

Abstract
Almost 30 years ago it was first appreciated that anti-apoptotic B-cell lymphoma-2 (BCL-2) prevents the induction of apoptosis not only in malignant cells, but also in normal cellular lineages. This critical observation has rapidly evolved from merely identifying new BCL-2 family members to understanding how their biochemical interactions trigger the cell death process, and, more recently, to pharmacological inhibition of anti-apoptotic BCL-2 function in disease. Indeed, the proper regulation of apoptosis is important in many aspects of life including development, homeostasis, and disease biology. To better understand these processes, scientists have used many tools to assess the contribution of individual anti-apoptotic BCL-2 family members. This review will focus on the prominent roles for BCL-2 and other pro-survival family members in promoting the development of mammals during early embryogenesis, neurogenesis, and hematopoiesis.
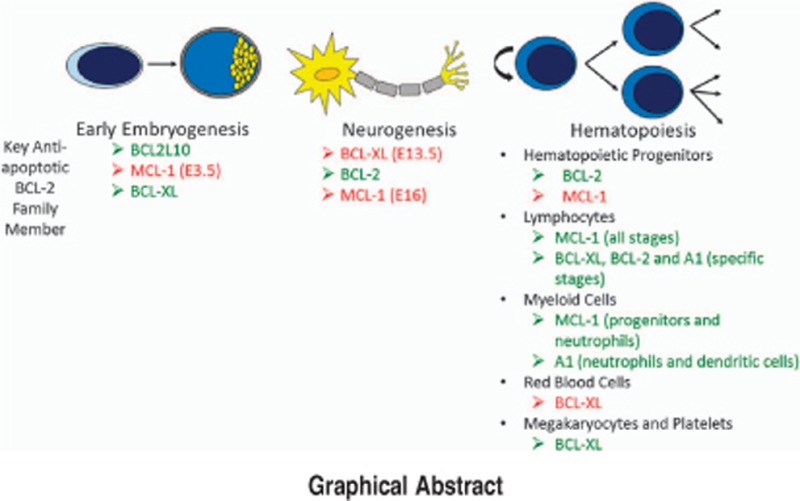
Facts
MCL-1 is essential for promoting development during early embryogenesis.
BCL-2 regulates kidney and melanocyte survival and restrains the action of pro-apoptotic BIM.
BCL-XL prevents the apoptotic death of post-mitotic immature neurons of the developing brain, spinal cord, and dorsal root ganglia.
During hematopoietic development, MCL-1 prevents cell death of hematopoietic progenitors, including hematopoietic stem cells, lymphocytes, and neutrophils.
BCL-XL, which is inhibited by the BH3-mimetic navitoclax, promotes the survival of mature platelets.
BCL-W functions to promote testicular survival, but otherwise appears redundant during development.
Open Questions
During embryogenesis do anti-apoptotic molecules play roles beyond inhibiting apoptosis?
Are extraneous, damaged, or obsolete cells in embryos cleared by non-apoptotic means?
What signals regulate the expression of anti-apoptotic molecules during nervous system development?
Which pro-survival member(s) are required to promote the survival of megakaryocyte-erythroid progenitors (MEPs) and mature macrophages?
Bcl-2 Family
The B cell lymphoma-2 (BCL-2) family of proteins are key regulators of programmed cell death or apoptosis.1 They are located predominantly on the mitochondria where the critical decision point, cytochrome c release, is regulated.2 Structurally, the family is characterized by evolutionarily conserved BCL-2 homology (BH) domains. It is divided into two major sub-groups, the anti-apoptotic proteins and the pro-apoptotic proteins. Anti-apoptotic proteins (eg, BCL-2, BCL-XL, MCL-1, BFL-1, BCL-W, and BCL2L10) possess BH 1–4 domains and they preserve outer mitochondrial membrane integrity by inhibiting the pro-apoptotic members (Table 1). The pro-apoptotic proteins can be either effector proteins with multiple BH domains (such as BAK, BAX, and BOK) or a wide number of BH3-only proteins, which are named for their single conserved BH3 domain (eg, BIM, PUMA, NOXA, BID, etc.). The fate of a cell depends on the balance of BCL-2 family protein levels through a dynamic process by which cellular signaling can modulate the expression of pro- or anti-apoptotic proteins to tip the equilibrium towards survival or death. When the balance favors death, the oligomerization of pro-apoptotic effector proteins on the outer mitochondrial membrane leads to mitochondrial outer membrane permeabilization and cytochrome c release. This triggers the activation of a cascade of caspases that cleave downstream substrates leading to cell death.
Table 1. Key anti-apoptotic BCL-2 family members and their roles in development.
| Key anti-apoptotic BCL-2 family protein | Common protein name | Mouse gene name | Early embryogenesis | Nervous system development | Hematopoiesis and immune system development |
|---|---|---|---|---|---|
| B-cell lymphoma 2 | BCL-2 | Bcl2 | 10–12 | 11, 12, 26, 28–33 | 11–13, 15, 41, 42, 74, 75 |
| B-cell lymphoma-extra large | BCL-XL | Bcl2l1 | 9, 10 | 9, 26, 28, 29, 34 | 9, 43, 66–68, 71–73, 79, 80 |
| Myeloid cell leukemia 1 | MCL-1 | Mcl1 | 8, 10 | 8, 27, 35, 36 | 44, 45, 50–55, 60–63, 72, 73 |
| BCL2 like 10 | BCL2L10, BCL-B or DIVA | Bcl2l10 | 6, 7, 10 | None reported | None reported |
| BCL2 related protein A1 | BFL-1 or A1 | Bcl2a1 | None reported | None reported | 57–59, 64, 65 |
| BCL2 like 2 | BCL-W | Bcl2l2 | None reported | No overt role | None reported |
This includes the common names, protein names, mouse genomic identity, and references to literature elucidating their roles in early embryogenesis, nervous system development, and hematopoiesis
Early Embryogenesis
Apoptotic death has long been considered to be crucial for proper development.3 For decades, studies of early vertebrate embryos detected the presence of apoptotic cells at various stages from the morula to the blastocyst. The blastocyst is composed of cells from the inner cell mass, which will become the embryo, as well as trophectoderm, which forms extra-embryonic tissues.4 It has long been postulated that the successful induction of apoptosis is critical to promote the appropriate differentiation and/or to eliminate damaged cells during the developmental process. Here we will explore the role for members of the BCL-2 family in modulating apoptosis during early embryonic development (Table 1).
During the earliest stages of embryogenesis (embryonic day 0–2, E0–2), gene expression in the embryo is largely controlled by the expression of maternally inherited mRNAs. By six to eight cell stage (E3), the embryonic genome becomes activated and the first signs of apoptosis are detectable.5 Analysis of early mRNA expression from early embryos indicates that the most prominent maternally inherited mRNA is BCL2L10 (also known as BCL-B or DIVA), which is highest in the mature oocyte, but decreases with division (Figure 1).6 It is hypothesized that BCL2L10 expression is necessary to prevent apoptosis during fertilization and through the earliest stages of embryogenesis. Despite the prominent expression of BCL2L10 in oocytes and early embryos, genetic ablation of Bcl2l10 in mice does not affect either oocyte survival or embryonic development, indicating that either BCL2L10 is dispensable for normal development or that other family members compensate for its absence.7 As maternal mRNA pools wane and embryonic gene expression initiates, the levels of anti-apoptotic MCL-1 and BCL-XL increase steadily (Figure 1). The importance of MCL-1 expression during early blastocyst was first appreciated from gene ablation studies. When Mcl-1 is deleted from the mouse germline it results in a peri-implantation embryonic lethality (E3.5).8 Unexpectedly, the loss of MCL-1 expression in mouse embryos was not accompanied by evidence of increased apoptosis, but rather it prevented the embryo from implanting in the uterine epithelium. This led to speculation that the loss of MCL-1 might actually render embryonic trophectoderm defective in adhesion causing defective implantation; however, it is unclear whether this defect is due to the loss of MCL-1’s anti-apoptotic function. Despite the concomitant induction of BCL-XL with MCL-1 in early embryos, gene ablation studies for Bcl2l1 (which encodes BCL-XL) have not revealed any overt defects in early embryogenesis (Figure 1).9 BCL-2 expression levels remain quite low during early embryo development and increase with development.10 Correspondingly, gene ablation studies have not implicated BCL-2 as playing a critical role in early embryogenesis.11, 12, 13, 14, 15 Thus, of the anti-apoptotic family members, MCL-1 has the most profound role in early embryonic development, but it is still unclear whether it plays roles beyond inhibiting cell death.
Figure 1.
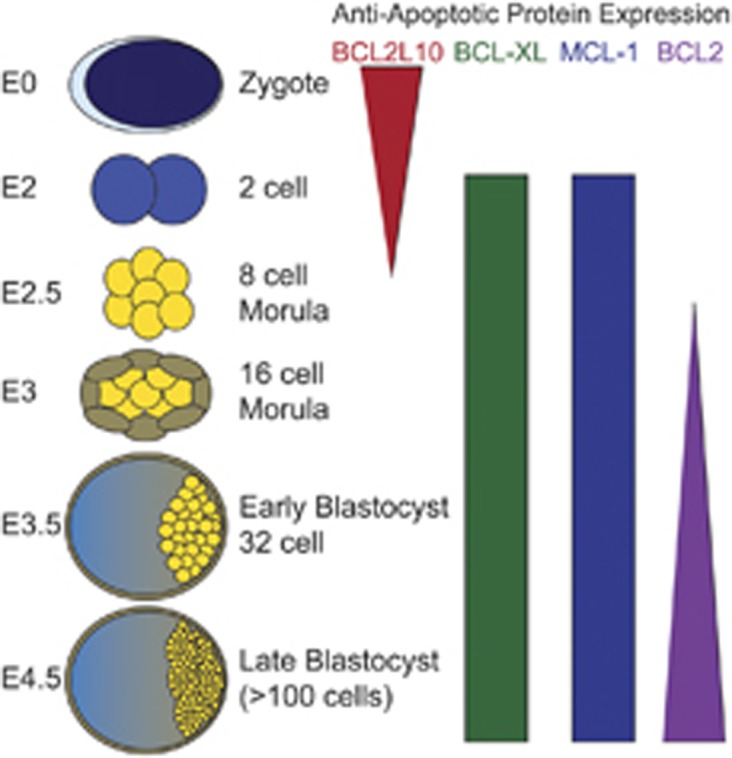
Anti-apoptotic expression levels during pre-implantation embryonic development. Fertilized eggs, known as zygotes, express proteins encoded for by maternally inherited mRNAs. The activation of gene expression from the embryonic genome occurs following E2. Early embryos rapidly divide becoming a mass of cells, known as a morula by E2.5. At the 16 cell morula stage, some of the cells (indicated in brown) destined to become extraembryonic tissues (eg, placenta, amniotic sac, etc.) start to segregate to the periphery of the embryo whereas the others (indicated in yellow) that will become the embryo coalesce. Around E3.5, the embryo undergoes blastogenesis where the trophoblasts (brown) that will become the extraembryonic endoderm form a thin layer around an open cavity (blastocoel). The cells that will become the embryo form the inner cell mass (yellow). Around E4–4.5 the blastocyst implants in the uterus. Gene expression analysis from isolated embryos has revealed the relative expression patterns of individual anti-apoptotic BCL-2 family members. Indicated are the anti-apoptotic molecules that have been implicated in pre-implantation development and the color-coded bars indicate their relative expression and timing by stage and the tapering indicates increased or decreased expression accordingly
Despite the almost canonical belief that apoptosis is essential to promote proper development, mice lacking both BAX and BAK, the critical effectors of apoptosis, have been successfully generated.16 Although Bax and Bak doubly-deficient mice are produced in sub-Mendelian ratios and suffer from a number of moderate to severe abnormalities as adults, early embryogenesis occurs without obvious defects despite the loss of both key apoptotic effectors.16 These data suggest that the induction of apoptosis may not actually be as critical as imagined for successful embryogenesis to occur. Alternatively, there could be compensation for loss of BAX and BAK that permits development by eliminating cells by other means. One such mechanism is linker cell death (LCD), a caspase-independent genetic program, initially identified in Caenorhabditis elegans, which kills cells during development.17 Phenotypically, LCD differs from apoptosis in that the chromatin remains uncondensed, nuclear membrane is crenellated, and intracellular organelles exhibit swelling.17 Indeed, in vertebrates some forms of developmental death exhibit LCD-like phenotypes.18 Therefore, it is tantalizing to speculate that non-apoptotic mechanisms, such as LCD, may have important roles in promoting proper development. We look forward to future efforts to untangle the contributions of different cell death mechanisms in early embryogenesis.
Nervous System Development
Nervous system development involves an enormous amount of cell death in many regions of the central and peripheral nervous system (PNS) during later embryonic and early postnatal stages.19 Most of this cell death is apoptotic and occurs during various stages of nervous system development including neurulation, synaptogenesis, and elimination of adult-generated central nervous system (CNS) cells. Importantly, wide-scale apoptosis during development, known as naturally occurring neuronal cell death (NOCD) regulates the progenitor population in the nervous system, selectively removes inaccurate neurons, and optimizes ‘size matching’ of neuronal populations to target size.20, 21, 22, 23, 24, 25 Imbalance in the extent of apoptosis is associated with various neurodevelopmental anomalies, neurodegenerative pathologies, and even tumorigenesis. Hence, BCL-2 family proteins are being investigated as therapeutic targets in diverse pathological neurobiological conditions.
B-cell lymphoma 2 (BCL-2)
The expression pattern of anti-apoptotic proteins in murine CNS has been determined mainly by immunohistochemistry and immunoblot (Table 2).26, 27 The induction of BCL-2 expression during early neurulation (E4.5–8) suggests that it has a role in preventing apoptosis at that stage. Once the neural tube is formed BCL-2 expression declines in the CNS, whereas high levels are maintained in the PNS.28, 29 BCL-2 is a promoter of cellular survival; therefore, it was not surprising that overexpression of BCL-2 under the control of the neuron-specific enolase promoter significantly increased the numbers of neurons in both the CNS and PNS in mice.30, 31 These data indicate that blocking apoptosis can rescue neuronal cells from developmental death, but they do not necessarily indicate which family member(s) are critical for preventing death under normal conditions. To address the specific contribution of individual anti-apoptotic BCL-2 family members, targeted gene ablation studies have been extremely informative (Table 1).
Table 2. Stages of nervous system development.
| Stages of nervous system development | Mouse age | Key features | Levels of pro-survival BCL-2 molecules |
|---|---|---|---|
| Early embryonic development | E4.5–8 | Onset of organogenesis and neurulation | BCL-2 ↑ |
| Period of NOCD | |||
| Early neural development | E8.5–11.5 | Neural plate and neural tube formation | BCL-2 ↑ MCL-1 ↑ |
| Early fetal life | E12–14.5 | Brain and spinal cord formation | BCL-2 ↓ BCL-XL ↑, MCL-1 ↑ |
| Later fetal life | E15.5–17.5 | Olfactory bulb formation and continued brain and spinal cord development | BCL-2 ↓ BCL-XL ↑, MCL-1 ↑ |
| Post-natal | P0–21 days | Synaptogenesis and astrogenesis | BCL-XL ↑, MCL-1 ↑ |
Abbreviation: NOCD, naturally occurring cell death
Stages of nervous system development with corresponding age of mice, along with their key features and correlation with key changes in expression levels of pro-survival BCL-2 family members. NOCD occurs from late embryonic to early post-natal stage
Developmentally, Bcl-2-deficient embryos do not exhibit any obvious phenotypic changes in neuronal development or aberrant signs of neuronal apoptosis.11, 12 However, immature sympathetic neurons from Bcl-2-deficient mice undergo cell death more rapidly upon withdrawal of nerve growth factor from their cultures.32 Thus, ex vivo BCL-2 regulates neuronal survival after trophic factor deprivation during the developmental window of NOCD. Interestingly, after birth the genetic deletion of Bcl-2 results in the loss of motor neurons of the facial nucleus, sensory neurons of dorsal root ganglia (DRGs) and sympathetic neurons of superior cervical ganglia.33 The delayed effects of BCL-2 loss in postnatal animals may indicate a more important role in post-natal development than during early neurogenesis. These observations are somewhat surprising considering BCL-2 expression levels are highest during early neurogenesis and wane postnatally.
BCL-XL
As BCL-2 expression wanes after neural tube formation (E10–11), BCL-XL expression increases and remains elevated throughout neuronal ontogeny with the highest levels in differentiating cells.28, 29 Genetic ablation of Bcl2l1, which encodes BCL-XL, results in embryonic death by E13.5 with substantial apoptosis occurring in both the developing nervous and hematopoietic systems.9 Notably, BCL-XL loss triggers the apoptotic death of post-mitotic immature neurons of the developing brain, spinal cord, and DRG, indicating that it is essential for promoting the survival of immature cells during development.9 However, in contrast to germline knockouts, a less severe phenotype was observed with a catecholaminergic-specific Bcl2l1 ablation.34 Conditional ablation of the Bcl2l1 gene in a subset of CNS neurons by the tyrosine hydroxylase-Cre resulted in viable mice, but the animals exhibited decreased numbers of catecholaminergic neurons when compared with wild-type mice, demonstrating that BCL-XL is required for the proper development of catecholaminergic cells. Furthermore, these mice were deficient in striatal dopamine production and had reduced total brain mass compared with controls.34 Despite the overt effects of BCL-XL loss, it is still unclear what signals regulate BCL-XL expression in the brain.
MCL-1
When the expression of MCL-1 in developing mouse brains during cortical neurogenesis (E11–E15.5) was assessed, high levels were detected in both proliferating neural precursor cells as well as post-mitotic neurons.27 In addition, MCL-1 expression has also been detected in the soma of sympathetic and DRG neurons in human tissues.35 Taken together, MCL-1 expression appears to be quite ubiquitous during neuronal development and maintenance. As described above, genetic ablation of Mcl-1 results in peri-implantation lethality at E3.5 necessitating conditional alleles to interrogate the function of MCL-1 in neurons.8 Conditional Mcl-1 mice have been used to study its role in many cellular lineages by driving Cre expression under the control of lineage specific promoters. Deletion of Mcl-1 by expression of Cre under the control of the Nestin promoter (specific for neural precursors) resulted in embryonic lethality (E16), with apoptosis observed in the Nestin-positive neural progenitors and Tuj1-positive (neuron-specific class III β-tubulin) newly committed neurons, indicating a prominent role for MCL-1 in survival of both of these populations.27 In cultured neurons, Mcl-1 deletion by adenoviral Cre-expression did not trigger immediate apoptosis, but rather sensitized neurons to acute DNA damaging insults such as campothecin.27
MCL-1 is a highly labile protein and therefore it is important to understand the mechanism by which MCL-1 protein expression is regulated. Using primary cultures it was revealed that MCL-1 is phosphorylated by glycogen synthase kinase 3 in KCI-deprived cerebellar granule neurons, a well characterized model of neuronal apoptosis. The MCL-1 phosphorylation promoted its interaction with the E3 ubiquitin-ligase, TRIM17, which catalyzed MCL-1’s ubiquitination and subsequent degradation.36 These data are an excellent example of how MCL-1 expression responds to cellular stimuli, allowing regulation of its survival function.
Hematopoiesis
The hematopoietic system is an excellent example of the need for homeostatic control over development and differentiation. All hematopoietic cells originate from hematopoietic stem cells (HSCs), which are capable of both self-renewal and the production of progeny that generate essential blood cell lineages (Figure 2).37 Many blood cell types, such as neutrophils, have relatively short life-spans and are constantly being removed and replaced. In many cases, the elimination of these obsolete, damaged, or defective cell types is controlled by apoptosis. Although apoptosis is important to prevent the inappropriate accumulation of cells, it is equally important that long-lived cell types and progenitor populations be protected and preserved, in some cases for the lifespan of the animal. Importantly, defective homeostasis in the hematopoietic system can lead to a number of pathological situations. For example, inappropriate cell death of hematopoietic progenitors can lead to immune deficiencies and anemias, whereas inappropriate survival can lead to dysplastic syndromes and contribute to malignancies. Therefore, understanding the contribution of members of the BCL-2 family to regulation of hematopoietic homeostasis has been the topic of intense research for many years (Table 1).
Figure 2.

Anti-apoptotic BCL-2 family member roles during hematopoiesis. All hematopoiesis arises from a common HSC that is capable of self-renewal as well as generating multi-potent progenitors (MPPs). Driven by cues, the MPP gives rise to CMPs and CLPs. CMPs produce two more progenitor populations, the MEP and the GMP. MEPs produce the megakaryocytes, which give rise to platelets, as well as the erythroid lineage. The GMPs give rise to the granulocytic lineages including neutrophils as well as monocytes, which become macrophages. CLPs give rise to B and T lymphocytes and natural killer (NK) cells. Indicated are the anti-apoptotic BCL-2 family members that have been demonstrated to be required at each stage or in each differentiation process. If multiple family members have been implicated, the one with a less prominent role is flanked by parentheses. Stages for which no family member has been identified to critically promote survival are indicated with a question mark (?). Arrows demonstrate the developmental pathways, but are not meant to indicate direct differentiation as in many cases intermediate developmental stages have been identified
During the earliest stages of embryogenesis, nutrients and oxygen are simply delivered by diffusion, but as the embryo gets larger the need for systemic delivery becomes necessary. The first orchestrated blood cells produced for this task are primitive erythroblasts that arise in the yolk sac (E7.5) and begin to circulate throughout the embryo along with yolk-sac derived myeloid cells. Little is known of the role of pro-survival BCL-2 family members in regulating primitive hematopoiesis nor is it clear whether intrinsic apoptosis is critical to the proper function of the primitive hematopoietic system. Primitive hematopoiesis is transient and these yolk sac-derived hematopoietic cells are soon replaced by definitive HSCs that arise from endothelial cells in the aorta-gonad-mesonephros (AGM) starting at E10.5.38 The AGM-derived HSCs then migrate to the fetal liver of developing mice starting at E12 where they continue to expand and give rise to differentiated definitive hematopoietic lineages. Throughout embryonic development these cells remain primarily in the fetal liver, but HSCs eventually migrate to the bone marrow by E17.5 where they primarily remain and give rise to differentiated blood cells throughout the life of the animal.39
Hematopoietic progenitors
HSC homeostasis is regulated by signaling events that occur in the bone marrow niche in adults.37 Normally, HSCs are quiescent and resist apoptotic stress, whereas cycling or mobilized HSCs are more sensitive to stress.40 A myriad of cell surface ligands and secreted growth factors have been implicated in promoting HSC viability. The specific action of these signals are diverse and include direct up-regulation of pro-survival molecules such as BCL-XL and MCL-1, as well as repression of pro-apoptotic modulators. The first evidence that HSC homeostasis was controlled by pro-survival members of the BCL-2 family came from experiments in which human BCL-2 was constitutively expressed under the control of the H2K promoter (encodes major histocompatibility complex I); this leads to the accumulation of HSCs numerically and enhanced their ability to both form colonies in culture and to reconstitute lethally irradiated recipients.41, 42 These data clearly indicated that increasing the balance of anti-apoptotic proteins can cause the accumulation of HSCs, but did not definitely define which pro-survival molecules were responsible for regulating cell death in the HSCs under normal physiological conditions. To gain insight into this question, gene ablation studies were performed to assess the contributions of specific molecules operating in HSC survival and function (Figure 2). Despite the ability of ectopic BCL-2 to augment HSC function, the germline deletion of Bcl-2 expression in mice did not lead to prominent defects in early hematopoietic progenitors as early hematopoiesis was essentially normal.11, 12 Similarly, germline ablation of Bcl2l1, which encodes BCL-XL, did not lead to deficiency of HSCs as chimeric mice could still give rise to definitive blood cell lineages.43 In contrast, hematopoietic-specific anti-apoptotic Mcl-1 ablation induced the rapid apoptosis of HSCs and resulted in a fatal bone marrow failure within three weeks after deletion.44 Indeed, MCL-1 is highly expressed in HSCs and its expression is in part triggered by the action of growth stimulating cytokines such as c-Kit ligand, also known as stem cell factor.44 Recent evidence demonstrated that even repressing MCL-1 expression, such as observed in heterozygous mice, hyper-sensitizes bone marrow progenitors to a number of stresses including 5-fluorouracil treatment and sub-lethal irradiation further emphasizing the important role for MCL-1 in protecting HSCs.45 These findings clearly indicate that MCL-1 is a critical modulator of HSC survival and highlights that an important consideration of effective and potent MCL-1 inhibitors might be their unanticipated sensitization of hematopoietic progenitors to stresses such as conventional chemotherapeutics.46
Lymphocytes
Both B and T lymphocytes arise from the common lymphocyte progenitor (CLP) and this differentiation is under tight homeostatic control (Figure 2). Evidence from conditional deletion in CLPs indicate that MCL-1 is the critical mediator of survival in these cells.44 Development from the CLP into both B and T lymphocytes depends on cell surface ligands and cytokine stimulation.47, 48, 49 These stimuli not only specify developmental fate, but promote survival by inducing the expression of anti-apoptotic genes such as MCL-1. As a result, conditional genetic ablation of Mcl-1 at virtually all stages of B- and T-cell ontogeny results in the rapid death of the deleted cells. During lymphocytic development, conditional deletion has revealed that both pro-B and pro-T cells require MCL-1 to promote their survival.50 In addition, naive, effector, T-regulatory, memory B and T cells, and NK cells are dependent on MCL-1 to promote their survival.50, 51, 52, 53, 54, 55 Despite the robust apoptosis induced upon Mcl-1 genetic ablation in effector and memory lymphocytes, co-ablation of pro-apoptotic BIM, the dominant pro-apoptotic molecule in lymphocytes, was unable to rescue T-lymphocyte survival.53
Notwithstanding the essential nature of MCL-1 for promoting survival at virtually all stages of lymphopoiesis, other anti-apoptotic BCL-2 family members have roles in promoting survival at specific stages of lymphoid development. For example, the loss of BCL-XL expression in immature (CD4+ CD8+) thymocytes increases their apoptosis and prevents efficient development of single-positive thymocytes in chimeric animals.43 Despite the normal early hematopoiesis in Bcl-2-deficient mice, within the first few weeks of life mature lymphocytes undergo massive apoptosis presumably due to the induction of endogenous pro-apoptotic stimuli that trigger the expression of pro-apoptotic BIM. This has been inferred since co-deletion of both Bcl-2 and Bcl2l11 (which encodes BIM) spares the induction of lymphocyte death.14 Evaluation of the contribution of BCL2A1, which in humans encodes a single protein known as BFL-1, has been challenging, as in mice the gene locus underwent quadruplication leading to 4 loci (A1-a, b, c, d of which c is a pseudogene) and therefore required multiple gene targeting events to effectively ablate all species.56 To bypass this conundrum, a transgenic, inducible short-hairpin RNA (shRNA) approach was used to express a shRNA that targets all A1 species in mice.57 This approach revealed that despite efficient knockdown, silencing of all A1 species does not alter T-lineage development or function. In contrast, silencing of the A1 species in the B-cell lineage revealed that mature B lymphocytes spontaneously underwent BIM-dependent apoptosis indicating that A1 has an important role in their survival.57 Recently, a conventional knockout of all three functional A1 alleles (A1-a, b and d) revealed that complete ablation of A1 resulted in only minor defects in the hematopoietic compartment, even when the mice were challenged with viral infection.58, 59 In fact, only conventional dendritic cells were substantially affected by A1 loss.59 Taken together, these data convey a complex portrait of how the anti-apoptotic molecules maintain survival during lymphopoiesis. Although MCL-1 is critical at multiple stages of lymphopoiesis, other BCL-2 family members clearly have important roles at specific stages or contribute to survival.
Myeloid cells
Myeloid cells, which contribute to the innate immune system, arise from HSC progenitors in the bone marrow (Figure 2). The common myeloid progenitor (CMP) gives rise to the granulocyte–monocyte progenitor (GMP). These progenitor populations are incapable of life-long self-renewal and require constant replenishment. They respond to growth factors, known as colony stimulating factors (CSFs) that drive their proliferation and differentiation into immature and mature myeloid cells. Conditional deletion studies have revealed that both the CMP and GMP require MCL-1 expression to promote their survival in mouse bone marrow and it has been demonstrated that CSF stimulation directly induces the expression of MCL-1.44, 60 Mature neutrophils are rapidly turned over and require constant replacement from the GMP. Although loss of BCL-2 and BCL-XL have not been reported to affect the number of peripheral neutrophils in mice, conditional ablation of Mcl-1 results in a severe blockade in neutrophil development with massive apoptosis of immature cells occurring in the bone marrow.61, 62 This death was specific to the mature neutrophil population, as monocytes and macrophages survived despite the conditional deletion of Mcl-1 in these cells. Although normal monocytes and macrophages do express MCL-1, the loss of MCL-1 expression did not trigger their death in vivo, probably due to the compensatory upregulation of BCL-XL and A1 expression.61, 63 Despite their survival in vivo, Mcl-1-deficient macrophages exhibited functional defects including a propensity to succumb to apoptosis induced by phagocytosis of bacteria.62 Similarly, either genetic ablation of A1-a (the most prominent A1 species expressed in myeloid cells) or expression of an inducible shRNA that silences all A1 species sensitized cultured neutrophils to spontaneous induction of apoptosis and reduced peripheral neutrophil numbers in mice.64, 65 Furthermore, the deletion of the functional A1-a,b and d alleles in mice impaired the survival of conventional dendritic cells.59 Thus, during myelopoiesis MCL-1 has essential roles in progenitor populations and in neutrophil differentiation, but in the monocytic lineage MCL-1 appears to be dispensable and can be compensated for by other pro-survival molecules.
Red blood cells
Erythrocytes and megakaryocytes arise from the megakaryocyte-erythroid progenitor (MEP) which, similar to the GMP, arise from the CMP (Figure 2). Unlike other progenitors, the specific pro-survival mediators for MEP are yet to be elucidated. Developmentally, MEPs give rise to proerythroblasts that develop into erythroblasts and, following extrusion of the nucleus, become reticulocytes. The final stages of erythroid maturation include elimination of all cellular organelles including ribosomes and mitochondria. Mature erythrocytes, which carry oxygen to the tissues, are relatively short-lived and under tight homeostatic control. This regulation is largely under control of the action of erythropoietin (EPO), which acts in part by inducing the expression of BCL-XL.66 These data correlate with the observation that genetic ablation of Bcl2l1 (which encodes BCL-XL) results in embryonic lethality in part due to a lack of mature erythrocytes, but not erythroid progenitors.9 Similarly, conditional deletion of Bcl2l1 in the hematopoietic lineage resulted in severe anemia with the spontaneous cell death of reticulocytes and compensatory extramedullary erythropoiesis of immature cells.67 Surprisingly, neither co-deletion of Bcl2l11 (encoding BIM) nor Bbc3 (encoding PUMA) were able to rescue the loss of erythrocytes triggered by deficiency of BCL-XL expression indicating that other or multiple BH3-only family members must be triggering the cell death in the erythroid lineage.68 To date, no other pro-survival proteins have been identified to have a key role in erythropoiesis, but it is possible that other family members may have still unrecognized roles mediating survival during the earlier stages of erythropoiesis.
Megakaryocytes and platelets
Megakaryocytes arise from the MEP and give rise to platelets, also known as thrombocytes, which are critical to clotting and coagulation. Platelets are anuclear cellular fragments that arise from multinucleated megakaryocytes and have a short lifespan and are under tight homeostatic control. The regulation of megakaryocyte survival is complex and appears to depend on multiple anti-apoptotic family members. Thrombopoeitin (TPO) is an important growth factor that induces the differentiation of megakaryocytes and promotes megakaryocyte survival.69, 70 Similar to EPO, TPO also induces the induction of BCL-XL expression.71 Although conditional loss of BCL-XL significantly reduced the numbers of mature megakaryocytes in mice, when combined with the deletion of Mcl-1 the numbers of megakaryocytes are even more drastically decreased resulting in the death of the mice from leaky vasculature and internal hemorrhaging.72, 73 Interestingly, the loss of Mcl-1 alone in megakaryocytes does not lead to overt loss of megakaryocyte numbers indicating that the function of MCL-1 is subservient to that of BCL-XL in this lineage.73 In megakaryocytes, the loss of BCL-2 does not affect megakaryocyte survival or function.74
The observation that platelets can undergo apoptosis was unexpected due to their being cellular fragments that lack a nucleus, but indeed platelets express members of the BCL-2 family and exhibit features of apoptosis such as phosphatidylserine exposure before their clearance in mice.75, 76, 77 Despite these early observations, it was not until the development of the BH3-mimetic agent ABT-737, which antagonizes BCL-2, BCL-XL, and BCL-W, that an unanticipated rapid, but transient thrombocytopenia was observed when animals were treated.78 This initial observation spurred interrogation into how ABT-737 and its orally bioavailable derivative ABT-263 (navitoclax) induce platelet death by inhibiting BCL-XL function and triggering a BAK-dependent apoptosis in mature platelets.79, 80 Coincidentally, a genome wide N-ethyl-N-nitrosourea mouse screen identified mutants exhibiting a heritable thrombocytopenia that were mapped to the Bcl2l1 locus and encoded destabilized BCL-XL proteins.79 Taken together, it is clear that both genetic loss of BCL-XL or pharmacological inhibition result in the rapid induction of apoptosis in mature platelets. As a result, the dose limiting toxicity of navitoclax is ‘on target’ thrombocytopenia. In contrast, treatment of animals with the selective BCL-2 inhibitor ABT-199 (venetoclax) does not affect platelet survival at all, further emphasizing the importance of BCL-XL in this process.81
Other Developmental Roles
Mice lacking BCL-2 suffer from a number of other developmental issues including polycystic kidney disease, which is observable from mice as young as 10 days and eventually results in the death of all knockout mice by 4–16 weeks of age.11, 12, 13 The onset of kidney dysfunction is associated with increased apoptosis in the kidney interstitium.11, 12, 13 In addition, Bcl-2-deficient mice, while born with normal hair color, rapidly turn to a gray coat color.11, 12, 13 This loss of pigmentation is due to the accelerated loss of melanocytes that produce melanin.15 By 5–6 weeks at the second hair follicle cycle, the hair of Bcl-2-deficient mice possess as little melanin as that found in albino mice.15 Interestingly, almost all of the developmental phenotypes (eg, hair color loss) associated with the loss of BCL-2 expression can be rescued by the loss pro-apoptotic BIM expression.14 In fact, even loss of only a single Bcl2l11 (encoding BIM) allele can restore normal kidney development and health of the mice.14 These data clearly indicate a prominent role for BIM in modulating the apoptosis during these developmental stages.
Mice lacking BCL-W (encoded for by the Bcl2l2 gene) display progressive testicular degeneration with apoptosis of Sertoli cells occurring soon after weaning.82, 83 Furthermore, Leydig cell death is detectable starting at 3 months of age. The consequence of this apoptosis is the complete defacement of the testes and sterility of male mice. Despite this profound effect on spermatogenesis, the BCL-W-deficient mice are otherwise normal in other lineages.83 Thus, BCL-W has a very selective role in promoting spermatogenesis.
When Mcl-1 is inducibly deleted in adult mice or when Mcl-1 is ablated using lineage-specific ablation from late embryogenesis it results in a fatal cardiomyopathy.84, 85 In these mice, the loss of MCL-1 was associated with a progressive decline in cardiac output and extensive cardiac fibrosis prior to heart failure and these phenotypes could be substantially reverted by co-deletion of pro-apoptotic effectors Bax and Bak.84 These data clearly demonstrate the critical role for MCL-1 in promoting the survival of cardiomyocytes.
Conclusions and Perspectives
The discovery of BCL-2 was a watershed event that has revolutionized our understanding of many aspects of normal and disease biology. As this event, knowledge of the roles and contributions of anti-apoptotic BCL-2 family members to the development of mammals has expanded dramatically (Table 1). Not only have multiple pro-survival family members been identified, but we now know that many of these are important factors during development and in disease biology. In many cases, the use of gene targeting has revolutionized our understanding of precisely which pro-survival family members are needed to maintain specific cell lineages during development. Although these studies have been definitive in assessing the requirement of individual family members, much is still to be learned about precisely how each family member is regulated and whether they have functions beyond merely inhibiting apoptosis. As pharmacologic inhibitors are advanced in the clinic, our basic understanding of the developmental requirement for individual anti-apoptotic BCL-2 family members will be important to foresee potential toxicities. After so much knowledge has been gained, we are fortunate to stand on the threshold of seeing the translation of this basic biological knowledge to the clinic where it will help treat malignancies; however, there is still much to be learned and new challenges, such as neurodegeneration, await.
Footnotes
Edited by F Pentimalli
The authors declare no conflict of interest.
References
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell 2010; 37: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 1993; 53: 4701–4714. [PubMed] [Google Scholar]
- Fabian D, Koppel J, Maddox-Hyttel P. Apoptotic processes during mammalian preimplantation development. Theriogenology 2005; 64: 221–231. [DOI] [PubMed] [Google Scholar]
- Hardy K. Cell death in the mammalian blastocyst. Mol Hum Reprod 1997; 3: 919–925. [DOI] [PubMed] [Google Scholar]
- Hardy K. Apoptosis in the human embryo. Rev Reprod 1999; 4: 125–134. [DOI] [PubMed] [Google Scholar]
- Boumela I, Assou S, Haouzi D, Dechaud H, Ait-Ahmed O, Hamamah S. Developmental regulated expression of anti- and pro-apoptotic BCL-2 family genes during human early embryonic development. Curr Med Chem 2014; 21: 1361–1369. [DOI] [PubMed] [Google Scholar]
- Russell HR, Lee Y, Miller HL, Zhao J, McKinnon PJ. Murine ovarian development is not affected by inactivation of the bcl-2 family member diva. Mol Cell Biol 2002; 22: 6866–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 2000; 14: 23–27. [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x- deficient mice. Science 1995; 267: 1506–1510. [DOI] [PubMed] [Google Scholar]
- Metcalfe AD, Hunter HR, Bloor DJ, Lieberman BA, Picton HM, Leese HJ et al. Expression of 11 members of the BCL-2 family of apoptosis regulatory molecules during human preimplantation embryo development and fragmentation. Mol Reprod Dev 2004; 68: 35–50. [DOI] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993; 75: 229–240. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2 alpha beta in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA 1994; 91: 3700–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T et al. bcl-2 deficiency in mice leads to pleiotropic abnormalities: accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res 1995; 55: 354–359. [PubMed] [Google Scholar]
- Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell 2001; 1: 645–653. [DOI] [PubMed] [Google Scholar]
- Yamamura K, Kamada S, Ito S, Nakagawa K, Ichihashi M, Tsujimoto Y. Accelerated disappearance of melanocytes in bcl-2-deficient mice. Cancer Res 1996; 56: 3546–3550. [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher LM, Shaham S. Non-apoptotic cell death in animal development. Cell Death Differ 2017; 24: 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaginuma H, Shiraiwa N, Shimada T, Nishiyama K, Hong J, Wang S et al. Caspase activity is involved in, but is dispensable for, early motoneuron death in the chick embryo cervical spinal cord. Mol Cell Neurosci 2001; 18: 168–182. [DOI] [PubMed] [Google Scholar]
- Dekkers MP, Nikoletopoulou V, Barde YA. Cell biology in neuroscience: death of developing neurons: new insights and implications for connectivity. J Cell Biol 2013; 203: 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR et al. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature 2005; 435: 1244–1250. [DOI] [PubMed] [Google Scholar]
- Haydar TF, Kuan CY, Flavell RA, Rakic P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex 1999; 9: 621–626. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci 2000; 23: 291–297. [DOI] [PubMed] [Google Scholar]
- Clarke PG, Posada A, Primi MP, Castagne V. Neuronal death in the central nervous system during development. Biomed Pharmacother 1998; 52: 356–362. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Pallas SL. Control of cell number in the developing mammalian visual system. Prog Neurobiol 1989; 32: 207–234. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci 1991; 14: 453–501. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Mai JK, Zapata JM, Ashwell KW, Schendel SL, Reed JC et al. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ 2002; 9: 145–157. [DOI] [PubMed] [Google Scholar]
- Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci 2008; 28: 6068–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe-Dohmae S, Harada N, Yamada K, Tanaka R. Bcl-2 gene is highly expressed during neurogenesis in the central nervous system. Biochem Biophys Res Commun 1993; 191: 915–921. [DOI] [PubMed] [Google Scholar]
- Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. Bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development 1994; 120: 301–311. [DOI] [PubMed] [Google Scholar]
- Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell 1993; 73: 295–307. [DOI] [PubMed] [Google Scholar]
- Farlie PG, Dringen R, Rees SM, Kannourakis G, Bernard O. bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA 1995; 92: 4397–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science 1992; 258: 302–304. [DOI] [PubMed] [Google Scholar]
- Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M et al. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron 1996; 17: 75–89. [DOI] [PubMed] [Google Scholar]
- Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM. Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci 2005; 25: 6721–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K et al. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol 1995; 146: 1309–1319. [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Mora S, Mojsa B, Robbins I, Lassot I, Desagher S. Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ 2013; 20: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008; 132: 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1994; 1: 291–301. [DOI] [PubMed] [Google Scholar]
- Delassus S, Cumano A. Circulation of hematopoietic progenitors in the mouse embryo. Immunity 1996; 4: 97–106. [DOI] [PubMed] [Google Scholar]
- Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008; 2: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med 2000; 191: 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domen J, Gandy KL, Weissman IL. Systemic overexpression of BCL-2 in the hematopoietic system protects transgenic mice from the consequences of lethal irradiation. Blood 1998; 91: 2272–2282. [PubMed] [Google Scholar]
- Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW et al. Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci USA 1995; 92: 4763–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307: 1101–1104. [DOI] [PubMed] [Google Scholar]
- Delbridge AR, Opferman JT, Grabow S, Strasser A. Antagonism between MCL-1 and PUMA governs stem/progenitor cell survival during hematopoietic recovery from stress. Blood 2015; 125: 3273–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016; 538: 477–482. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Liu ZJ, Kawahara A, Minami Y, Yamada K, Tsujimoto Y et al. Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic cell proliferation. Cell 1995; 81: 223–231. [DOI] [PubMed] [Google Scholar]
- Akbar AN, Borthwick NJ, Wickremasinghe RG, Panayoitidis P, Pilling D, Bofill M et al. Interleukin-2 receptor common gamma-chain signaling cytokines regulate activated T cell apoptosis in response to growth factor withdrawal: selective induction of anti-apoptotic (bcl-2, bcl-xL) but not pro- apoptotic (bax, bcl-xS) gene expression. Eur J Immunol 1996; 26: 294–299. [DOI] [PubMed] [Google Scholar]
- von Freeden-Jeffry U, Solvason N, Howard M, Murray R. The earliest T lineage-committed cells depend on IL-7 for Bcl-2 expression and normal cell cycle progression. Immunity 1997; 7: 147–154. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003; 426: 671–676. [DOI] [PubMed] [Google Scholar]
- Dzhagalov I, Dunkle A, He YW. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol 2008; 181: 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikstrom I, Carotta S, Luthje K, Peperzak V, Jost PJ, Glaser S et al. Mcl-1 is essential for germinal center formation and B cell memory. Science 2010; 330: 1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P, Koss B, Opferman JT, Hildeman DA. Mcl-1 antagonizes Bax/Bak to promote effector CD4(+) and CD8(+) T-cell responses. Cell Death Differ 2013; 20: 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol 2013; 14: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun 2014; 5: 4539. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Hamasaki A, Negishi I, Loh DY, Sendo F, Nakayama K et al. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int Immunol 1998; 10: 631–637. [DOI] [PubMed] [Google Scholar]
- Sochalska M, Ottina E, Tuzlak S, Herzog S, Herold M, Villunger A. Conditional knockdown of BCL2A1 reveals rate-limiting roles in BCR-dependent B-cell survival. Cell Death Differ 2016; 23: 628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D et al. The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ 2017; 24: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE et al. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ 2017; 24: 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JR, Wang JM, Lee SF, Peng HW, Lin YH, Chou CH et al. mcl-1 is an immediate-early gene activated by the granulocyte- macrophage colony-stimulating factor (GM-CSF) signaling pathway and is one component of the GM-CSF viability response. Mol Cell Biol 1998; 18: 4883–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhagalov I St, John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 2007; 109: 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 2009; 113: 2805–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 2014; 41: 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlofsky A, Weiss LM, Kawachi N, Prystowsky MB. Deficiency in the anti-apoptotic protein A1-a results in a diminished acute inflammatory response. J Immunol 2002; 168: 1840–1846. [DOI] [PubMed] [Google Scholar]
- Ottina E, Grespi F, Tischner D, Soratroi C, Geley S, Ploner A et al. Targeting antiapoptotic A1/Bfl-1 by in vivo RNAi reveals multiple roles in leukocyte development in mice. Blood 2012; 119: 6032–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz C, Benet I, Richard C, Badia B, Andreu EJ, Prosper F et al. Antiapoptotic protein Bcl-x(L) is up-regulated during megakaryocytic differentiation of CD34(+) progenitors but is absent from senescent megakaryocytes. Exp Hematol 2001; 29: 728–735. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Claudio E, Rucker EB 3rd, Riedlinger G, Broussard C, Schwartzberg PL et al. Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development 2000; 127: 4949–4958. [DOI] [PubMed] [Google Scholar]
- Delbridge AR, Aubrey BJ, Hyland C, Bernardini JP, Di Rago L, Garnier JM et al. The BH3-only proteins BIM and PUMA are not critical for the reticulocyte apoptosis caused by loss of the pro-survival protein BCL-XL. Cell Death Dis 2017; 8: e2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen SE, Borge OJ, Ramsfjell V, Cui L, Cardier JE, Veiby OP et al. Thrombopoietin, a direct stimulator of viability and multilineage growth of primitive bone marrow progenitor cells. Stem Cells 1996; 14(Suppl 1): 173–180. [DOI] [PubMed] [Google Scholar]
- Borge OJ, Ramsfjell V, Veiby OP, Murphy MJ Jr., Lok S, Jacobsen SE. Thrombopoietin, but not erythropoietin promotes viability and inhibits apoptosis of multipotent murine hematopoietic progenitor cells in vitro. Blood 1996; 88: 2859–2870. [PubMed] [Google Scholar]
- Kirito K, Watanabe T, Sawada K, Endo H, Ozawa K, Komatsu N. Thrombopoietin regulates Bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J Biol Chem 2002; 277: 8329–8337. [DOI] [PubMed] [Google Scholar]
- Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR et al. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med 2011; 208: 2017–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrincat MA, Josefsson EC, James C, Henley KJ, Ellis S, Lebois M et al. Mcl-1 and Bcl-xL coordinately regulate megakaryocyte survival. Blood 2012; 119: 5850–5858. [DOI] [PubMed] [Google Scholar]
- Debrincat MA, Pleines I, Lebois M, Lane RM, Holmes ML, Corbin J et al. BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis 2015; 6: e1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanags DM, Orrenius S, Aguilar-Santelises M. Alterations in Bcl-2/Bax protein levels in platelets form part of an ionomycin-induced process that resembles apoptosis. Br J Haematol 1997; 99: 824–831. [DOI] [PubMed] [Google Scholar]
- Brown SB, Clarke MC, Magowan L, Sanderson H, Savill J. Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. A caspase-independent cell clearance program. J Biol Chem 2000; 275: 5987–5996. [DOI] [PubMed] [Google Scholar]
- Pereira J, Soto M, Palomo I, Ocqueteau M, Coetzee LM, Astudillo S et al. Platelet aging in vivo is associated with activation of apoptotic pathways: studies in a model of suppressed thrombopoiesis in dogs. Thromb Haemost 2002; 87: 905–909. [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681. [DOI] [PubMed] [Google Scholar]
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173–1186. [DOI] [PubMed] [Google Scholar]
- Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 2007; 14: 943–951. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19: 202–208. [DOI] [PubMed] [Google Scholar]
- Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD et al. Testicular degeneration in Bclw-deficient mice. Nat Genet 1998; 18: 251–256. [DOI] [PubMed] [Google Scholar]
- Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA 1998; 95: 12424–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 2013; 27: 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 2013; 27: 1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]