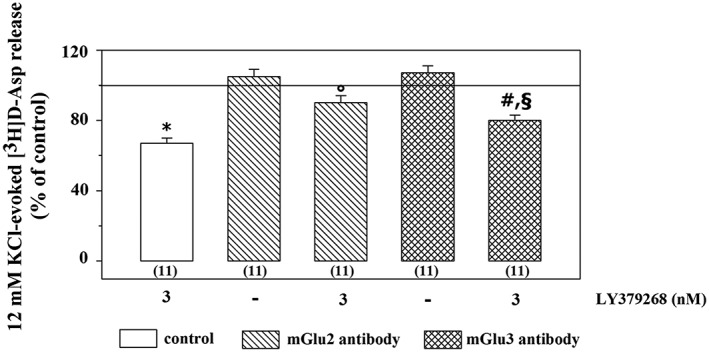
Effects of the incubation of cortical synaptosomes with an anti‐mGlu2 receptor or anti‐mGlu3 receptor antibody on the 12 mM KCl/LY379268‐evoked release of [3H]‐D‐Asp. The 12 mM KCl‐evoked release of tritium amounted to the following: control synaptosomes, 1.48 ± 0.43 nCi; anti‐mGlu2 antibody‐incubated synaptosomes, 1.63 ± 0.27 nCi; and anti‐mGlu3 antibody‐incubated synaptosomes 1.48 ± 0.29 nCi. Results are expressed as percentage of the 12 mM KCl‐evoked [3H]‐D‐Asp release (% of control). Data are the mean ± SEM of n (number within brackets) experiments * P < 0.05 versus the 12 mM KCl‐evoked tritium overflow from control cortical synaptosomes; °P < 0.05 versus the 12 mM KCl/3 nM LY379268‐evoked tritium overflow from control cortical synaptosomes; # P < 0.05 versus the 12 mM KCl‐evoked tritium overflow from cortical synaptosomes incubated with the anti‐mGlu3 receptor antibody; § P < 0.05 versus the 12 mM KCl/3 nM LY379268‐evoked tritium overflow from control cortical synaptosomes.
mGlu2 and mGlu3 receptor antibodies prevent the LY379268‐induced inhibition of the 15 mM KCl‐evoked [3H]‐D‐Asp exocytosis from spinal cord glutamatergic synaptosomes
Incubation of synaptosomes with antibodies failed to significantly affect the 15 mM KCl‐evoked release of [3H]‐D‐Asp (control synaptosomes: 4.52 ± 0.72; synaptosomes incubated with the anti‐mGlu2 receptor antibody: 4.61 ± 0.52; synaptosomes incubated with the anti‐mGlu3 receptor antibody: 4.83 ± 0.39, n = 8, results expressed as induced overflow; mean ± SEM). The inhibition of the 15 mM KCl‐evoked [3H]‐D‐Asp exocytosis elicited by LY379268 (30 pM) was unmodified in synaptosomes pre‐exposed to the anti‐mGlu2 receptor antibody, but it was significantly abolished in synaptosomes pre‐exposed to the anti‐mGlu3 receptor antibody (Figure 9).
Figure 9.
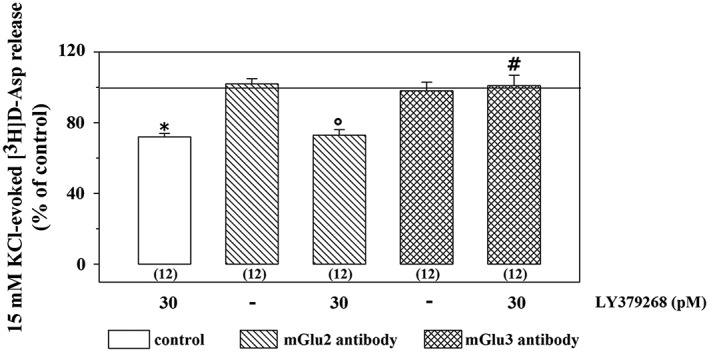
Effects of the incubation of spinal cord synaptosomes with anti‐mGlu2 receptor or anti‐mGlu3 receptor antibody on the 15 mM KCl/LY379268‐evoked release of [3H]‐D‐Asp. The 15 mM KCl‐evoked release of tritium amounted to the following: control synaptosomes, 4.76 ± 0.69 nCi; anti‐mGlu2 antibody‐incubated synaptosomes, 3.21 ± 0.55 nCi; and anti‐mGlu3 antibody‐incubated synaptosomes 3.98 ± 0.42 nCi. Results are expressed as percentage of the 15 mM KCl‐evoked [3H]‐D‐Asp release. Data are the mean ± SEM of n (number within brackets) experiments * P < 0.05 versus the 15 mM KCl‐evoked tritium overflow from control spinal cord synaptosomes; °P < 0.05 versus the 15 mM KCl‐evoked tritium overflow from spinal cord synaptosomes incubated with the anti‐mGlu2 receptor antibody; # P < 0.05 versus the 15 mM KCl/30 pM LY379268‐evoked tritium overflow from control spinal cord synaptosomes.
Discussion
Since the 90s, novel ligands for the group II mGlu receptors have been synthesized and their activity characterized. These compounds are typified by selectivity and by nanomolar affinities for these receptors and might represent the prototype of new neuroprotective agents (Nickols and Conn, 2014; Yin and Niswender, 2014). The novel compounds include ligands displaying mixed agonist/antagonist pharmacological effects on both mGlu2 and mGlu3 receptors, The highly conserved sequence of the two receptor proteins makes it difficult to compare the pharmacological profile of these mixed agents, in terms of affinity, activity and efficacy at each receptor subtype, and also to identify selective native neuronal and glial systems to be used as cellular models for their pharmacological characterization (but see Schoepp, 2001; Hanna et al., 2013; Sanger et al., 2013; Durand et al., 2014).
We used new molecules and established compounds to characterize the pharmacological profile of native mGlu2/3 autoreceptors in cortical and spinal cord terminals. We recently proposed that these native autoreceptors belong to different receptor subtypes, having a different receptor composition and pharmacological profile, and might, therefore, provide native receptor models for studying the pharmacological profile of new drugs (Di Prisco et al., 2016a). This conclusion is now further supported by the functional and immunochemical studies described in the present work.
As a first approach, we focussed on two novel allosteric modulators able to discriminate between mGlu2 and mGlu3 receptors: the mGlu2 receptor PAM LY566332 and the mGlu3 receptor NAM LY2389575. It is known that PAMs preferentially amplify receptor‐mediated events elicited by submaximal concentrations of orthosteric agonists, while NAMs impede receptor‐mediated events regardless of the amount of agonist applied. To study the impact of PAM and NAM, mGlu2/3 receptor‐mediated inhibition of glutamate exocytosis was elicited by exposing synaptosomes to concentrations of LY379268 that assure the submaximal activation of both of the two autoreceptors. When choosing the LY379268 concentrations to inhibit glutamate exocytosis from the two synaptosomal preparations, we took into consideration the strikingly different potency (about 4 order of magnitude) of LY379268 in cerebral cortex when compared to spinal cord synaptosomes highlighted in our previous study (Di Prisco et al., 2016a). A molecular explanation of this striking difference is not yet available, and future studies will be required to address this aspect. We found that LY566332 amplified the inhibitory activity of LY379268 at presynaptic cortical autoreceptors, leaving unchanged the LY379268‐mediated effect in spinal cord terminals. In spinal cord synaptosomes, the positive allosteric effects of LY566332 could not be revealed even if the mGlu2/3 agonist was present at a lower concentration, further confirming that the mGlu2 receptor subtype does not participate in the LY379268‐mediated inhibition of glutamate exocytosis in these terminals. In contrast, LY2389575 significantly recovered the inhibitory activity exerted by LY379268 at spinal cord mGlu3‐preferring autoreceptors, while it slightly, although not significantly, modified the efficacy of a submaximal amount of LY379268 at presynaptic mGlu2‐preferring autoreceptors in cortical synaptosomes. The antagonist‐like activity of the mGlu3 NAM, however, was disclosed when cortical synaptosomes were exposed to a concentration of LY379268 eliciting the almost maximal inhibitory effect at these terminals. Comparable results were obtained when studying the impact of the mGlu2 PAM BINA and the mGlu3 NAM ML337. Altogether, these observations largely corroborated the hypothesis that mGlu3 receptor subtypes account for presynaptic control of glutamate exocytosis from spinal cord terminals, while both mGlu2 and mGlu3 receptors participate in the control of glutamate overflow from cortical terminals.
To further support the data obtained with group II NAMs and PAMs, we approached the pharmacological characterization of these receptors by using selective anti‐mGlu2 and anti‐mGlu3 receptor antibodies as receptor ligands. Classically, antibodies have been used to characterize the subunit composition of receptors in immunochemical studies. More recently, these reagents were proposed as tools to define the pharmacological profile of receptors, in particular GPCRs (Gupta et al., 2008; Musante et al., 2008b and references therein, Di Prisco et al., 2012, 2016b; Merega et al., 2015). Actually, the binding of an antibody to the ligand‐binding pocket of the receptor or to its extraterminal region(s) causes either activation and/or blockade of the receptor‐mediated functions, mimicking the effect of an agonist or antagonist at these structures (Musante et al., 2008b; Di Prisco et al., 2012).
Antibodies raised against the extracellular epitope of the mGlu receptors have different effects on the LY379268‐mediated inhibition of glutamate exocytosis in the two synaptosomal preparations. In particular, the anti‐mGlu2 receptor antibody impeded the inhibitory activity of LY379268 on glutamate exocytosis in cortical synaptosomes, but not in spinal cord terminals.Whereas, the LY379268‐induced inhibition of glutamate exocytosis was impeded by the anti‐mGlu3 receptor antibody in spinal cord synaptosomes, and it was slightly, although significantly, reduced in cortical terminals pretreated with the antibody.
Before drawing any conclusions from these findings, these observations deserve some brief comments. Firstly, the possibility that antibody cross‐reactivity might affect our results is excluded on the basis of the high selectivity of the two antibodies, revealed in the Western blot analysis carried out with cortical homogenates from mGlu2 and/or mGlu3 receptor knockout mice. In our previous studies, we demonstrated that incubation of synaptosomes with antibodies raised against cytosolic proteins (i.e. β actin) did not affect the spontaneous and the K+‐evoked release of glutamate from both cortical and spinal cord terminals, ruling out the possibility that unspecific events might account for the effects observed (Musante et al., 2008b; Di Prisco et al., 2012; Merega et al., 2015). Secondly, the fact that both antibodies failed to modify on their own the KCl‐evoked glutamate exocytosis in both synaptosomal preparations allows us to rule out the occurrence of unspecific effects and also suggest that the antibodies do not mimic mGlu2/3 receptor agonist(s) and/or PAM(s) at mGlu2/3 receptors. Rather, our results indicate that antibodies preferentially act as selective antagonist and/or NAM at these targets. Finally, even if monoclonal antibodies are thought to be the best candidates as receptor agonists/antagonists, our results demonstrate that polyclonal antibodies can also be applied to function as receptor agonists or antagonists (reviewed by Gupta et al., 2008). Notably, the results obtained with the two antibodies were superimposable on those obtained with selective PAM and NAM and suggest that in spinal cord terminals, mGlu3‐preferring autoreceptors are pivotal for LY379268‐induced inhibition of glutamate exocytosis, while in cortical synaptosomes, mGlu2 autoreceptors play the main role, although the mGlu3 receptors also participate in this effect.
Quite interestingly, the use of the selective antibodies in immunochemical analysis unveiled a scenario well in line with the functional conclusions from release studies. In fact, Western blot analysis revealed the presence of both anti‐mGlu2 and anti‐mGlu3 receptor immunoreactivities in cortical (see also Ferraguti and Shigemoto, 2006) and spinal cord synaptosomal lysates; this is compatible with the idea that synaptosomal particles originating from both CNS regions are endowed with the two mGlu receptor proteins. These results, however, do not provide information on where the receptors are located (i.e. on glutamatergic or non‐glutamatergic spinal cord terminals). This conclusion is proposed on the basis of the results obtained with confocal microscopy, which was carried out to highlight the co‐localization of anti‐mGlu2 and anti‐mGlu3 receptor immunoreactivities with VGLUT1, used here as a selective marker of the glutamatergic particles.
Confocal analysis suggested that within the cortical synaptosomal preparations, there are glutamatergic synaptosomes endowed with group II mGlu receptors containing the mGlu2 receptor protein and synaptosomes possessing group II mGlu receptors containing the mGlu3 receptor protein. Here, the high co‐localization of mGlu2 positivity with VGLUT1 when compared to mGlu3 receptor indicates the predominant expression of mGlu2 receptors in these terminals. However, the high co‐localization of mGlu3 receptor reactivity with the mGlu2 receptor‐positive particles suggests that most of the cortical glutamatergic terminals possessing mGlu2‐preferring autoreceptors also express co‐localized mGlu3 receptors. In contrast in the spinal chord, the largest population of glutamatergic terminals was endowed with mGlu3 receptors, but mGlu3 immunopositivity (i.e. of the receptor that shows the largest distribution in glutamatergic terminals) slightly co‐localized with the less diffuse mGlu2 receptor immunoreactivity, suggesting that most of the spinal cord terminals do not possess both receptor proteins.
As a whole, based on both the functional and non‐functional observations described here, we confirm our original working hypothesis that cortical terminals are endowed with mGlu2‐preferring receptors, the releasing activity of which is also influenced, but not obviously so, by co‐localized mGlu3 receptors, while spinal cord terminals possess mGlu3‐preferring receptors that control glutamate release. We propose the selective antibodies, that recognize the N‐terminal of mGlu receptor protein, as new pharmacological tools for characterizing these receptors. Also we confirm that the group II mGlu autoreceptors controlling glutamate exocytosis in spinal cord terminals represent an appropriate model of native mGlu3‐preferring receptor for studying the detailed pharmacological profile of ligands at this receptor subtype.
Author contributions
A.P. was involved in the supervision, planning and execution of the research. T.B. and G.O. were involved in animal care and release experiments. G.O. and M.V. were involved in Western blot analysis. C.U. performed confocal microscopy analysis. B.R. and G.B. analysed the specificity of anti mGlu2 and anti‐mGlu3 receptor antibodies. G.B. and F.N. provided the transgenic mice. A.P. wrote the manuscript. A.P., G.B. and F.N. discussed the results and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the University of Genoa (Fondi per la Ricerca di Ateneo). The authors thank Dr James Monn and Dr David L. McKinzie for their useful scientific discussion. The authors thank Maura Agate and Silvia Smith, PhD (University of Utah, School of Medicine) for editorial assistance.
Olivero, G. , Bonfiglio, T. , Vergassola, M. , Usai, C. , Riozzi, B. , Battaglia, G. , Nicoletti, F. , and Pittaluga, A. (2017) Immuno‐pharmacological characterization of group II metabotropic glutamate receptors controlling glutamate exocytosis in mouse cortex and spinal cord. British Journal of Pharmacology, 174: 4785–4796. doi: 10.1111/bph.14061.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benneyworth MA, Xiang Z, Smith RL, Garcia EE, Conn PJ, Sanders‐Bush E (2007). A selective positive allosteric modulator of metabotropic glutamate receptor subtype 2 blocks a hallucinogenic drug model of psychosis. Mol Pharmacol 72: 477–484. [DOI] [PubMed] [Google Scholar]
- Caraci F, Molinaro G, Battaglia G, Giuffrida ML, Riozzi B, Traficante A et al (2011). Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer's disease: selective activation of mGlu2 receptors amplifies beta‐amyloid toxicity in cultured neurons, whereas dual activation of mGlu2 and mGlu3 receptors is neuroprotective. Mol Pharmacol 79: 618–626. [DOI] [PubMed] [Google Scholar]
- Copeland CS, Wall TM, Sims RE, Neale SA, Nisenbaum E, Parri HR et al (2017). Astrocytes modulate thalamic sensory processing via mGlu2 receptor activation. Neuropharmacology 121: 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti C, Battaglia G, Molinaro G, Riozzi B, Pittaluga A, Corsi M et al (2007). The use of knock‐out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci 27: 8297–8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes S, Daelemans D, Cho E, Dobbin Z, Pavlakis G, Lockett S (2004). Automatic and quantitative measurement of protein–protein colocalization in live cells. Biophys J 86: 3993–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prisco S, Summa M, Chellackudam V, Rossi PI, Pittaluga A (2012). RANTES‐mediated control of excitatory amino acid release in mouse spinal cord. J Neurochem 121: 428–437. [DOI] [PubMed] [Google Scholar]
- Di Prisco S, Merega E, Milanese M, Summa M, Casazza S, Raffaghello L et al (2013). CCL5‐glutamate interaction in central nervous system: early and acute presynaptic defects in EAE mice. Neuropharmacology 75: 337–346. [DOI] [PubMed] [Google Scholar]
- Di Prisco S, Merega E, Bonfiglio T, Olivero G, Cervetto C, Grilli M et al (2016a). Presynaptic, release‐regulating mGlu2 ‐preferring and mGlu3 ‐preferring autoreceptors in CNS: pharmacological profiles and functional roles in demyelinating disease. Br J Pharmacol 173: 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prisco S, Olivero G, Merega E, Bonfiglio T, Marchi M, Pittaluga A (2016b). CXCR4 and NMDA receptors are functionally coupled in rat hippocampal noradrenergic and glutamatergic nerve endings. J Neuroimmune Pharmacol 11: 645–656. [DOI] [PubMed] [Google Scholar]
- Durand D, Carniglia L, Beauquis J, Caruso C, Saravia F, Lasaga M (2014). Astroglial mGlu3 receptors promote alpha‐secretase‐mediated amyloid precursor protein cleavage. Neuropharmacology 79: 180–189. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Shigemoto R (2006). Metabotropic glutamate receptors. Cell Tissue Res 326: 483–504. [DOI] [PubMed] [Google Scholar]
- Grilli M, Raiteri L, Pittaluga A (2004). Somatostatin inhibits glutamate release from mouse cerebrocortical nerve endings trough presynaptic sst2 receptor linked to the adenylyl cyclase‐protein kinase A pathway. Neuropharmacology 46: 388–396. [DOI] [PubMed] [Google Scholar]
- Gu G, Lorrain DS, Wei H, Cole RL, Zhang X, Daggett LP et al (2008). Distribution of metabotropic glutamate 2 and 3 receptors in the rat forebrain: implication in emotional responses and central disinhibition. Brain Res 1197: 47–62. [DOI] [PubMed] [Google Scholar]
- Gupta A, Heimann AS, Gomes I, Devi LA (2008). Antibodies against G‐protein coupled receptors: novel uses in screening and drug development. Comb Chem High Throughput Screen 11: 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna L, Ceolin L, Lucas S, Monn J, Johnson B, Collingridge G et al (2013). Differentiating the roles of mGlu2 and mGlu3 receptors using LY541850, an mGlu2 agonist/mGlu3 antagonist. Neuropharmacology 66: 114–121. [DOI] [PubMed] [Google Scholar]
- Kew JN, Pflimlin MC, Kemp JA, Mutel V (2002). Differential regulation of synaptic transmission by mGlu2 and mGlu3 at the perforant path inputs to the dentate gyrus and CA1 revealed in mGlu2−/− mice. Neuropharmacology 43: 215–221. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden AM, Shannon H, Baez M, Yu JL, Koester A, Schoepp DD (2005). Anxiolytic‐like activity of the mGlu2/3 receptor agonist LY354740 in the elevated plus maze test is disrupted in metabotropic glutamate receptor 2 and 3 knock‐out mice. Psychopharmacology (Berl) 179: 284–291. [DOI] [PubMed] [Google Scholar]
- Manders EM, Verbeek FJ, Aten JA (1993). Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc 169: 375–382. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merega E, Prisco SD, Severi P, Kalfas F, Pittaluga A (2015). Antibody/receptor protein immunocomplex in human and mouse cortical nerve endings amplifies complement‐induced glutamate release. Neurosci Lett 600: 50–55. [DOI] [PubMed] [Google Scholar]
- Molinaro G, Traficante A, Riozzi B, Di Menna L, Curto M, Pallottino S et al (2009). Activation of mGlu2/3 metabotropic glutamate receptors negatively regulates the stimulation of inositol phospholipid hydrolysis mediated by 5‐hydroxytryptamine2A serotonin receptors in the frontal cortex of living mice. Mol Pharmacol 76: 379–387. [DOI] [PubMed] [Google Scholar]
- Musante V, Longordo F, Neri E, Pedrazzi M, Kalfas F, Severi P et al (2008a). RANTES modulates the release of glutamate in human neocortex. J Neurosci 28: 12231–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musante V, Neri E, Feligioni M, Puliti A, Pedrazzi M, Conti V et al (2008b). Presynaptic mGlu1 and mGlu5 autoreceptors facilitate glutamate exocytosis from mouse cortical nerve endings. Neuropharmacology 55: 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musante V, Summa M, Cunha RA, Raiteri M, Pittaluga A (2011). Pre‐synaptic glycine GlyT1 transporter–NMDA receptor interaction: relevance to NMDA autoreceptor activation in the presence of Mg2+ ions. J Neurochem 117: 516–527. [DOI] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Zuo D, Janczura KJ, Profaci CP, Lavin KM et al (2011). Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J Neurochem 118: 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols HH, Conn PJ (2014). Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis 61: 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD et al (2011). Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60: 1017–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Ngomba RT, Gradini R, Battaglia G (2015). Metabotropic glutamate receptors as drug targets: what's new? Curr Opin Pharmacol 20: 89–94. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50: 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N (1993). Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol 335: 252–266. [DOI] [PubMed] [Google Scholar]
- Pittaluga A (2016). Presynaptic release‐regulating mGlu1 receptors in central nervous system. Front Pharmacol 7: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiteri M, Angelini F, Levi G (1974). A simple apparatus for studying the release of neurotransmitters from synaptosomes. Eur J Pharmacol 25: 411–414. [DOI] [PubMed] [Google Scholar]
- Romei C, Raiteri M, Raiteri L (2012). Glycine release is regulated by metabotropic glutamate receptors sensitive to mGluR2/3 ligands and activated by N‐acetylaspartylglutamate (NAAG). Neuropharmacology 66: 311–316. [DOI] [PubMed] [Google Scholar]
- Rorick‐Kehn LM, Hart JC, McKinzie DL (2005). Pharmacological characterization of stress‐induced hyperthermia in DBA/2 mice using metabotropic and ionotropic glutamate receptor ligands. Psychopharmacology (Berl) 183: 226–240. [DOI] [PubMed] [Google Scholar]
- Sanger H, Hanna L, Colvin EM, Grubisha O, Ursu D, Heinz BA et al (2013). Pharmacological profiling of native group II metabotropic glutamate receptors in primary cortical neuronal cultures using a FLIPR. Neuropharmacology 66: 264–273. [DOI] [PubMed] [Google Scholar]
- Schoepp DD (2001). Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299: 12–20. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa M, Di Prisco S, Grilli M, Usai C, Marchi M, Pittaluga A (2013). Presynaptic mGlu7 receptors control GABA release in mouse hippocampus. Neuropharmacology 66: 215–224. [DOI] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R (2001). Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre‐ and postsynaptic sites. Neuroscience 106: 481–503. [DOI] [PubMed] [Google Scholar]
- Wenthur CJ, Morrison R, Felts AS, Smith KA, Engers JL, Byers FW et al (2013). Discovery of (R)‐(2‐fluoro‐4‐((−4‐methoxyphenyl)ethynyl)phenyl) (3‐hydroxypiperidin‐1‐yl)methanone (ML337), an mGlu3 selective and CNS penetrant negative allosteric modulator (NAM). J Med Chem 56: 5208–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RA, Johnson BG, Zhang C, Salhoff C, Kingston AE, Calligaro DO et al (2013). CNS distribution of metabotropic glutamate 2 and 3 receptors: transgenic mice and [3H]LY459477 autoradiography. Neuropharmacology 66: 89–98. [DOI] [PubMed] [Google Scholar]
- Yin S, Niswender CM (2014). Progress toward advanced understanding of metabotropic glutamate receptors: structure, signaling and therapeutic indications. Cell Signal 26: 2284–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini S, Pittaluga A, Summa M, Brocca‐Cofano E, Fabris M, De Michele R et al (2013). Seizure susceptibility in tat‐transgenic mice: implications for the role of tat in human immunodeficiency virus type 1 associated encephalopathy. Neurobiol Dis 62: 354–359.24141021 [Google Scholar]