

Abstract
Severe lupus nephritis in the absence of systemic lupus erythematosus (SLE) is a rare condition with an unclear clinical presentation and outcome.
We conducted a historical observational study of 12 adult (age >18 years) patients with biopsy-proven severe lupus nephritis or lupus-like nephritis without SLE immunological markers at diagnosis or during follow-up. Excluded were patients with chronic infections with HIV or hepatitis B or C; patients with a bacterial infectious disease; and patients with pure membranous nephropathy. Electron microscopy was retrospectively performed when the material was available. End points were the proportion of patients with a complete response (urine protein to creatinine ratio <0.5 g/day and a normal or near-normal eGFR), partial response (≥50% reduction in proteinuria to subnephrotic levels and a normal or near-normal eGFR), or nonresponse at 12 months or later after the initiation of the treatment.
The study included 12 patients (66% female) with a median age of 36.5 years. At diagnosis, median creatinine and proteinuria levels were 1.21 mg/dL (range 0.5–11.6) and 7.5 g/day (1.4–26.7), respectively. Six patients had nephrotic syndrome and acute kidney injury. Renal biopsy examinations revealed class III or class IV A/C lupus nephritis in all cases. Electron microscopy was performed on samples from 5 patients. The results showed mesangial and subendothelial dense deposits consistent with LN in 4 cases, and a retrospective diagnosis of pseudo-amyloid fibrillary glomerulonephritis was made in 1 patient.
Patients received immunosuppressive therapy consisting of induction therapy followed by maintenance therapy, similar to treatment for severe lupus nephritis. Remission was recorded in 10 patients at 12 months after the initiation of treatment. One patient reached end-stage renal disease. After a median follow-up of 24 months, 2 patients relapsed.
Lupus nephritis in the absence of overt SLE is a nosological entity requiring careful etiological investigation, including systematic electron microscopy examination of renal biopsies to rule out fibrillary glomerulonephritis. In this series, most patients presented with severe glomerulonephritis, which was highly similar to lupus nephritis at presentation and in terms of response to immunosuppressive therapy.
Keywords: lupus nephritis, glomerulonephritis, fibrillary glomerulonephritis
1. Introduction
Renal involvement in lupus includes a broad range of morphological lesions that are not individually pathognomonic. However, certain features are highly suggestive of lupus nephritis (LN). This includes the Full House immunofluorescence pattern (detection of IgM, IgA, IgM, C3, and C1q deposits), membranous nephropathy, and cytoplasmic tubuloreticular inclusions observed by electron microscopy. The presence of these histological patterns raises the possibility of systemic lupus erythematosus (SLE).
According to the Systemic Lupus International Collaborating Clinics (SLICC) criteria, SLE can be diagnosed in patients with LN who have positive immunological markers (anti-nuclear antibodies or anti-dsDNA antibodies), even in the absence of extra-renal manifestations of SLE.[1,2] These markers, however, may be absent upon presentation, either due to the sensitivity of the test or because of the delayed onset of lupus.[2,3]
Due to the similarity of renal lesions to those observed in LN, clinicians have often considered these patients as having “lupus-like” or “renal-limited” lupus nephritis or “Full House nephropathy” (FHN), and usually treat them with the classical immunosuppressive regimen used in SLE.
A recent study of childhood FHN without serologic evidence of SLE has reported encouraging results of 90% remission (complete + partial) and few relapses with immunosuppressive drug treatments.[3] However, data on the outcome in adult patients have shown conflicting results from small cohorts.[4–10] To date, the clinical presentation and outcome of adult FHN remain unclear.
Herein, we report the clinical/histological features and the outcomes in 12 adult patients diagnosed with severe proliferative “lupus-like” nephropathy. We also provide electron microscopy analysis of their biopsies and emphasize the possibility of differential diagnoses of the same phenotype.
2. Methods
2.1. Study population
This was a multicenter, observational, historical study of adult (>17 years old) patients diagnosed with FHN in the absence of overt systemic lupus. Inclusion criteria were (1) glomerular disease diagnosed by renal biopsy as observed in LN according to the ISN/RPS classification and extensive complement (C3, C1q) and immunoglobulin ((IgG, IgM +/– IgA) glomerular staining by immunofluorescence (IF); and (2) the absence of immunological criteria for SLE: ANA <1/160e and anti-ds DNA antibody negative.
Patients were excluded if the following criteria were present: (1) actual or suspected auto-immune disorder; (2) delayed onset SLE: development of positive ANA (>1/160e) and/or anti-ds DNA antibody; (3) HIV or hepatitis B or C positivity; or (4) isolated membranous nephropathy (corresponding to a possible pure class V diagnosis according to the ISN/RPS classification of LN).
This historical study was conducted according to the principles expressed in the Declaration of Helsinki. The ethics committee of the Groupe Coopératif sur le Lupus Rénal approved the study.
2.2. Histology
2.2.1. Light microscopy
Renal biopsy was performed at diagnosis. Renal specimens were evaluated by light microscopy (sections were stained with hematoxylin and eosin, trichrome, and Jones silver stains) and immunofluorescence (sections were stained with anti-C3, anti-C1q, IgG, IgA, IgM, λ and κ antibodies) by the local pathologist. Renal lesions were evaluated according to the ISN/RPS classification for lupus nephritis.[11]
2.2.2. Electron microscopy
Evaluations were retrospectively performed with IF samples as described previously.[12]
2.3. Definitions
The 4 variable Modification of Diet in Renal Disease (MDRD) study equations were used for the estimation of the glomerular filtration rate (eGFR). Proteinuria was measured using a 24-h urine collection.
The nephrotic syndrome was defined as proteinuria (> 3.5 g/d) or creatinuria (> 3.5 g/g) and a serum albumin concentration < 30 g/L. Hypertension was defined as a blood pressure > 140/90 mmHg.
The stage of chronic kidney disease (CKD) was defined according to the K/DOQI guidelines.[13]
SLE activity was evaluated according to the ACR classification and the Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index.[1,2]
2.4. Outcome measures
Complete renal remission (CR) was defined as a urine protein to creatinine ratio (UPCR) <0.5 g/g or g/d and normal or near-normal (within 10% of the normal eGFR if previously abnormal) eGFR. A partial renal response (PR) was defined as a ≥50% reduction in proteinuria to subnephrotic levels and a normal or near-normal eGFR. Relapse was defined as an increase of more than 50% in 24-h proteinuria and a decrease in the eGFR> 25% from the baseline eGFR following a CR or PR.
2.5. Statistical analysis
Each result was reported as the mean ± SD or a percentage. Comparisons between patients were performed using the paired T-test or Wilcoxon test, as appropriate. Renal function tests (creatinine and eGFR) and proteinuria were compared using the paired Wilcoxon test. Statistical significance was established for a P value < .05. Graphical representations were created, and statistical analyses were performed with Prism GraphPad software.
3. Results
3.1. Baseline Characteristics
The study included 12 patients (4 men/8 women) from 8 Nephrology Departments from 2003 to 2014. Demographic characteristics are shown in Table 1. The median age at presentation was 36.5 years (range 18–48). The baseline median creatinine level was 1.21 mg/dL (range 0.5–11.6 mg/dL), and acute kidney injury was recorded at presentation in 6 patients. The median proteinuria level was 7.5 g/d, and the nephrotic syndrome was diagnosed in 6 patients. One patient was diagnosed with an irreversible acute kidney injury (AKI) that required immediate renal replacement therapy (RRT).
Table 1.
Cohort characteristics at baseline.
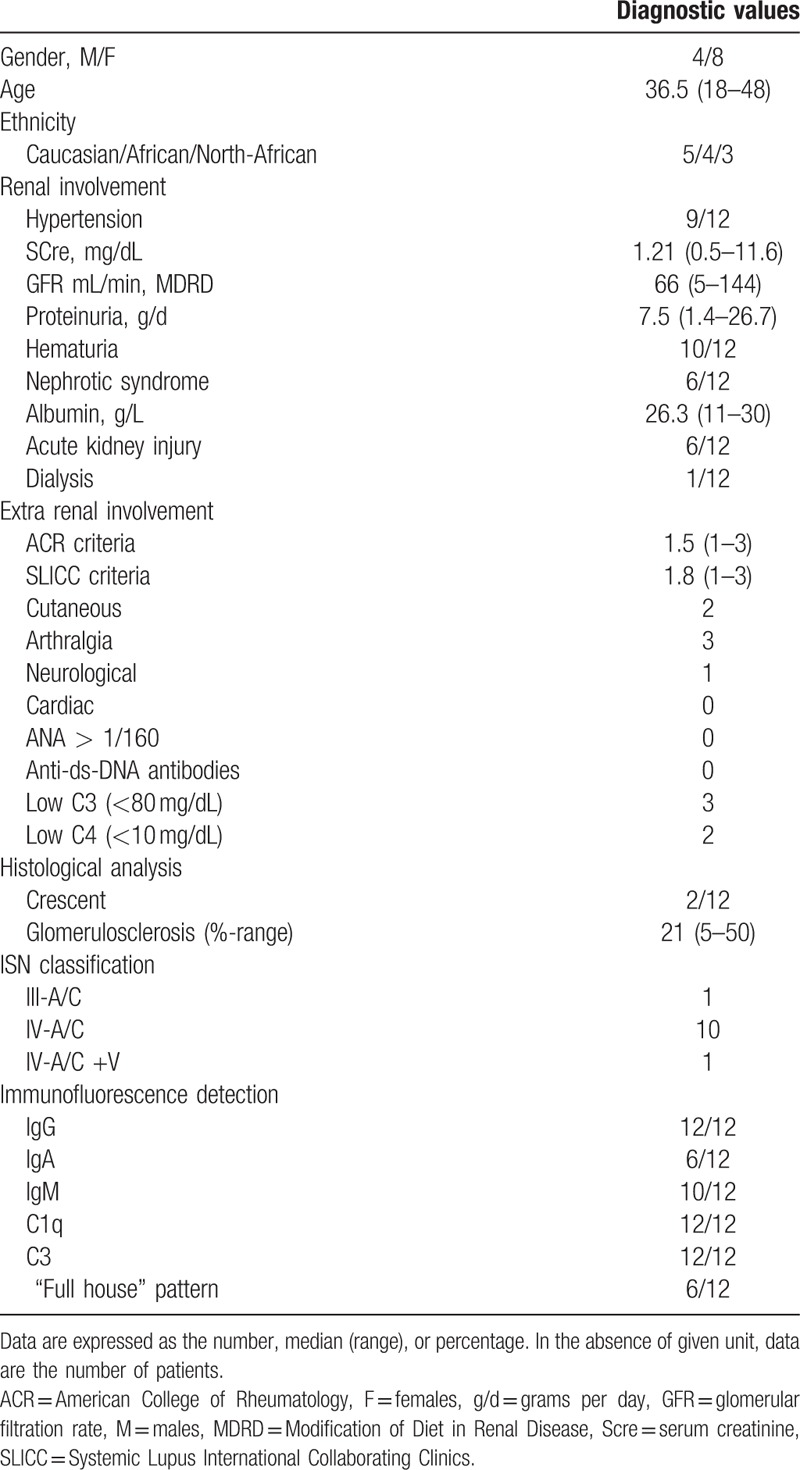
Few patients (N = 4) had extra-renal manifestations, with mean ACR criteria values and a SLICC score of 1.25 (range 1–3) and 1.5 (range 1–3), respectively. Skin rash and polyarthritis were the most common extrarenal manifestations (Table 1).
3.2. Renal pathology features
The main histological pattern observed was diffuse proliferative glomerulonephritis similar to a class IV A/C in 11/12 patients, with associated class V in 1 patient (Table 1). Class III A/C was diagnosed in 1/12 patients. Extracellular crescentic proliferation was observed in 2 patients. IgG, C3, and C1q mesangio-parietal deposits were present in all samples. IgM deposits were present in 10 patients. The complete Full House pattern was observed in 6 out of 12 biopsies. One patient had concomitant membranous nephropathy. Glomerulosclerosis was present in 5 biopsies, with a mean proportion of sclerotic glomeruli of 21% (range 5–50%).
Electron microscopy analysis was retrospectively performed with the available frozen samples used for IF. Eight samples were obtained, but only 5 were of sufficient quality for analysis. Four patients had typical nonorganized mesangial and subendothelial electron-dense deposits (Fig. 1A and B), as observed in LN. No tubuloreticular inclusions were observed in the samples. One patient (Patient 2) diagnosed with class III-A/C lupus nephritis by light microscopy presented with pseudo amyloid fibrillary deposits infiltrating the mesangium, suggesting fibrillar glomerulonephritis (Fig. 1C and D). It should be noted that this patient had concomitant cutaneous manifestations similar to skin lesions observed in SLE.
Figure 1.
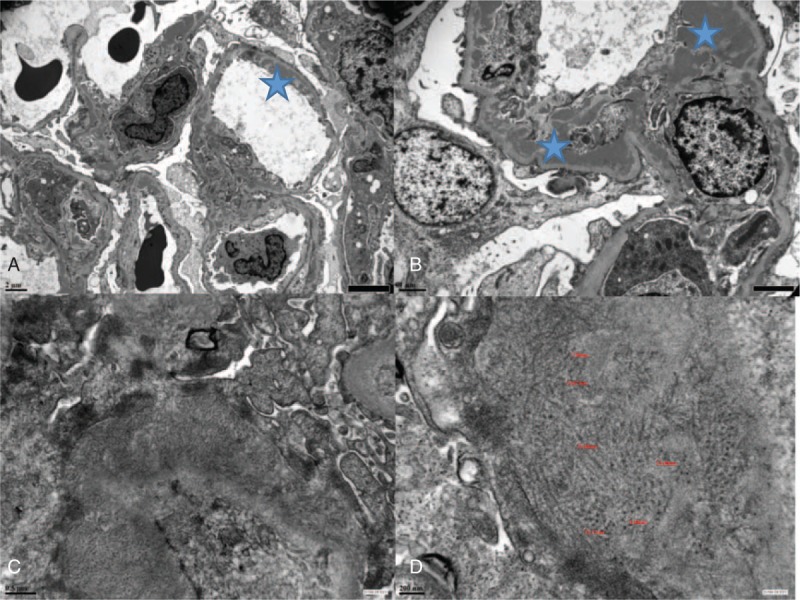
Electron microscopy. (A) Full House nephropathy, magnification ×5000: subendothelial electron-dense deposits in patient P4; (B) Full House nephropathy, magnification ×12,000: mesangial and subendothelial electron-dense deposits in patient P5; (C) fibrillary glomerulonephritis, magnification × 30,000: fibrils extending through the lamina densa of the glomerular basement membrane; (D) fibrillary glomerulonephritis, magnification × 60,000: deposits organized into unbranched, randomly oriented extracellular fibrils of 7–18 nm in diameter.
3.3. Treatment
Treatment modalities depend on the local practice, but all patients received an immunosuppressive treatment comprising an induction regimen for 3 to 6 months, followed by a maintenance regimen for at least 12 to 18 months (Table 2).
Table 2.
Clinical and biological characteristics of patients at diagnosis and at the last follow-up.
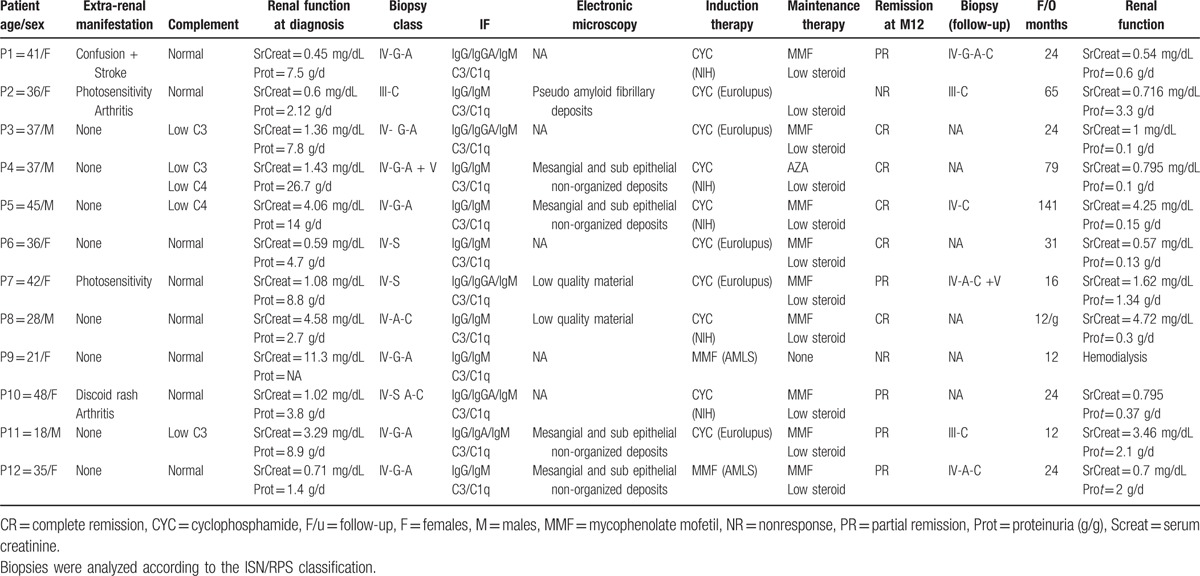
Regarding the induction protocol, 5 patients were treated with the Eurolupus Nephritis Trial regimen,[14] 5 were treated with the short-NIH regimen, and 2 received mycophenolate mofetil (MMF) and high-dose steroids as given in the ALMS trial.[15] Maintenance therapy included a low dose of prednisone and MMF or azathioprine. After 3 to 6 months of induction, all patients except for 2 were switched to purine inhibitors, either MMF (9/12) or azathioprine (1/12). Low-dose steroids (<10 mg/d) were given to 10 out of 12 patients during the maintenance period. For relapse or refractory disease, 2 patients were treated with a calcineurin inhibitor (cyclosporine A) or 4 weekly pulses of 375 mg/m2 rituximab.
All patients received pneumocystis prophylaxis with sulfamethoxazole/trimethoprim during the induction phase.
3.4. Outcome results
The median follow-up was 24 months (range 12 to 134 months). Twelve months after the initiation of induction therapy, 10 patients were in remission (5 CR and 5 PR).
Excluding the patient with RRT at diagnosis, renal function remained stable, with median creatinine levels of 0.92 mg/dL (range 0.59–4.75) and 1.21 mg/dL (range 0.5–11.6). Median proteinuria levels decreased from 7.7 (range 1.4–26) to 0.76 g/d (range 0.15–3), P < .001 (Fig. 2). All patients received renin-angiotensin-aldosterone system blockers. Immunological markers of SLE were still negative at 12 months and during the follow-up period.
Figure 2.
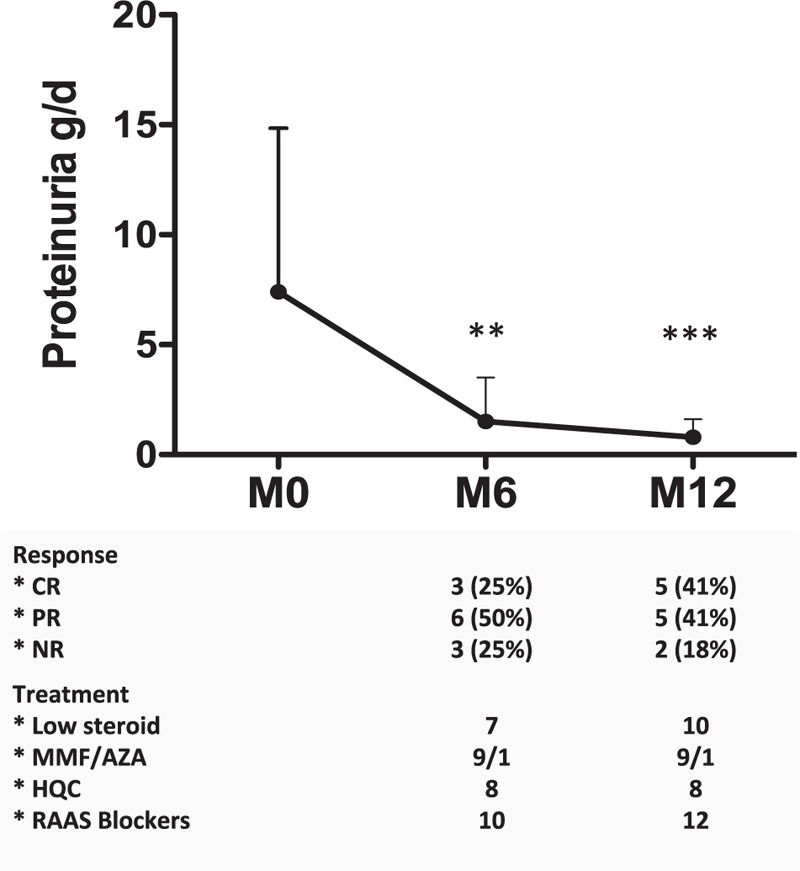
Evolution of proteinuria during the first year of treatment. ∗, P < .05, Wilcoxon test. Complete response (CR), partial response (PR), and nonresponse (NR) are shown at 6 and 12 months after the initiation of the immunosuppressive induction treatment. AZA = azathioprine, CR = complete response, HCQ = hydroxychloroquine, MMF = mycophenolate mofetil, NR = nonresponse, PR = partial response, RAAS = renin-aldosterone angiotensin system; low steroid therapy: <10 mg/d prednisone.
An additional renal biopsy was performed in 6 patients after a median follow-up of 12 months (range 12–127 months). The indication for biopsy was persistent proteinuria (N = 4) or proteinuria relapse (N = 2). When compared to baseline, the repeated biopsies revealed decreased but persistent activity and an increase in chronic lesions (Table 2). All these patients received an additional treatment of a combination of high-dose steroids with CYC, MMF, the monoclonal anti-CD20 antibody rituximab, or ciclosporin A. One patient had a CR, 4 had a PR, and 1 maintained an NR. Patient 3, who had a fibrillary glomerulonephritis, maintained a PR despite multiple IS treatments.
Patient 9, who had RRT, did not recover during follow-up. Progression to CKD stage 5 was recorded in 1 patient (Patient 5), 106 months after a relapse. At the last follow-up, 10 patients were still in remission (6 CR, 4 PR). Their renal function was stable when compared individually to their renal function at the first record of remission. Their median creatinine level was 0.79 mg/dL (range 0.54–4.72 mg/dL; 1 patient had a stage 3 CKD, 3 patients had stage 4 CKD, and 1 patient had stage 5 CKD). No serious adverse events, such as infections, cardiovascular events, or death, were observed.
4. Discussion
In the present paper, we report an observational series of 12 patients with severe proliferative lupus nephritis, or “lupus-like” nephritis, not fulfilling the ACR or SLICC classification criteria for SLE at diagnosis and during follow-up. Most of the patients showed considerable proteinuria in the nephrotic range and AKI at presentation, which correspond to the most severe presentation of LN.[14,15] Patients were also older, and a higher proportion of males was observed than what is found in a typical SLE with renal involvement population. According to the ISN/RPS LN classification, most of the patients had diffuse proliferative glomerulonephritis (Class IV with activity) and were treated with an immunosuppressive regimen similar to that used in severe LN (induction + maintenance). The overall response (CR+PR) was good after 12 months and during follow-up.
Severe lupus nephritis in the absence of overt SLE or “lupus-like” nephritis represents a challenge for clinicians because of 2 important issues. The first is whether this presentation should be considered as a distinct disease (referred to as “seronegative” or “incomplete” LN) or as a syndromic entity that includes several underlying diseases that are infection-related glomerulonephritis (viral or bacterial), fibrillar glomerulonephritis, or immune-complex mediated glomerulonephritis.[9,16] The second issue is related to the treatment used.
Since the mid-1980s, isolated cases or small case series have been published on “renal-limited LN” that lacks immunological evidence of SLE at diagnosis.[3,4,6,8,9,17] Some patients may later develop specific SLE immunological markers, and this is referred to as “delayed SLE.”[3] In most patients with delayed SLE or renal-limited LN, tubuloreticular inclusions are observed by electron microscopy, suggesting that this phenotype is part of “incomplete/seronegative” LN.[3,4,6] To avoid this bias, we excluded patients who developed SLE immunological markers during the follow-up. Interestingly, no tubuloreticular inclusions were observed in the present cases.
Some authors also refer to “lupus-like” nephropathy as “C1q nephropathy” in cases of FHN with dominant C1q staining.[5,18] However, in C1q nephropathies, C1q deposits are specifically localized in the mesangial area of the glomeruli, which was not the case for patients in our series.[18]
Unfortunately, only 5 biopsies were analyzed by electron microscopy due to the quantity and quality of the materials. Four patients had deposits consistent with LN, but 1 patient presented with pseudo amyloid fibrillary glomerulonephritis. In this patient, light microscopy and IF examination revealed class III A/C LN, a diagnosis that was validated by 2 renal pathologists. This patient also had extra-renal manifestations (cutaneous) consistent with seronegative SLE. Pseudo amyloid fibrillar glomerulonephritis has already been described in 2 SLE patients by Nasr et al,[19] but the renal biopsies in that study lacked the characteristic histological features of LN, particularly Full House immunostaining providing less ambiguity for ruling out the LN diagnosis. Nonetheless, this report and our experience highlight the need for electron microscopy validation of fibrillar deposits when the associated renal morphological findings do not fit with the clinical context of SLE or, conversely, in the case of possible lupus nephritis in the absence of overt systemic lupus.
No tubuloreticular inclusions were observed in our patients. Although tubuloreticular inclusions assessed by electron microscopy are currently considered a specific marker of LN, they are not detected in all cases of LN and are not mandated for this diagnosis. In addition, a recent study showed that tubuloreticular inclusions are detected in renal tissue in the context of virus-associated glomerulonephritis (HIV, HBV, and HCV) and to a lesser extent in membranous nephropathy or IgA nephropathy.[20] Thus, the absence of tubuloreticular inclusions is not helpful for the diagnostic classification of patients.
Therefore, we can conclude that FHN or “lupus-like nephritis” represents a syndromic entity that requires electron microscopy analysis of renal biopsies in addition to extensive etiological investigations, and in the absence of the fibrillar organization of deposits and in the absence of an underlying disease (e.g., in the absence of viral or other infectious diseases and in the absence of auto-immunity), physicians may consider a diagnosis of severe lupus nephritis in the absence of overt SLE.
The renal outcomes of severe “lupus-like” nephritis remain unclear. In the largest series of 42 pediatric patients presenting with FHN and no SLE criteria, the overall renal response was 91% after immunosuppressive treatment.[16] The authors used the same criteria for CR and PR that were used in our study. Recurrence was limited (18% at 4 years), allowing the progressive withdrawal of immunosuppression. However, only half of the cohort had proliferative glomerulonephritis (class IIIA or IVA), which could hamper the conclusion of a favorable outcome.
In adult patients, few reports or small series suggested a poor prognosis of the proliferative “lupus-like” nephritis.[4,8,9,21]
More recently, Rijnink and colleagues[10] also reported worse outcomes for idiopathic non-lupus FHN. Heterogeneity in the histological findings at diagnosis (presence of pure class V lupus nephritis), as well as heterogeneity in the immunosuppressive regimen, may explain the discrepancy with our results.
The treatment of severe lupus nephritis in the absence of overt SLE is not standardized. In our cohort, all patients received induction therapy, which is used as the standard of care for severe LN, followed by maintenance therapy with azathioprine or MMF. Overall outcome was good. Thus, we conclude that severe lupus nephritis in the absence of overt SLE should be treated as severe LN. The early recognition of fibrillary glomerulonephritis by ME should expand the treatment discussion to include other IS drugs, such as B cell depleting therapy (e.g., anti CD20 monoclonal antibodies). However, the prognosis is usually poor, despite such therapy.[12,19]
Our study has several limitations. It is a small, historical observational cohort. There was no central re-evaluation of renal biopsies. Less than half of the biopsies were analyzed by electron microscopy due the quality and quantity of the material available.
5. Conclusion
FHN, or proliferative “lupus-like” nephritis, is a syndromic entity that requires a careful etiological investigation, including the systematic electron microscopy evaluation of renal biopsies to rule out fibrillary glomerulonephritis. If negative, a diagnosis of severe lupus nephritis in the absence of overt SLE should be considered. The severity of the inflammatory process in the kidney seems to justify an aggressive immunosuppressive regimen. Remission can be achieved with the same immunosuppressive regimen used as a first-line therapy in SLE patients.
Acknowledgment
The authors thank the HUPNVS of Assistance Publique - Hôpitaux de Paris for funding publication fees.
Footnotes
Abbreviations: CKD = chronic kidney disease, CR = complete response, eGFR = estimated glomerular filtration rate, FHN = Full House nephropathy, LN = lupus nephritis, MMF = mycophenolate mofetil, PR = partial response, SLE = systemic lupus erythematosus.
Contributions: MT and ED contributed to the conception of the study. MT and CTP collected the data. MT analyzed the data. NQ, JMG, and GT performed the electron microscopy. MT drafted the manuscript. MT, NJC, JMG, and ED contributed to writing the article.
Funding: The HUPNVS of Assistance Publique—Hôpitaux de Paris funded publication fees.
The authors have no conflicts of interest to disclose.
References
- [1].Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- [2].Petri M, Orbai A-M, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ruggiero B, Vivarelli M, Gianviti A, et al. Lupus nephritis in children and adolescents: results of the Italian Collaborative Study. Nephrol Dial Transplant 2013;28:1487–96. [DOI] [PubMed] [Google Scholar]
- [4].Huerta A, Bomback AS, Liakopoulos V, et al. Renal-limited “lupus-like” nephritis. Nephrol Dial Transplant 2012;27:2337–42. [DOI] [PubMed] [Google Scholar]
- [5].Sharman A, Furness P, Feehally J. Distinguishing C1q nephropathy from lupus nephritis. Nephrol Dial Transplant 2004;19:1420–6. [DOI] [PubMed] [Google Scholar]
- [6].Baskin E, Agras PI, Menekşe N, et al. Full-house nephropathy in a patient with negative serology for lupus. Rheumatol Int 2007;27:281–4. [DOI] [PubMed] [Google Scholar]
- [7].Caltik A, Demircin G, Bülbül M, et al. An unusual case of ANA negative systemic lupus erythematosus presented with vasculitis, long-standing serositis and full-house nephropathy. Rheumatol Int 2013;33:219–22. [DOI] [PubMed] [Google Scholar]
- [8].Gianviti A, Barsotti P, Barbera V, et al. Delayed onset of systemic lupus erythematosus in patients with “full-house” nephropathy. Pediatr Nephrol 1999;13:683–7. [DOI] [PubMed] [Google Scholar]
- [9].Wen Y-K, Chen M-L. Clinicopathological study of originally non-lupus “full-house” nephropathy. Ren Fail 2010;32:1025–30. [DOI] [PubMed] [Google Scholar]
- [10].Rijnink EC, Teng YKO, Kraaij T, et al. Idiopathic non-lupus full-house nephropathy is associated with poor renal outcome. Nephrol Dial Transplant 2017;32:654–62. [DOI] [PubMed] [Google Scholar]
- [11].Weening JJ, D’Agati VD, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004;15:241–50. [DOI] [PubMed] [Google Scholar]
- [12].Javaugue V, Karras A, Glowacki F, et al. Long-term kidney disease outcomes in fibrillary glomerulonephritis: a case series of 27 patients. Am J Kidney Dis 2013;62:679–90. [DOI] [PubMed] [Google Scholar]
- [13].Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004;43(5 suppl 1):S1–290. [PubMed] [Google Scholar]
- [14].Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002;46:2121–31. [DOI] [PubMed] [Google Scholar]
- [15].Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009;20:1103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ruggiero B, Vivarelli M, Gianviti A, et al. Outcome of childhood-onset full-house nephropathy. Nephrol Dial Transplant 2017;32:1194–204. [DOI] [PubMed] [Google Scholar]
- [17].Jones E, Magil A. Nonsystemic mesangiopathic glomerulonephritis with “full house” immunofluorescence. Pathological and clinical observation in five patients. Am J Clin Pathol 1982;78:29–34. [DOI] [PubMed] [Google Scholar]
- [18].Devasahayam J, Erode-Singaravelu G, Bhat Z, et al. C1q nephropathy: the unique underrecognized pathological entity. Anal Cell Pathol 2015;2015:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nasr SH, Valeri AM, Cornell LD, et al. Fibrillary glomerulonephritis: a report of 66 cases from a single institution. Clin J Am Soc Nephrol 2011;6:775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee C-J, Suh K-S, Kim K-H, et al. The clinicopathologic significance of endothelial tubuloreticular inclusions in glomerular diseases. Ultrastruct Pathol 2013;37:386–94. [DOI] [PubMed] [Google Scholar]
- [21].Baskin E, Agras PI, Menekşe N, et al. Full-house nephropathy in a patient with negative serology for lupus. Rheumatol In 2006;27:281–4. [DOI] [PubMed] [Google Scholar]