
Fig. 2. SOAP-GAP predictions for a molecular database.
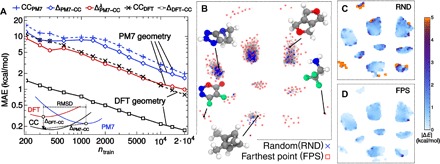
(A) Learning curves for the CC atomization energy of molecules in the GDB9 data set, using the average-kernel SOAP with a cutoff of 3 Å. Black lines correspond to using DFT geometries to predict CC energies for the DFT-optimized geometry. Using the DFT energies as a baseline and learning ΔDFT − CC = ECC − EDFT lead to a fivefold reduction of the test error compared to learning CC energies directly as the target property (CCDFT). The other curves correspond to using PM7-optimized geometries as the input to the prediction of CC energies of the DFT geometries. There is little improvement when learning the energy correction (ΔPM7 − CC) compared to direct training on the CC energies (CCPM7). However, using information on the structural discrepancy between PM7 and DFT geometries in the training set brings the prediction error down to 1 kcal/mol mean absolute error (MAE) (). (B) A sketch-map representation of the GDB9 (each gray point corresponding to one structure) highlights the importance of selecting training configurations to uniformly cover configuration space. The average prediction error for different portions of the map is markedly different when using a random selection (C) and FPS (D). The latter is much better behaved in the peripheral, poorly populated regions.