

SUMMARY
Biased agonism has been proposed as a means to separate desirable and adverse drug responses downstream of G protein-coupled receptor (GPCR) targets. Herein we describe structural features of a series of mu opioid receptor (MOR)-selective agonists that preferentially activate receptor to couple to G proteins or to recruit βarrestin proteins. By comparing relative bias for MOR-mediated signaling in each pathway, we demonstrate a strong correlation between the respiratory suppression/antinociception therapeutic window in a series of compounds spanning a wide range of signaling bias. We find that βarrestin-biased compounds, such as fentanyl, are more likely to induce respiratory suppression at weak analgesic doses, while G protein signaling-bias broadens the therapeutic window, allowing for antinociception in the absence of respiratory suppression.
Introduction
Opioids such as morphine and fentanyl are highly efficacious for the treatment of severe pain (Melnikova, 2010); however, the number of deaths due to overdose caused by respiratory distress have drastically increased over the past decade due to the misuse of prescription and illicit narcotics (Frank and Pollack, 2017; Rudd et al., 2016). Both the analgesic and respiratory suppressive effects of opioids are due the activation of the mu opioid receptor (MOR) (Dahan et al., 2001; Matthes et al., 1996). As a G protein-coupled receptor (GPCR), the MOR also interacts with βarrestins, scaffolding proteins that serve to regulate or facilitate subsequent GPCR signaling. In studies spanning more than a decade, researchers have shown that the interaction between MOR and βarrestin2 may drive many of the unwanted side effects of MOR activation (Bohn et al., 2000; Bohn et al., 1999; Bu et al., 2015; Li et al., 2009; Raehal, 2011; Raehal and Bohn, 2005). βArrestin2-KO mice, for example, display enhanced and prolonged morphine-induced antinociception yet are protected from morphine-induced respiratory suppression (Bohn et al., 1999; Raehal et al., 2005). These findings suggest that activating the MOR without engaging βarrestin2 regulation may be critically important for developing safer opioid analgesics.
A recent development in pharmacological theory and practice is the concept that the structure of a GPCR ligand may be systematically modified to confer alternative receptor conformations upon binding, each displaying a unique pattern of activation of intracellular signaling cascades (Rankovic et al., 2016; Urban et al., 2007). This concept of functional selectivity, or biased agonism, can be quantified by comparing drug potency and efficacy in cell-based signaling assays to the performance of a reference agonist (a compound that fully activates the system, and thus, defines the full potential of what one might hope to measure in the assay). Application of the operational model, described by Black and Leff (Black and Leff, 1983), allows one to simultaneously compare the relative potency and efficacy of a test agonist to the reference agonist, allowing for normalization within an assay, (derivation of the parameter: ΔLog τ/KA). After normalization, the performance of the compound can then be compared to its performance within another assay. The result of the comparison can be calculated as a “bias factor,” which essentially defines the extent of difference in relative agonist activity between two assays (the bias factor is 10^ΔΔLogτ/KA(assay1-assay2)) (Kenakin et al., 2012). The higher the bias factor, the greater the separation between an agonist’s performance in the two assays, relative to the performance of the reference agonist.
Biased agonism at the MOR is a promising avenue for therapeutic development, as late phase clinical studies are demonstrating encouraging effects of biased agonism in human patients. TRV-130, or Oliceridine®, the clinical candidate from Trevena, Inc., has shown efficacy in providing pain relief with modest improvement in preventing respiratory events (Singla et al., 2016; Soergel et al., 2014). In early preclinical studies, TRV-130 was shown to be biased towards activating G protein-mediated inhibition of adenylyl cyclase while displaying only marginally detectable signaling in βarrestin2 recruitment assays (DeWire et al., 2013). Applying analysis based on the operational model and taking account the relative affinity of the agonist, the group assigned a “bias factor” of 3 to TRV-130, as an indicator of its preference for the G protein signaling pathway over the βarrestin pathway. Studies in rodents showed that at certain doses, TRV130 could induce antinociception in the rodent thermal pain tests without respiratory suppression; however, comparisons between ED50 values and a quantified assessment of therapeutic window was not determined. Clinical trials reveal that the compound serves as a potent analgesic in humans; however, in human patients the therapeutic window is not as broad as originally anticipated (Singla et al., 2016; Soergel et al., 2014). The question remains as to whether it is simply enough to have biased signaling or if the degree of bias (i.e., the magnitude of the bias factor) will impact the separation of analgesia and respiratory side effects.
In addition to TRV-130, other recent examples of G protein-biased MOR agonists have emerged in the preclinical literature. While no calculation of the degree of bias that each of these individual compounds possess was provided, promising results were obtained showing that they could induce antinociception with fewer side effects in mice (Kruegel et al., 2016; Manglik et al., 2016). However, there has not been a comprehensive evaluation of the contribution of the degree of bias to the separation of the therapeutic window; nor have there been reports on agonists that preferentially recruit βarrestin2 over G protein signaling pathways. In this study, we show, in a series of new, but structurally related compounds, how agonists can be driven to promote one pathway over another. We also present preclinical studies in mice that establish a direct correlation of increasing “bias factor” with an improved separation of therapeutic benefit from respiratory side effects.
RESULTS
Development of MOR agonists with functional selectivity between G protein signaling βarrestin2 recruitment
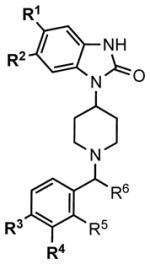
In order to fully address whether the degree of signaling bias effects the therapeutic window, we developed a series of compounds with a piperidine core structure, as shown in Table 1 and Figure S1. While distantly related compounds had been shown to be opioid agonists (e.g., bezitramide) (Janssen et al., 1971), no studies of how substituent modifications may effect bias have emerged (Table 1). As shown with a subset of the series, several of these compounds have high affinity for MOR (0.2 – 3.0 nM) and are highly selective for MOR over other opioid receptors (kappa (KOR) or delta (DOR) opioid receptors) as shown by radioligand competition binding assays (Table S1). While some affinity at KOR and DOR was detected, subsequent assays show no functional effect on these receptors for any of the compounds; further, no functional impact on the opioid-like receptor (nociception receptor, NOP) could be detected (Figure S2). MOR selectivity is therefore conferred, which is a very important consideration when studying biased agonist effects in vivo.
Table 1. Structural features influencing GTPγS binding and βarrestin2 recruitment.
Potency (EC50) and efficacy (EMAX, percent of DAMGO) values from GTPγS binding assays performed on CHO-hMOR membranes and βarrestin2 recruitment determined by EFC βarrestin2 assay with the U2OS-βarrestin2-hMOR-PathHunter cells. Data are presented as mean ± S.E.M. of 3 or more assays run in duplicate or triplicate. See also: Figure S1 for chemical synthesis; Figure S2 for counter-screens against other opioid receptors; and Table S1 for binding affinities for the compounds below the dotted line.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Agonist | Entry | Substituentsa | GTPγS binding | βarrestin2 recruitment | ||||||
|
|
|
|
||||||||
| R1 | R2 | R3 | R4 | EC50 nM | EMAX % | EC50 nM | EMAX % | |||
|
| ||||||||||
|
DAMGO | 33 ± 1.4 | 100 | 229 ± 12 | 100 | |||||
| Morphine | 64 ± 4.2 | 81 ± 1 | 372 ± 20 | 24 ± 1 | ||||||
| Sufentanil | 1.3 ± 0.18 | 78 ± 1 | 1.5±0.6 | 73±11 | ||||||
| Fentanyl | 43 ± 9.7 | 80 ± 5 | 53 ± 6.8 | 60 ± 2 | ||||||
|
| ||||||||||
| (±) SR-8595 | 1 | H | H | H | Me | 102 ± 10 | 89 ± 7 | 447 ± 52 | 67 ± 3.8 | |
| (±) SR-11065 | 2 | H | H | Cl | Me | 16 ± 1.2 | 97 ± 7 | 253 ± 88 | 76 ± 8.8 | |
| SR-20382 | 3 | H | H | Cl | H | 563 ± 104 | 62 ± 4 | 7656 ± 1469 | 67 ± 9.8 | |
| SR-20437 | 4 | Cl | Cl | H | H | 171 ± 24 | 60 ± 5 | 1092 ± 273 | 24 ± 3.6 | |
|
| ||||||||||
| SR-17018 | 5 | Cl | Cl | Cl | H | 97 ± 13 | 75 ± 4 | >10,000 | 10 ± 6c | |
| SR-15099 | 6 | Cl | Cl | Br | H | 155 ± 11 | 81 ± 4 | >10,000 | 3 ± 1c | |
| (±) SR-14968 | 7 | Cl | Cl | Br | Me | 8.9 ± 3.8 | 92 ± 1 | 2438 ± 710 | 64 ± 5 | |
| (±) SR-14969a | 8 | Cl | Cl | Cl | Me | 28 ± 7.7 | 88 ± 2 | 2949 ± 789 | 81 ± 6 | |
| SR-15098a | 9 | Cl | Cl | Cl | H | 179 ± 24 | 68 ± 4 | >10,000 | 12 ± 5c | |
| (±) SR-11501 | 10 | H | H | b | Me | 106 ± 9.1 | 70 ± 1 | 374 ± 60 | 59 ± 2 | |
|
| ||||||||||
R5 = F;
(R3 + R4) = (-OCH2CH2O-); for all other compounds, R4 = R5 = H;
percent of maximum stimulation at the 10 μM concentration is presented rather than EMAX.
Functional activity at human MOR was characterized using cell-based assays designed to measure G protein signaling or βarrestin2 recruitment in comparison to the enkephalin analogue, [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin (DAMGO), as the reference agonist (Figure 1, Table 2). Initially, 35S-GTPγS binding assays were performed in membranes from CHO-hMOR cells (Figure 1A), while a commercially available enzyme fragment complementation (EFC) assay was used to assess βarrestin2 recruitment (Figure 1C). The compounds performed as partial or full agonists in the GTPγS binding assay with a range of potencies between 9 – 563 nM. Their ability to induce βarrestin2 recruitment to the MOR varied more substantially, with some compounds, such as SR-15098, SR-15099 and SR-17018, revealing no significant efficacy in the βarrestin2 EFC assay until the 10 μM concentration.
Figure 1. SR compounds are potent activators of GTPγS binding, but have differential βarrestin2 signaling profiles at the human MOR.

(A–C) Cell-based assays assessing (A) stimulation of GTPγS binding in membranes and (B) inhibition of forskolin-stimulated cAMP accumulation in CHO-hMOR cells and (C) stimulation of βarrestin2 recruitment in the U2OS-βarrestin-hMOR-PathHunter via the EFC assay. For SR-15098, SR-15099 and SR-17018, βarrestin2 EFC concentration response curves were also performed in the presence of e-6.5 M DAMGO (open symbols) to test for partial agonism. For all three assays, the data were normalized to the % maximal response for DAMGO and are presented as mean ± S.E.M. of 3 or more assays run in duplicate or triplicate.
(D–E) The ΔΔLog(τ/KA) bias values with 95% confidence intervals with for the (D) human MOR and (E) mouse MOR. The G protein signaling was determined by either the GTPγS binding assay in CHO-hMOR or CHO-mMOR cells or mouse brainstem or by inhibition of forskolin-stimulated cAMP in CHO-hMOR cells. βarrestin2 recruitment to the MOR was determined by the EFC assay in U2OS-βarrestin-hMOR-PathHunter cells for the human receptor and by the βarrestin2 imaging based assay using the U2OS-βarrestin2-GFP-mMOR cell line for the mouse receptor. In all assays, DAMGO served as the reference agonist.
See also: Table 2 for the Log(τ/KA) and ΔΔLog(τ/KA) values with statistical comparison and Figure S3 for the concentration response curves for the mouse MOR assays (cells and brainstem).
Table 2. SR compounds display a range of bias at the human and mouse MOR.
Bias factor parameters of MOR agonists are presented across functional assays. GTPγS binding was determined in membranes from CHO-hMOR or -mMOR cells or from brainstems of C57BL/6J mice. Inhibition of forskolin-stimulated cAMP accumulation was measured in CHO-hMOR cells. βArrestin2 recruitment to the human MOR was determined by the EFC assay performed in the U2OS-βarrestin-hMOR-PathHunter cells and to the mouse MOR by the βarrestin2-eGFP translocation imaging based assay in U20S-βarrestin2-eGFP-mMOR cells. Data are presented as mean ± S.E.M. for ΔLog(τ/KA) values and with 95% confidence intervals for ΔΔlog(τ/KA) values. Assays were run in duplicate or triplicate, n ≥ 3. ΔLog(τ/KA) values were calculated relative to DAMGO within each individual experiment. ΔΔLog(τ/KA) values were then calculated between the indicated assays as described in the methods to propagate error (95% CI). See also: Figures 1 and S3 for the corresponding concentration response curves and Table S2 for corresponding EC50 and EMAX values.
| Agonist | Human MOR | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Δlog(τ/KA) | GTPγS (CHO)/βarr2 | cAMP (CHO)/βarr2 | |||||
|
|
|
|
|||||
| GTPγS (CHO) | cAMP (CHO) | βarr2 (EFC) | ΔΔlog(τ/KA) | Bias | Δδlog(τ/KA) | Bias | |
| DAMGO | 1 | 1 | |||||
| Sufentanil | 1.42±0.01 | 2.24±0.10 | 2.21±0.16 | −0.78 (−1.28 to −0.29)b | 0.16 | 0.02 (−0.40 to 0.44) | 1.1 |
| Fentanyl | −0.19±0.08 | 1.00±0.13 | 0.56±0.04 | −0.75 (−0.92 to −0.58)d | 0.18 | 0.45 (0.11 to 0.79)a | 2.8 |
| SR-11501 | −0.58±0.02 | −0.28±0.12 | −0.19±0.06 | −0.39 (−0.53 to −0.26)c | 0.41 | −0.09 (−0.39 to 0.21) | 0.81 |
| Morphine | −0.35±0.02 | −0.67±0.07 | −0.46±0.03 | 0.11 (0.02 to 0.20)a | 1.3 | −0.21 (−0.34 to −0.08)b | 0.62 |
| SR-14969 | 0.14±0.11 | −0.48±0.08 | −0.88±0.12 | 1.03 (0.66 to 1.39)d | 11 | 0.40 (0.04 to 0.76)a | 2.5 |
| SR-14968 | 0.70±0.13 | −0.15±0.09 | −0.85±0.07 | 1.55 (1.25 to 1.85)d | 36 | 0.71 (0.46 to 0.96)d | 5.1 |
| SR-15098 | −0.88±0.04 | −1.04±0.11 | −2.34±0.11 | 1.47 (1.18 to 1.75)d | 29 | 1.28 (0.91 to 1.66)d | 19 |
| SR-15099 | −0.81±0.05 | −1.05±0.22 | −2.48±0.06 | 1.68 (1.47 to 1.89)d | 47 | 1.44 (1.10 to 1.77)d | 27 |
| SR-17018 | −0.64±0.06 | −0.92±0.29 | −2.56±0.20 | 1.93 (1.22 to 2.63)d | 85 | 1.60 (0.84 to 2.35)c | 40 |
|
| |||||||
| Mouse MOR | |||||||
|
| |||||||
| Δlog(τ/KA) | GTPγS (CHO)/βarr2 | GTPγS (brain)/βarr2 | |||||
|
|
|
|
|||||
| GTPγS (CHO) | GTPγS (brain) | βarr2 (imaging) | Δδlog(τ/KA) | Bias | Δδlog(τ/KA) | Bias | |
|
| |||||||
| DAMGO | 1 | 1 | |||||
| Sufentanil | 1.08±0.07 | 1.48±0.22 | 2.33±0.06 | −1.25 (−1.48 to −1.02)d | 0.06 | −0.85 (−1.45 to −0.25)a | 0.14 |
| Fentanyl | −0.44±0.03 | 0.003±0.26 | 0.83±0.08 | −1.27 (−1.56 to −0.99)d | 0.05 | −0.83 (−1.30 to −0.35)b | 0.15 |
| SR-11501 | −0.68±0.02 | −0.41±0.12 | 0.23±0.05 | −0.91 (−1.06 to −0.77)d | 0.12 | −0.64 (−0.99 to −0.29)b | 0.23 |
| Morphine | −0.42±0.07 | −0.04±0.05 | −0.33±0.07 | −0.09 (−0.37 to 0.18) | 0.80 | 0.29 (0.11 to 0.46)b | 1.9 |
| SR-14969 | −0.06±0.05 | 0.41±0.09 | −0.52±0.09 | 0.46 (0.21 to 0.71)b | 2.9 | 0.93 (0.63 to 1.24)c | 8.6 |
| SR-14968 | 0.50±0.08 | 1.22±0.10 | −0.32±0.15 | 0.83 (0.40 to 1.25)b | 6.7 | 1.54 (1.13 to 1.94)d | 34 |
| SR-15098 | −0.94±0.02 | −0.22±0.05 | −1.96±0.19 | 1.03 (0.44 to 1.61)b | 11 | 1.74 (1.36 to 2.12)d | 55 |
| SR-15099 | −0.92±0.02 | −0.25±0.08 | −1.99±0.19 | 1.07 (0.51 to 1.63)b | 12 | 1.74 (1.33 to 2.15)d | 55 |
| SR-17018 | −0.86±0.03 | −0.32±0.08 | −2.33±0.12 | 1.47 (1.12 to 1.82)d | 30 | 2.01 (1.67 to 2.35)d | 102 |
Unpaired, two-tailed t test to vs. DAMGO:
p<0.05,
p<0.01,
p<0.001,
p<0.0001.
The evaluation of analogues in our series of MOR agonists suggests structural origins for MOR potency that directed further modifications to the molecules to produce compounds with greater G protein signaling potency and less βarrestin2 recruitment. The substituents R1-R6 markedly impact the properties of MOR agonists. As shown in Table 1, the compound SR-8595 (entry 1) lacks substituents on each aryl ring and is a full MOR agonist showing modest preference towards recruiting βarrestin2 over G protein signaling. Adding a chlorine atom at R3 (entry 2) improves potency for G protein signaling with little effect on βarrestin2 recruitment. Omission of the methyl group at R6 (entry 3) erodes potency in both assays. Having chlorine atoms at positions R1 and R2 rather than at R3 (entry 4) modestly improves potency but does not impart separation between the signaling assays. Importantly, when the chlorine (or alternatively bromine) substituents R1-R3 are used in combination (entries 5–9), a greater separation between G protein signaling and βarrestin2 recruitment is observed in MOR agonists. Compounds with non-halogen substituents at R1-R5, such as SR-11501 (entry 10) which has an ethylenedioxy group spanning the R3 and R4 positions, show improved potency in βarrestin2 recruitment relative to G protein signaling.
To quantitatively compare the differences observed between the two signaling assays, the operational model was used to calculate ΔΔlog(τ/KA) values with confidence intervals (bias factor = 10^ΔΔlog(τ/KA), Table 2). The series of compounds was narrowed to six SR candidates (Table 1, entries 5–10) selected to capture a wide range of bias factors; these were further compared to morphine, fentanyl and sufentanil, as examples of clinically relevant opioid analgesics. We find that fentanyl, sufentanil, and SR-11501 promote bias towards βarrestin2 recruitment in the EFC βarrestin2 assay when compared to GTPγS binding assay, relative to DAMGO (Table 2, Figure 1). At the other end of the spectrum, SR-15098, 15099 and 17018 promote preferential signaling towards GTPγS binding. Morphine, SR-14968 and SR-14969 fall between these two extremes. Examination of structural features within this series of compounds shows that halogen substituents at R1, R2, R3, and/or R5 apparently favor MOR conformations that promote robust GTPγS binding while disfavoring βarrestin2 signaling (Table 1).
It is important to note that, for an appropriate fit, the operational model requires a reasonable estimate of functional affinity, which cannot be produced in a curve that does not reach a maximum response. The inactivity of SR-15098, 15099 and 17018 in the βarrestin2 EFC assay therefore, poses the complication that no reliable potency can be inferred from a flat line. Modifications were hence made to the model based on two observations. First, to ensure that the compounds are not merely weakly efficacious, potent partial agonists, we tested whether they could block a stimulatory dose of DAMGO (a potent, weakly efficacious partial agonist will behave as an antagonist under these conditions and allow for derivation of potency). Previously, we determined that this approach could be used to refine the assessment of bias when conditions of an assay preclude the detection of an effect, or give the appearance of “extreme bias” (Stahl et al., 2015). As shown in Figure 1C, the compounds do not compete with DAMGO at the doses tested, suggesting that their potency in the βarrestin2 assay must be greater than 10 μM. This, taken together with their high affinity for the MOR in the 3H-DAMGO competition binding assays (Table S1), led us to apply a conservative constraint to the operational model to limit the calculated functional affinity; the log(KA) values were constrained to fall between 1 and 10−15 M and the Δlog(τ/KA) values, calculated relative to DAMGO, were constrained to be less than 10 (Brust et al., 2016; Stahl et al., 2015).
Since context greatly influences the determination of the numerical bias factor, we opted to evaluate the compounds in multiple systems to determine if the rank order of signaling preference is maintained. As a secondary measure of MOR signaling through inhibitory G proteins, we measured the inhibition of forskolin-stimulated cAMP accumulation in CHO-hMOR cells (Figure 1B, Table S2). G protein signaling bias was preserved for morphine and SR compounds, with the exception of SR-11501, regardless of whether the GTPγS binding assay or the cAMP accumulation assay was used. Though the bias factors decreased, ranging from 2.5 to 40 for the cyclase assay compared to 11 to 85 for the GTPγS binding assay, the rank-order remains the same (Figure 1D, Table 2). However, sufentanil and SR-11507 no longer calculate as significantly biased towards βarrestin2 when cyclase inhibition is used the measure of G protein signaling; meanwhile, fentanyl’s bias profile switches from being biased against GTPγS binding to being biased towards inhibition of cAMP accumulation.
Collectively, these findings showcase the utility of bias factors as a means of comparing relative performance of compounds in cell-based assays but also underscore the fact that a calculated bias factor is a function of the cellular context. Therefore, bias factors may serve as guiding parameters, but not as numeric constants that define a ligand in the absence of context. Since context plays an important role in determining relative signaling preference, we asked whether the rank order of bias would be maintained at the mouse MOR, since mice would be used for in vivo studies. In cells expressing the mouse MOR, agonism in GTPγS binding assays (Figure S3A) were compared to effects in an imaging-based βarrestin2-eGFP translocation assay (Figure S3C). Although the cell lines, assays and the species of the receptor changed, the overall bias profiles for these compounds is similar to that measured for human MOR (GTPγS binding and βarrestin2 EFC) (Figure 1E; Table S2).
Moving directly to the mouse model, agonists were shown to stimulate GTPγS binding in mouse brainstem, a region rich in pain regulatory neurons. DAMGO, maximally stimulated GTPγS binding with a potency of 400 ± 33 nM; while morphine performed as a partial agonist (41% EMAX) relative to DAMGO, with a potency of 159 ± 19 nM (Figure S3B, Table S2). While most of the agonists performed as nearly full agonists in the cell line G protein signaling assays, partial agonism (38–41%) could be more readily observed for some compounds, including fentanyl and sufentanil (32–33%), in the endogenous environment. The SR compounds showed a range in potency in this assay with SR-14968 as the most potent (26 ± 2 nM) and SR-11501 as the least potent (396 ± 68 nM) (Table S2). Importantly, MOR agonist-stimulated GTPγS binding is absent in brainstem from MOR knockout mice (Figure S3D) demonstrating selectivity of the compounds in vivo. When a bias factor is calculated using the parameters from the brainstem GTPγS binding assays compared to the mouse MOR βarrestin2 imaging assay, we find that the rank order bias profiles, while differing in absolute value, are again maintained (Figure 1E; Table S2).
Therefore, regardless of the assay used, or the species of receptor, the relative rank order of bias, in reference to DAMGO, is mostly conserved for the compounds: SR-14969 and 14968 show moderate bias while SR-15098, 15099 and 17018 display high bias toward G protein signaling compared to βarrestin2 recruitment (Figure 1D and 1E, Table 2). The ΔΔlog(τ/KA) for the βarrestin2 preferring compounds (fentanyl, sufentanil and SR-11501) are mostly conserved among the assays, except for when the inhibition of cAMP accumulation is used as a measure of their ability to promote G protein signaling. Morphine’s ΔΔlog(τ/KA) values vacillate around zero, suggesting that it is relatively unbiased, or balanced, when comparing G protein signaling and βarrestin2 recruitment.
Biased MOR agonists are long lasting and brain penetrant with systemic delivery
Since an important question is whether a pharmacological parameter (i.e., bias factor) that is broadly influenced by cellular context can be used to predict response profiles in vivo, we set out to compare the physiological effects of the compounds in mice to those induced by fentanyl and morphine. First, we determined that the SR MOR agonists could enter the brain by systemic drug delivery, like fentanyl and morphine. Morphine and the SR compounds were administered intraperitoneally (i.p.) at 6 mg/kg; plasma (Figure 2A) and brain (Figure 2B) levels were determined over 6 hours. The plasma levels for SR-15099 and SR-17018 rise more slowly than for morphine, suggesting a slower absorption rate and all of the SR compounds peak at the same level in the plasma as morphine within 30 minutes of injection. Importantly, all of the SR compounds are present in brain one hour following systemic injection, while some of them remain at high levels in the brain and plasma for the duration of the 6-hour period. Fentanyl, known to be very potent and brain penetrant, was injected at 1 mg/kg i.p. and brain levels were determined after 15 minutes for comparison. As anticipated, fentanyl was quickly cleared from brain and plasma, with no detection after 2 hours (Kalvass et al., 2007). When tested at higher doses, fentanyl and morphine brain levels escalate; however, the levels of the more biased compounds, SR-15098, 15099 and 17018, appear to level off, indicating that a maximum concentration may have been reached in the brain by 24 mg/kg despite higher dosing (Figure 2C). Plasma protein binding assays determined that the SR compounds are between 90–95% bound which allows for an estimation of approximately 200 nM freely circulating compound at 6 mg/kg, i.p. dosing (Table S3); notably, this is within range of the EC50 values calculated for GTPγS binding (26–400 nM) in membranes from the mouse brainstem (Table S2). These studies demonstrate that the drugs used in vivo have a comparable opportunity to activate MOR in the brain of mice and further informed the dosing used in the behavioral studies.
Figure 2. SR agonists cross the blood brain barrier and are present in plasma 6 hours after injection.

C57BL/6J mice were systemically (i.p.) injected with 6 mg/kg of each agonist (or 1 mg/kg for fentanyl) and (A) plasma and (B) brain levels were determined at the indicated time-points by LC/MS analysis. (A) While morphine and SR-11501 levels decrease over time, the other SR compounds remain at elevated levels up to 6 hours after injection (Dunnett’s multiple comparisons test: morphine (15 minutes) vs: aSR-15099 or SR-17018, p < 0.05; bSR-15098, p < 0.05). (B) The SR compounds can be detected in the brain at higher concentrations than morphine which persist 6 hours following treatment (Dunnett’s multiple comparisons test: morphine (1 hour) vs: aSR-11501, p < 0.01; bSR-14968 or SR-14969, p < 0.0001; cSR-15098 or SR-15099 or SR-17018, p < 0.01).
(C) C57BL/6J mice were administered the indicated dose of compound and brain levels were determined 1 hour after injection (i.p.). Increasing the dose of morphine or fentanyl increases the amount of drug in the brain, but there is no difference between the amount of drug in the brain at the 24 and 48 mg/kg doses of the SR compounds tested (One-way ANOVA, followed by Tukey’s post-hoc analysis within each treatment: p < 0.05 when compared to a0.5 mg/kg, b1 mg/kg, c6 mg/kg, d24 mg/kg, e50 mg/kg; ffor SR15-098, 6 versus 48 p < 0.5). Data are presented as mean ± S.E.M. of 3 or more mice. The limits of detection (LOD) are indicated for plasma (1 ng/mL) and brain homogenates (10 ng/mL).
See also: Table S3 for the plasma protein binding and estimated free plasma concentrations and Figure S4 for the antinociceptive and respiratory responses that correspond to these doses of the drugs.
G protein signaling-biased MOR agonist promote antinociception without respiratory suppression
Given that the SR compounds are brain penetrant following systemic dosing, we then compared them to morphine and fentanyl in mouse thermal nociception assays. All of the compounds produce antinociception, in both the hot plate and warm water tail withdrawal (tail flick) assays, that are on par with morphine and fentanyl (maximum efficacious doses are shown for comparison in Figure 3A). However, when these doses are tested for effects on respiration (% arterial oxygen saturation and breathing frequency), a clear delineation becomes apparent (Figure 3B). Remarkably, the SR compounds with the greater preference for stimulating GTPγS binding over βarrestin2 recruitment produce the least respiratory suppression, compared to an equi-antinociceptive dose of morphine tested.
Figure 3. Agonists that displayed G protein signaling bias in the cell based assays promote antincocicpetion without respiratory suppression.

(A) Antinociceptive responses were measured in male C57BL/6J mice in (A, top) hot plate (52 °C, top) and (A, bottom) warm water tail flick (49 °C) assays over 6 hours at doses (mg/kg, i.p.) of compounds that produce antinociceptive responses on par with morphine in male C57BL/6J mice.
(B) Respiratory responses were tested at the same doses in male C57BL/6J mice fit with a pulse oximeter to detect (B, top) % arterial oxygen saturation and (B, bottom) breath rate changes over 1 hour. The data are presented as mean ± S.E.M. of the % maximal possible effect (100% MPE), with basal responses determined for each mouse (A) prior to injection or (B) as the average response for 30 minutes prior to injection (at time 0, arrow) and setting the maximum thresholds at 20 sec for hot plate, 30 seconds for tail flick, 70% for oxygen saturation and 75 breaths per minute for breath rate measures.
(C) Therapeutic windows were calculated by dividing the ED50 values for the respiratory measures (%O2, arterial oxygen saturation or BR, breath rate) by the ED50 values for the antinociception measures (HP, hot plate or TF, tail flick) presented in SFig4 and Table 2. To show comparison to morphine, the values for morphine were then subtracted from each compound (morphine therapeutic window = 0).
See also: Figure S4 for dose response curves for all the compounds in both the antinociceptive and respiratory assays, as well as single dose in MOR-KO mice and in female mice; Table S4 for the number of mice used in each study; and Table 3 for the calculated ED50 values and therapeutic windows.
To fully investigate this apparent separation in therapeutic efficacies between the responses, we performed dose response studies for all of the compounds (Figure S4A–D, Table 3). Since the different compounds have different pharmacokinetic properties, we calculated ED50 values for each response by analyzing the area under the curve (AUC) for 1 hour following drug treatment in order to capture the peak effect of each drug in each assay as well as to account for each drug being present in the brain within that time frame. It can be readily seen that in both the hot plate and tail withdrawal assays, all compounds induce dose-dependent antinociception that is very similar if not more potent than that produced by morphine. However, as the G protein bias factors increase (as determined from the cellular assays), we begin to see less respiratory suppression at the higher doses. In contrast, fentanyl and SR-11501, which are calculated to be biased towards βarrestin2 recruitment over GTPγS binding, produce robust respiratory suppression at low doses. Additionally, it should be noted that the maximal doses of each of the compounds have no effect in MOR-KO mice demonstrating MOR selectivity for mediating these effects. Further, the same separation between antinociception and respiratory responses can be observed in female mice (Figure S4E).
Table 3. ED50 values and therapeutic windows for SR compounds.
Hot plate (HP), tail flick (TF), % arterial oxygen saturation (O2) and breath rate (BR, breaths per minute) ED50 values (mg/kg) were calculated from the hyperbolic fits (maximum value shared at 100%) of the area-under-the-curve analysis of dose response studies performed in male C57BL6/J mice (Figure S4 (A–D)) for the 1 hour period following drug treatment. The ratios of ED50 values for respiratory suppression over antinociception are represented by the therapeutic windows. Mean and 95% confidence intervals are provided. %MPE for all assays are defined on the lower limit by the baseline (BL) response and an upper limit (max, 100%) designed to limit the severity of each assay: Hot Plate (52°C): BL: 6.78 ± 0.14 sec, max: 20 sec; Tail Flick (49°C): BL: 2.95 ± 0.07 sec, max: 30 sec; %O2: BL: 95.11 ± 0.12 %, max: 70%; Breath Rate: BL: 165.0 ± 0.2, max: 75). See also: Figure S4 for the behavioral dose response curves and Figure 3C for a graphical representation of the therapeutic windows.
| Agonist | ED50 (95% confidence intervals) | Therapeutic Window | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Hot Plate | Tail Flick | %O2 Saturation | Breath Rate | O2/HP | O2/TF | BR/HP | BR/TF | |
|
|
|
|
|
|
|
|||
| Fentanyl | 0.24 (0.16 to 0.32) | 0.14 (0.07 to 0.20) | 0.71 (0.60 to 0.85) | 0.52 (0.37 to 0.67) | 3 | 5 | 2 | 4 |
| SR-11501 | 4.6 (2.9 to 6.3) | 11 (5.6 to 16) | 18 (13 to 27) | 7.6 (4.6 to 11) | 4 | 2 | 2 | 1 |
| Morphine | 5.9 (4.4 to 7.5) | 3.8 (2.5 to 5.2) | 79 (62 to 104) | 33 (23 to 42) | 13 | 21 | 5 | 9 |
| SR-14969 | 1.7 (0.88 to 2.5) | 2.3 (1.4 to 3.2) | 21 (16 to 29) | 20 (9.8 to 30) | 13 | 9 | 12 | 9 |
| SR-14968 | 0.44 (0.31to 0.57) | 0.61 (0.32 to 0.89) | 14 (11 to 18) | 11 (5.6 to 17) | 31 | 23 | 28 | 20 |
| SR-15098 | 12 (9.5 to 15) | 13 (8.9 to 18) | 538 (276 to 3359) | 174 (70 to 277) | 44 | 40 | 14 | 13 |
| SR-15099 | 8.4 (6.2 to 11) | 7.4 (4.9 to 9.9) | 560 (260 to >5000) | 206 (25 to 386) | 67 | 75 | 28 | 28 |
| SR-17018 | 6.9 (4.7 to 9.1) | 7.7 (4.1 to 11.3) | 719 (278 to >5000) | 197 (24 to 370) | 105 | 93 | 29 | 26 |
For the highly G protein signaling-biased compounds, SR-15098, 15099 and 17018, very little respiratory suppression is detected compared to vehicle treatment even when administered at 48 mg/kg. Since we did not define a maximum response in these animals, we performed the estimation of ED50 values for these measures by imposing a maximum threshold (for arterial oxygen saturation, data were normalized to a maximum response threshold of a 70% O2 and for breath rate, 75 breaths per minute). In this manner, we make a very conservative assumption that at high enough doses, the maximum suppression could be obtained, and thereby calculate ED50 values for SR-15098, SR-15099, and SR-17018 (Table 3).
G protein signaling-biased MOR agonists produce wider therapeutic windows
The calculated potencies were then used to generate therapeutic windows by dividing the ED50 values obtained in each of the two respiratory measures by the ED50 values from the two antinociception assays; whereby, a greater value indicates a higher degree of separation between the two responses (i.e. more pain relief with less respiratory suppression) (Table 3). As with the bias factor calculations, there are no absolute values for the therapeutic windows, but there are general trends. For instance, we find that fentanyl and SR-11501, the compounds that show βarrestin2 bias over G protein signaling, are more likely to induce respiratory suppression at lower doses and have very narrow therapeutic windows, compared to morphine. The compounds that show bias for G protein signaling over βarrestin2 recruitment prove to have much broader therapeutic windows than morphine. This profile is preserved regardless of which respiratory or antinociception measure is used; for graphical representation, the therapeutic windows relative to morphine are presented in Figure 3C.
Bias factor correlates with therapeutic window
A linear correlation analysis of the bias factor (hMOR GTPγS binding over βarrestin2 EFC) against the therapeutic window (% arterial oxygen saturation and hot plate antinociception) reveals a high coefficient of determination (R2= 0.96, Figure 4A, left panel), suggesting that increasing bias, as determined in cell-based signaling assays, can be predictive of an improved therapeutic window for analgesia without respiratory suppression in the mouse model. A similar correlation between a compound’s bias and its therapeutic window is observed when inhibition of cAMP accumulation is used as the measure of G protein signaling (R2=0.95, Figure 4A, right panel). When we compare other cell-based signaling biased factors, whether it be the parameters derived from assays using mouse MOR or mouse brainstem, the correlation remains high when compared to the therapeutic window is calculated from hot plate and arterial oxygen saturation potencies (R2>0.82, Table S5). In general, the therapeutic windows derived from the tail flick potencies were less correlative, with R2 values ranging from 0.64–0.94. In the calculation of bias factors, we noted that fentanyl and SR-11501 produced differently directed bias profiles depending upon the G protein signaling assay used (Figure 1D and E); therefore, we performed a correlation analysis on this subset of compounds, including morphine, to determine which G protein assay is more correlative with the therapeutic window. We find that a bias towards βarrestin2 recruitment over GTPγS binding, but not cAMP accumulation, highly correlates with a narrow therapeutic window (hot plate/%O2) compared to morphine (R2= 0.99 for GTPγS compared to R2= 0.41 for cAMP; Figure 4B). As such, for compounds that had more narrow safety margins, the bias factors calculated from comparing the GTPγS assays to βarrestin2 recruitment were more correlative than those derived from comparing the cAMP accumulation assay to βarrestin2 recruitment.
Figure 4. Bias factors positively correlate with therapeutic window.

(A) The bias factors determined in the hMOR cell lines (GTPγS binding in CHO-hMOR membranes (left) or inhibition of forskolin-stimulated cAMP accumulation in the CHO-hMOR cells (right), versus βarrestin2 recruitment in the U2OS-βarrestin-hMOR-PathHunter EFC assay) when plotted against the therapeutic windows calculated from the in vivo studies (O2 ED50: % arterial oxygen saturation over HP ED50: hot plate antinociception) produce linear correlations: GTPγS/βarr2: R2= 0.9589; cAMP/βarr2: R2= 0.9525.
(B) Correlation analysis of compounds that display bias towards βarrestin2 from (A) (i.e., fentanyl and SR-11501) with morphine when plotted against the therapeutic window (HP/O2) produce reveals a strong correlation when GTPγS/βarr2 bias factors (R2=0.99) are plotted, but not with cAMP/βarr2 bias factors (R2=0.4140).
See also: Table S5 for the correlation analysis between bias factor and therapeutic window for the other bias factors calculated for the compounds (CHO-mMOR and brainstem) and the therapeutic windows for the other behavioral measures (breath rate and tail flick).
Discussion
In this study, we have used several cell-based signaling assays to compare a series of structurally related MOR agonists as well as prescription opiates, for their ability to activate MOR in comparison to a reference compound, DAMGO. Using these data, we normalized their potencies and efficacies with respect to the efficiency of DAMGO in each assay using the classic operational model to determine bias factors (Black and Leff, 1983; Kenakin et al., 2012). We then tested each compound in mice, using conventional antinociception assays and a mouse pulse oximeter to determine in vivo efficacies. Upon calculating potency (ED50) values from the mouse studies, we then calculated therapeutic windows by dividing the potencies observed in the respiratory assays by the potencies measured in the pain assays. A comparison between the degree of separation measured in the cell-based G protein signaling and βarrestin2 recruitment assays (bias factors) positively correlates with the ability to separate antinociception and respiratory suppression in vivo (increase in the therapeutic window). Using these parameters, we show for the first time that fentanyl induces bias towards promoting βarrestin2 recruitment over inducing GTPγS binding and that this correlates with an increase propensity for respiratory suppression at low doses. Since the therapeutic window of fentanyl is known to be more narrow than morphine in humans, these findings may be important for understanding fentanyl-related overdose fatalities.
One of the goals of this study was to understand how changing the signaling assay and the cellular context impacts the calculation of bias (while maintaining the reference agonist constant). Interestingly, compounds in our studies that display bias towards βarrestin2 over GTPγS do not maintain this bias profile when inhibition of cAMP accumulation is measured as a surrogate for G protein signaling. Moreover, we found that the GTPγS binding assay-derived bias factors were more predictive of the calculated therapeutic windows, especially of those for fentanyl and SR-11501, than the cAMP assay-derived bias factors (Figure 4 and Table S5). Overall, we determined that once we reached bias factors exceeding 10 using the hMOR GTPγS/βarrestin2 measures, that the type of assay used to subsequently test G protein signaling had little bearing on the confidence in bias calculation. However, as that value approaches zero, we found greater variation in bias calculations when other G protein signaling assays were used (i.e. inhibition of cAMP accumulation).
In the early βarrestin2-KO mouse studies, morphine was shown to induce pronounced antinociception when compared to WT mice (Bohn et al., 1999). In subsequent studies, it was shown that the βarrestin2-KO mice were resistant to morphine-induced respiratory suppression (Raehal et al., 2005). Together the studies in the knockout mice along with the biased agonists strongly support the hypothesis that compounds that do not recruit βarrestin2 may prove to be safer (i.e. have a wider therapeutic window) than current clinical opioids. However, it is important to acknowledge that to date, it has not been directly demonstrated that βarrestins are indeed mediating the respiratory side effects in the mice. Moreover, it also remains to be seen whether bias factors correlate with the expression of other opiate side effects, such as constipation, analgesic tolerance and the development of physical dependence and addiction, as these physiological responses and adaptations are complex and are resultant of multiple converging biological systems. Going forward, the G protein-biased compounds will be useful for elucidating how MOR signals in vivo to promote these effects. Moreover, compounds like fentanyl, sufentanil and SR-11501 may serve as important tools to challenge the counter hypothesis.
Another important aspect of this study is the emphasis that bias factors, as are therapeutic windows, are highly context dependent. The use of the operational model to estimate the degree of separation of functional performance between two cellular assays is an attempt to normalize the contributions of within assay variances. The normalization is made possible by use of a reference agonist that serves to define the maximum potential of the receptor to produce a response in that system. Therefore, the correlation plots of response parameters are not to be viewed as a universal scale upon which independently generated values could be plotted. For new compounds to be assessed, a defining data set should be produced; further, the different assays should all be performed in parallel and including the appropriate reference agonists (one that produces the maximum response in the system) prior to calculation of bias between assays. Bias factors calculated for compounds wherein different mathematical models are used to calculate bias, different assays are run to determine G protein or βarrestin2 signaling potential or different reference agonists are used could not be appropriately plotted with the data reported here. Similarly, one should not expect to plot therapeutic windows derived from other species, other pain tests or other side effect measures and infer that a linear correlation would ensue. Such conclusions must be empirically determined.
With these limitations in mind, this study still remains the first of its kind to systematically assess a chemical series of agonists for bias across multiple signaling assays and to also perform thorough analyses of behavioral responses in a dose dependent manner. The demonstration of a correlation between the bias observed to the width of the therapeutic window is highly encouraging for using these signaling assays to predict favorable outcomes in the mouse models used here. Moreover, we have demonstrated that key regions of the chemical scaffold can be mindfully modified to direct signaling between the different assays in cell culture and that this recapitulates as differences in potencies in vivo. Finally, this study introduces a new series of G protein signaling biased MOR agonists that have the highest degree of separation between respiratory suppression and antinociception in rodent models reported to date. Our hope is that this work may aid the pharmaceutical development of safer alternatives to current opioid therapeutics.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA Tag Monoclonal Antibody (16B12), Alexa Fluor 594 | ThermoFisher Scientific | Cat# A-21288 RRID:AB_2535830 |
| Hoechst 33342, Trihydrochloride, Trihydrate | ThermoFisher Scientific | Cat# H3570 |
| Chemicals, Peptides and Recombinant Proteins | ||
| DAMGO | Tocris | Cat# 1171 |
| DTT | Fisher Scientific | Cat# BP172-5 |
| Fentanyl Citrate | Sigma-Aldrich | Cat# F3886 |
| Forskolin | Sigma-Aldrich | Cat# F6886 |
| GDP, disodium salt | Fisher Scientific | Cat# ICN15121325 |
| [Tyrosyl-3,5-3H(N)]-DAMGO | PerkinElmer | Cat# NET902250UC |
| [Phenyl-3,4-3H]-U-69,593 | PerkinElmer | Cat# NET952250UC |
| [15,16-3H]-Diprenorphine | PerkinElmer | Cat# NET1121250UC |
| [35S]-GTPγS | PerkinElmer | Cat# NEG030H250UC |
| Morphine sulfate pentahydrate | NIDA Drug Supply Program | Cat# 9300-001 |
| Naloxone hydrochloride dihydrate | Sigma-Aldrich | Cat# N7758 |
| Naltrindole | Tocris | Cat# 0740 |
| Nociceptin | Tocris | Cat# 0910 |
| NorBNI | Sigma-Aldrich | Cat# N-1771 |
| [Nphe1]Nociceptin(1-13)NH2 | Tocris | Cat#1308 |
| Poly(ethyleneimine) solution (PEI) | Sigma-Aldrich | Cat# 03880 |
| Ro-20-1724 | Sigma-Aldrich | Cat# B8279 |
| SNC80 | Enzo Life Sciences | Cat# ALX-550-172-M005 |
| Solution D | ThermoFisher Scientific | Cat# K1156 |
| SR-8595 | This study | N/A |
| SR-11065 | This study | N/A |
| SR-11501 | This study | N/A |
| SR-14968 | This study | N/A |
| SR-14969 | This study | N/A |
| SR-15098 | This study | N/A |
| SR-15099 | This study | N/A |
| SR-17018 | This study | N/A |
| SR-20382 | This study | N/A |
| SR-20437 | This study | N/A |
| Sufentanil Citrate | NIDA Drug Supply Program | Cat# 9740-001 |
| U69,593 | Sigma-Aldrich | Cat#U103 |
| Critical Commercial Assays | ||
| cAMP HiRange Kit | Cisbio | Cat# 62AM6PEC |
| LiveBLAzer FRET-B/G loading kit | ThermoFisher Scientific | Cat#K1095 |
| PathHunter Detection Kit | DiscoveRx | Cat# 93-0001 |
| Rapid Equilibrium Dialysis Device | ThermoFisher Scientific | Cat#90006 |
| Experimental Models: Cell Lines | ||
| CHO-K1 | ATCC | Cat# CCL-61, RRID:CVCL_0214 |
| CHO-hMOR | R.B. Rothman: Ananthan et al., 2012 | N/A |
| CHO-mMOR | This study | N/A |
| CHO-hDOR | L.M. Bohn: Zhou et al., 2013 | N/A |
| CHO-hKOR | L.M. Bohn: Schmid et al., 2013 | N/A |
| Phoenix-Ampho | ATCC | Cat# SD-3443, RRID:CVCL_H716 |
| U2OS-βarrestin-hMOR PathHunter | DiscoveRx | Cat# 93-0213C3 |
| U2OS-βarrestin2-GFP | L. Barak: Addiction Research GPCR Assay Bank, NIDA | N/A |
| U2OS-βarrestin2-GFP-mMOR | This study | N/A |
| U2OS-Tango-hOPRL1-bla | ThermoFisher Scientific | Cat# K1786 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: MOR-KO: B6.129S2-Oprm1tm1kff/J | The Jackson Laboratory | RRID:IMSR_JAX:007559 |
| Recombinant DNA | ||
| HA-mMOR-MSCV | L.M.Bohn: Groer et al., 2011 | N/A |
| Software and Algorithms | ||
| HCS Studio 2.0 Cell Analysis Software | ThermoFisher Scientific | https://www.thermofisher.com |
| MouseOx Plus Revision 1.5.1b | Starr Life Sciences Corporation | http://www.starrlifesciences.com |
| Prism v. 6.0h | GraphPad Software | https://www.graphpad.com |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Laura Bohn (lbohn@scripps.edu). The Scripps Research Institute requires that a material transfer agreement (MTA) be signed for the transfer of materials.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Male and female C57BL/6J and male MOR-KO mice were purchased from The Jackson Laboratory and propagated by homozygous breeding in-house. Mice were group housed (3–5 mice per cage) and maintained on a 12-hour light/dark cycle with food and water ad libitum. Experiments were performed on naïve adult mice between 10–14 weeks of age. Same sex littermates were randomly assigned to experimental groups; males and females were separately tested and their responses are separately reported. Experiments were performed by investigators who were blinded to the treatment assignments. Mice were dosed i.p. at a volume of 10 μl/g mouse, except all 48 mg/kg injections were dosed at a volume of 20 μl/g mouse to adjust for compound solubility. The number of mice used in each assay are indicated in Table S4. All mice were used in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals with approval by The Scripps Research Institute Animal Care and Use Committee.
Cell lines
Chinese Hamster Ovary (CHO-K1) and Phoenix-AMPHO® cells were purchased from ATCC; U2OS-βarrestin-hMOR PathHunter cells were purchased from DiscoveRx and the U2OS-Tango-hOPRL1-bla cells were purchased from ThermoFisher Scientific. The U2OS-βarrestin2-GFP cells were provided by the Addiction Research GPCR Assay Bank. Based on the other reports, all of the parent cell lines are female (query: http://web.expasy.org/cellosaurus/). Receptor levels in the CHO-hMOR, hKOR and hDOR cells have been described previously as well as in the current manuscript (Ananthan et al., 2012; Schmid et al., 2013; Zhou et al., 2013). To make the mMOR lines, the HA (haemagglutinin)-N-terminus tagged mMOR was packaged into murine stem cell retroviral particles via the phoenix packaging system and then CHO-K1 and U2OS-βarrestin2-GFP cells were transduced with the particles. A BD FACSAria3 flow cytometer was used to select for high expressing cells using an anti-HA AlexaFluor 594 conjugate antibody (1:100). All cells lines were cultured according to standard protocols at 37° in the indicated media with 10% fetal bovine serum (FBS) and 1% pen/strep: DMEM/F12 all CHO-K1 lines; DMEM Phoenix- AMPHO®; MEM all U2OS cell lines. CHO-hMOR, -hKOR and -hDOR cell lines were grown under geneticin selection (500 μg/μl). U2OS mMOR βarrestin2 cells lines were grown under puromycin selection (500 μg/μl). The U2OS-βarrestin-hMOR PathHunter cell line (in which the MOR retains its natural C-terminal tail, tagged with the enzyme fragment) was cultured according to the manufacturer’s protocol (DiscoveRx), as was the U2OS-Tango-OPRL1-bla cell line (ThermoFisher Scientific).
METHOD DETAILS
Synthesis of MOR ligands
All reagents and anhydrous solvents were used as obtained from commercial vendors. 1H NMR spectra were recorded at 400 MHz, with chemical shifts are reported in parts per million (ppm) using an internal standard, CHCl3 (δ 7.26), MeOH (δ 3.34) or DMSO (δ 2.54). Mass spectra were recorded by ESI Ion trap. Analytical HPLC retention times were measured using reverse phase conditions with a Zorbax® 5 micron column, model Eclipse-XDB-C18 80Å (155 x 4.6 mm), column temperature = 40°C, flow rate = 3.00 mL/min. The method incorporates a gradient elution, beginning with 98% H2O / 2% acetonitrile, each with 0.1% TFA. After 1 minute, hydrophobicity was increased to 5% acetonitrile and then linearly in a gradient to 95% acetonitrile over an additional 5 minutes. Purity assessment (>95%) was made LC using UV absorbance at multiple wavelengths, typically 215, 254, and 280 nm.
General synthesis methods: the synthesis of all compounds followed the standard methods depicted in Figure S1. Nucleophilic aromatic substitution followed by nitro group reduction (Obase et al., 1983), urea formation (Budzik et al., 2010), standard Boc deprotection, and finally direct alkylation (Lindsley et al., 2005) or reductive amination of an aldehyde (Zhao et al., 2005) or ketone (Patel et al., 2014) gave the indicated SR compounds. The compounds were isolated and characterized in free base form unless indicated (overall yields 15–40% for 5 steps) and then the compounds were evaluated in all biological and pharmacological assays as their mono mesylate salts.
Representative nucleophilic aromatic substitution procedure, R1 = R2 = Cl: 1,2-Dichloro-4-fluoro-5-nitrobenzene (0.43 mL, 3.3 mmol) was added to a mixture of tert-butyl 4-aminopiperidine-1-carboxylate (0.66 g, 3.3 mmol) and K2CO3 (0.50 g, 3.6 mmol) in DMSO (5 mL). The reaction mixture was stirred overnight at room temperature under argon. Water was added and the organic layer extracted with EtOAc, dried over Na2SO4, and concentrated under reduced pressure. Purification was achieved by flash column chromatography on silica gel using a gradient of EtOAc:hexanes as the eluent to give an orange solid (0.79 g, 62% yield). 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.01 (d, J = 7.2 Hz, 1H), 6.97 (s, 1H), 4.03 (d, J = 13.6 Hz, 2H), 3.65-3.56 (m, 1H), 3.05 (td, J = 12.4, 2.8 Hz, 2H), 2.05 (dd, J = 13.0, 3.4 Hz, 2H), 1.63-1.49 (m, 2H), 1.47 (s, 9H); MS(m/z): [M + H] calc’d for C16H21Cl2N3O4 is 390.26, found 389.49.
Representative nitro group reduction procedure, R1 = R2 = Cl: Tert-butyl 4-((4,5-dichloro-2-nitrophenyl)amino)piperidine-1-carboxylate (0.79 g, 2.0 mmol) was dissolved in EtOH (40 mL) and a 50% aqueous suspension of Raney nickel (5 mL) was added. Hydrazine hydrate (0.98 mL, 20 mmol) was then added dropwise. The mixture was heated to 45 °C and maintained at that temperature for 10 min. The mixture was filtered through a pad of Celite® which was washed with MeOH. The filtrate was concentrated under reduced pressure. Purification was achieved by flash column chromatography on silica gel using a gradient of EtOAc: hexanes as the eluent to give the diamine product (0.55 g, 76% yield). 1H NMR (400 MHz, CD3OD) δ 6.75 (s, 1H), 6.62 (s, 1H), 4.46 (s, 1H), 4.01 (d, J = 13.2 Hz, 2H), 3.36 (tt, J = 10.0, 4.0 Hz, 1H), 2.95 (t, J = 12.2 Hz, 2H), 2.00 (d, J = 13.6 Hz, 2H), 1.45 (s, 9H), 1.36 (qd, J = 12.0, 4.0 Hz, 2H); MS(m/z): [M + H] calc’d for C16H23Cl2N3O2 is 360.28, found 359.58.
Representative urea formation and Boc removal procedure, R1 = R2 = Cl: Tert-butyl 4-((2-amino-4,5-dichloro-phenyl)amino)piperidine-1-carboxylate (0.55 g, 1.5 mmol) was dissolved in THF (15 mL) under argon. CDI (0.35 g, 2.1 mmol) was added and the reaction mixture was stirred at room temperature overnight. Upon completion, the solvent was removed under reduced pressure and the residue was dissolved in EtOAc. This mixture was washed with 1M HCl (aq) followed by brine. The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. Purification was achieved by flash column chromatography on silica gel using a gradient of EtOAc: hexanes as the eluent to give tert-butyl 4-(5,6-dichloro-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)piperidine-1-carboxylate (0.54 g, 91% yield). 1H NMR (400 MHz, CDCl3) δ 9.78 (s, 1H), 7.20 (s, 1H), 7.19 (s, 1H), 4.44-4.34 (m, 3 H), 2.85 (t, J = 11.2 Hz, 2H), 2.26 (qd, J = 12.6, 4.2 Hz, 2H), 1.82 (d, J = 10.8 Hz, 2H), 1.52 (s, 9H); MS(m/z): [M + H] calc’d for C17H21Cl2N3O3 is 386.27, found 385.33. This product was dissolved in a 33% solution of TFA in CH2Cl2 (4 mL). Upon completion, the solvent was removed under reduced pressure and the residue was dissolved in a minimal amount of water-acetonitrile (1:1). The solution was frozen and was then subjected to lyophilization overnight, giving the amine product as a TFA salt (86% crude yield). 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.17 (s, 1H), 4.36 (tt, J = 12.6, 4.0 Hz, 1H), 3.27 (d, J = 12.0 Hz, 2H), 2.79 (td, J = 12.2, 2.0 Hz, 2H), 2.27 (qd, J = 12.4, 4.0 Hz, 2H), 1.83 (dd, J = 12.0, 2.0 Hz, 2H); MS(m/z): [M + H] calc’d for C12H13Cl2N3O is 286.16, found 286.12.
Representative reductive amination procedure, R1 = R2 = Cl, R3 = Br: NaBH(OAc)3 (97 mg, 0.44 mmol) was added to an anhydrous DCE (3 mL) solution of 5,6-Dichloro-1-(piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (59 mg, 0.15 mmol), and 4-bromobenzaldehyde (85 mg, 0.44 mmol). A few drops of AcOH were added to the solution and the reaction mixture was stirred overnight at room temperature under argon. Upon completion, saturated aq. NaHCO3 was added to the reaction mixture, which was then diluted with CH2Cl2. The aqueous layer was extracted with CH2Cl2 and the combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. Purification was achieved by flash column chromatography on silica gel using with CH2Cl2:MeOH as the eluent to give the desired product SR-15099 (36 mg, 55% yield). Methanesulfonic acid (5.2 μL, 0.08 mmol) was added to a suspension of SR-15099 free base in EtOH (1 mL). The mixture was heated to 60°C for 30 min. The solvent was evaporated under reduced pressure and the residue was dissolved in a minimal amount of water-acetonitrile (1:1). The solution was frozen and subjected to lyophilization overnight, giving 5,6-Dichloro-1-(1-(4-bromobenzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate as an white powder. Analytical data for this tested compound is given later in this section. Note: for reductive amination reactions of methyl ketones rather than aldehydes, the ketone (3 equiv.) and amine was treated with 10 eq. of Ti(Oi-Pr)4, heated to 60 °C, then 8.75 equiv. of NaBH3CN in ethanol was added. After overnight reaction, the mixture was processed as described above. Added note: the unsubstituted N-benzyl compound SR-20437 was prepared by alkyation rather than by reductive amination. To the amine in minimal DMF was added 1.1 equiv. of benzyl bromide, 1.1 equiv. of K2CO3, 1.0 equiv. of NaI. Heating at 60 °C overnight, standard workup, and salt formation gave the desired material in 53% yield.
Analytical data for final compounds
SR-8595 (Table S1, entry 1), (±)-1-(1-(1-phenylethyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one
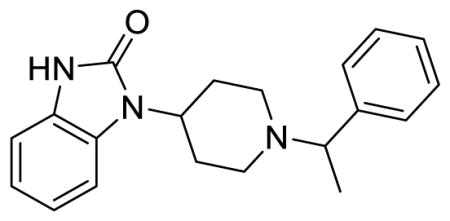
1H NMR (400 MHz, (CD3)2SO) δ 10.80 (s, 1H), 7.34 (t, J = 3.0 Hz, 4H), 7.26-7.19 (m, 2H), 6.99-6.95 (m, 3H), 4.05 (tt, J = 12.6, 4.0 Hz, 1H), 3.53 (q, J = 6.8 Hz, 1H), 3.10 (d, J = 10.0 Hz, 1H), 2.89 (d, J = 9.6 Hz, 1H), 2.45-2.23 (m, 2H), 2.07 (td, J = 10.4, 2.4 Hz, 1H), 1.97 (td, J = 10.8, 2.0 Hz, 1H), 1.66 (d, J = 10.8 Hz, 1H), 1.58 (d, J = 10.4 Hz, 1H), 1.33 (d, J = 6.8 Hz, 3H); MS(m/z): [M + H] calculated for C20H23N3O is 321.42, found 321.96; HPLC tR = 3.52 min.
SR-11065 (Table S1, entry 2), (±)-1-(1-(1-(4-chlorophenyl)ethyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one
1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.19 (td, J = 7.6, 1.2 Hz, 1H), 7.09-7.02 (m, 2H), 4.62-4.54 (m, 1H), 4.21-4.18 (m, 1H), 3.89 (d, J = 10.4 Hz, 1H), 3.52 (d, J = 12.8 Hz, 2H), 3.27 (qd, J = 11.6, 4.0 Hz, 1H), 2.78 (q, J = 10.4 Hz, 1H), 2.65 (q, J = 11.2 Hz, 1H), 1.97 (d, J = 6.8 Hz, 3H), 1.93-1.89 (m, 2H); MS(m/z): [M + H] calculated for C20H22ClN3O is 355.87, found 355.92; HPLC tR = 3.76 min.
SR-20382 (Table S1, entry 3), 1-(1-(4-chlorobenzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
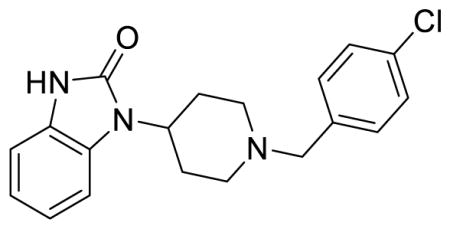
1H NMR of the mesylate salt (400 MHz, CD3OD) 7.56-7.54 (m, 4H), 7.29-7.26 (m, 1H), 7.10-7.07 (m, 3H), 4.56 (tt, J = 12.4, 4.0 Hz, 1H), 4.39 (s, 2H), 3.66 (dd, J = 10.6, 1.8 Hz, 2H), 3.27-3.23 (m, 2H), 2.80 (qd, J = 13.4, 3.8 Hz, 2H), 2.72 (s, 3H), 2.09 (d, J = 14.8 Hz, 2H); MS(m/z): [M + H] calculated for C19H20ClN3O is 341.84, found 342.02; HPLC tR = 3.67 min.
SR-20437 (Table S1, entry 4), 5,6-dichloro-1-(1-(benzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
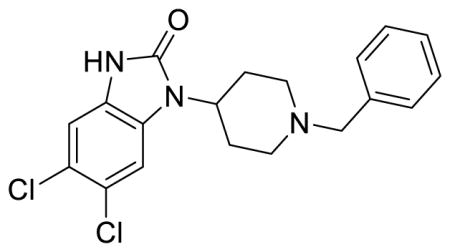
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.56-7.52 (m, 5H), 7.48 (s, 1H), 7.19 (s, 1H), 4.53 (tt, J = 12.4, 4.0 Hz, 1H), 4.39 (s, 2H), 3.65 (d, J = 12.8 Hz, 2H), 3.28-3.22 (m, 2H), 2.77 (qd, J = 13.4, 4.0 Hz, 2H), 2.72 (s, 3H), 2.08 (d, J = 14.8 Hz, 2H); MS(m/z): [M + H] calculated for C19H19Cl2N3O is 376.28, found 375.98; HPLC tR = 3.95 min.
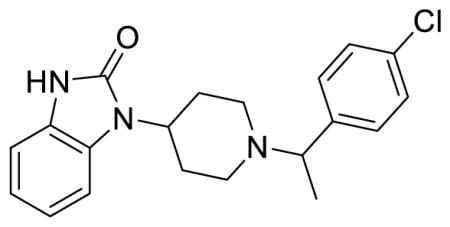
SR-17018 (Table 1; Table S1, entry 5), 5,6-dichloro-1-(1-(4-chlorobenzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
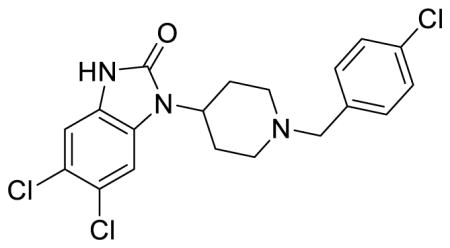
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.58-7.53 (m, 4H), 7.48 (s, 1H), 7.19 (s, 1H), 4.52 (tt, J = 12.4, 3.8 Hz, 1H), 4.38 (s, 2H), 3.65 (d, J = 12.8 Hz, 2H), 3.25 (t, J = 12.2 Hz, 2H), 2.80-2.69 (m, 5H), 2.08 (d, J = 13.6 Hz, 2H); MS(m/z): [M + H] calculated for C19H18Cl3N3O is 410.72, found 410.01; HPLC tR = 4.12 min.
SR-15099 (Table 1; Table S1, entry 6), 5,6-dichloro-1-(1-(4-bromobenzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
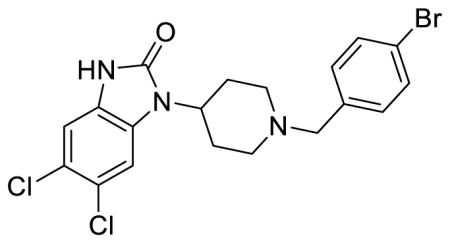
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.70 (dt, J = 8.4, 2.1 Hz, 2H), 7.50 (dd, J = 6.6, 2.0 Hz, 3H), 7.19 (s, 1H), 4.53 (tt, J = 12.4, 4.0 Hz, 1H), 4.37 (s, 2H), 3.63 (d, J = 12.8 Hz, 2H), 3.24 (t, J = 12.4 Hz, 2H), 2.79-2.70 (m, 6H), 2.07 (d, J = 12.4 Hz, 2H); MS(m/z): [M + H] calculated for C19H18BrCl2N3O is 455.18, found 456.23; HPLC tR = 3.76 min.
SR-14968 (Table 1; Table S1, entry 7), (±)-5,6-dichloro-1-(1-(1-(4-bromophenyl)ethyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
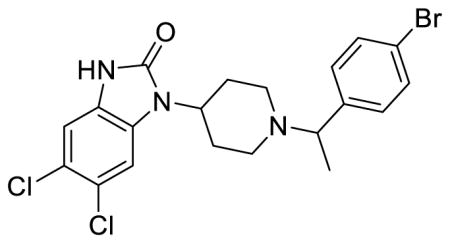
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.71 (d, J = 8.4 Hz, 2H), 7.49 (t, J = 8.4 Hz, 3H), 7.19 (s, 1H), 4.57 (q, J = 6.8 Hz, 1H), 4.44 (tt, J = 12.4, 4.0 Hz, 1H), 3.87 (dd, J = 11.0, 1.8 Hz, 1H), 3.49 (dd, J = 11.2, 2.0 Hz, 1H), 3.14 (td, J = 13.0, 2.8 Hz, 1H), 3.04 (td, J = 13.0, 2.8 Hz, 1H), 2.84-2.66 (m, 6H), 2.12-2.02 (m, 2H), 1.80 (d, J = 6.8 Hz, 3H); MS(m/z): [M + H] calculated for C20H20BrCl2N3O is 469.20, found 469.89; HPLC tR = 4.00 min.
SR-14969 (Table 1; Table S1, entry 8), (±)-5,6-dichloro-1-(1-(1-(4-chloro-2-fluorophenyl)ethyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
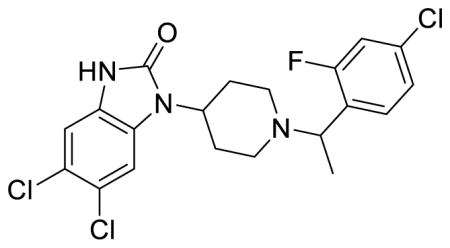
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.67 (t, J = 7.8 Hz, 1H), 7.48-7.43 (m, 3H), 7.19 (s, 1H), 4.93-4.89 (m, 1H), 4.47 (tt, J = 12.2, 4.0 Hz, 1H), 3.89 (d, J = 12.0 Hz, 1H), 3.60 (d, J = 10.4 Hz, 1H), 3.19 (td, J = 13.0, 2.2 Hz, 1H), 3.10 (td, J = 13.0, 2.2 Hz, 1H), 2.88-2.72 (m, 6H), 2.12-2.04 (m, 2H), 1.83 (d, J = 6.8 Hz, 3H); MS(m/z): [M + H] calculated for C20H19Cl3FN3O is 442.74, found 441.87; HPLC tR = 3.94 min.
SR-15098 (Table 1; Table S1, entry 9) 5,6-dichloro-1-(1-(4-chloro-2-fluorobenzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
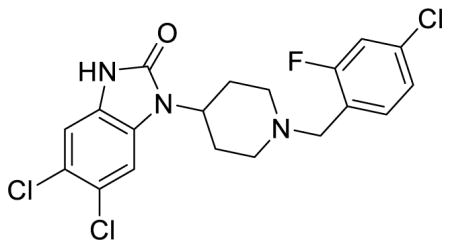
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.67 (t, J = 8.2 Hz, 1H), 7.50 (s, 1H), 7.43 (qd, J = 7.8, 2.0 Hz, 2H), 7.19 (s, 1H), 4.55 (tt, J = 12.4, 4.0 Hz, 1H), 4.46 (s, 2H), 3.70 (d, J = 12.4 Hz, 2H), 3.36-3.30 (m, 2H), 2.82-2.71 (m, 5H), 2.09 (d, J = 13.6 Hz, 2H); MS(m/z): [M + H] calculated for C19H17Cl3FN3O is 428.71, found 427.96; HPLC tR = 4.15 min.
SR-11501 (Table 1; Table S1, entry 10), (±)-1-(1-(1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one methanesulfonate
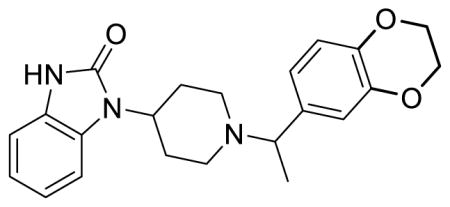
1H NMR of the mesylate salt (400 MHz, CD3OD) δ 7.30-7.27 (m, 1H), 7.09-7.06 (m, 4H), 7.01 (dd, J = 8.4, 2.0 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 4.50-4.41 (m, 2H), 4.28 (s, 4H), 3.84 (dt, J = 12.8, 1.9 Hz, 1H), 3.49 (dt, J = 11.8, 1.4 Hz, 1H), 3.14 (td, J = 13.0, 2.8 Hz, 1H), 3.04 (td, J = 13.0, 2.8 Hz, 1H), 2.92-2.72 (m, 6H), 2.12-2.01 (m2H), 1.77 (d, J = 7.2 Hz, 3H); MS(m/z): [M + H] calculated for C22H25N3O3 is 379.46, found 379.87; HPLC tR = 4.58 min.
Preparation of drug solutions
For the in vitro studies, the reference compounds DAMGO, morphine sulfate and nociceptin were prepared in water as a 10 mM stock and a 10 mM stock of U69,593 was prepared in ethanol. All of the other compounds were prepared in DMSO at concentrations spanning from 32 nM to 10 mM, for dilutions. For all assays, the final DMSO concentration was 1%. For the in vivo studies, compounds were dissolved from powder immediately prior to use. Morphine sulfate and the test compounds were prepared in a vehicle of 1:1:8 DMSO: Tween 80: dH2O. Fentanyl citrate was dissolved in 0.9% saline for the studies in C57BL/6J male mice. For studies where only one dose was tested (females and MOR-KO mice) all compounds were made in the same vehicle to facilitate blinding of drug preparation and experimenter handling. Compounds were administered intraperitoneally (i.p.) at a concentration of 10 μl per gram mouse, except for the 48 mg/kg dose of the test compounds. In this case, the drugs were administered at a volume of 20 μl per gram mouse due to limited solubility. Morphine sulfate and fentanyl citrate dosing is based on the salt weight of the drugs, while the SR compounds dosing is based on the free base weight.
Saturation and competition radioligand binding
Receptor binding assays were performed on CHO-hMOR, CHO-hDOR and CHO-hKOR cell lines as previously described (Groer et al., 2011; Schmid et al., 2013). Cells were serum-starved for 30 minutes, cells were collected and membrane pellets were prepared by Teflon-on-glass dounce homogenization in membrane buffer containing (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA), followed by centrifugation at 20,000 x g for 30 minutes at 4 °C. Membranes were resuspended in assay buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl). Binding reactions (200 μl volume) were performed on 10 μg membranes with the appropriate radioligand (MOR, 3H-DAMGO; KOR, 3H-U69,593; DOR, 3H-diprenorphine) for 2 hours at 25 °C. For competition experiments, the concentration of each of the radioligands was approximately 1 nM (0.96–1.10 nM 3H-DAMGO; 1.06–1.19 nM 3H-U69,593; 0.92–0.98 nM 3H-diprenorphine). Nonspecific binding was determined in the presence of 10 μM DAMGO (MOR), 10 μM U69,593 (KOR) or 10 μM Naloxone (DOR). Reactions were terminated by filtration through GF/B glass fiber filter plates (PerkinElmer), which had been pre-incubated with 0.1% polyethyleneimine, on a Brandel cell harvester. Radioactivity was counted with Microscint on a TopCount NXT Scintillation Counter (PerkinElmer). Saturation binding assays and hyperbolic curve fitting of specific binding was used to determine radioligand binding affinities and receptor numbers for the CHO cell lines (hMOR, 1.02 ± 0.10 nM for 3H-DAMGO and 1.58 ± 0.11 pmol/mg; hDOR, 0.70 ± 0.11 nM [3H]-Diprenorphine and 1.46 ± 0.26 pmol/mg; hKOR, 1.07 ± 0.01 nM [3H]-U69,593 and 0.71 ± 0.12 pmol/mg).
35S-GTPγS binding to membranes
35S-GTPγS binding was determined in membranes prepared from CHO-hMOR and CHO-mMOR cells and brainstems isolated from adult male C57BL/6J and MOR-KO mice as described previously (Schmid et al., 2013). CHO-hMOR and CHO-mMOR cellular membranes, collected and prepared as described above with in GTPγS binding membrane buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA). Reactions (200 μl volume) were performed for 1 hour at 25 °C on 10 μg membranes suspended in assay buffer (50 mM Tris-Cl, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA) with 50 μM Guanosine-5”-diphosphate (GDP) and 0.1 nM 35S-GTPγS. Reactions were terminated by filtration through GF/B filter plates and radioactivity was counted as described above. For [35S]-GTPγS binding on brainstems isolated from C57BL/6J and MOR-KO mice, tissues were homogenized by polytronic tissue tearor and membranes were prepared as described above. Binding reactions, containing 2.5 μg protein, 1 mM dithiothreitol (DTT), 20 μM GPD and 0.1 nM 35S-GTPγS, were incubated at room temperature for 2 hours prior to harvesting. The average vehicle value for the CHO-hMOR membranes was 786 ± 78 cpm and the average fold over vehicle for DAMGO was 4.6± 0.26. The average vehicle value for the CHO-mMOR cell membranes was 694 ± 28 cpm and the average fold over vehicle for DAMGO was 5.9 ± 0.57. The average vehicle for the C57BL/6J brainstem membranes was 657 ± 62 cpm and the average fold over vehicle for DAMGO was 1.9 ± 0.03. The average vehicle for the MOR-KO brainstem membranes was 1647 ± 507 cpm.
cAMP accumumlation assay
CHO-hMOR, -hDOR and -hKOR cells were plated at a density of 4,000 cells per well of a 384-well, white-walled, 30 μl-volume microplate (Greiner Bio-One) in Opti-MEM containing 1% FBS 4 hours prior to assaying. Cells were treated with 20 μM forskolin, 25 μM 4-(3-Butoxy-4-methoxybenzyl)imidazolidin-2-one (Ro-20-1724) and increasing concentrations of test compounds for 30 minutes at 25 °C. Inhibition of cAMP was then determined using the Homogenous Time-Resolved Fluorescence resonance energy transfer (FRET) cAMP HiRange assay by Cisbio (Cisbio-62AM6PEC). Fluorescence was measured at 620 and 665 nm using an Envision Multilabel Reader (PerkinElmer). FRET was calculated by the ratio of 665 nm / 620 nm. The average vehicle ratio for CHO-hMOR cells was 3134 ± 99 and the average fold over vehicle for DAMGO was 2.2 ± 0.04. The average vehicle ratio for CHO-hDOR cells was 2962 ± 181 and the average fold over vehicle for SNC80 was 1.6 ± 0.04. The average vehicle ratio for CHO-hKOR cells was 2965 ± 153 and the average fold over vehicle for U69,593 was 1.9 ± 0.12.
βArrestin2 recruitment assays
To determine βarrestin2 recruitment to the human MOR a commercial enzyme fragment complementation assay (β-galactosidase) was used. U2OS-βarrestin-hMOR PathHunter® cells were plated at a density of 5,000 cells per well of a 384-well, white-walled assay microplate (Greiner Bio-One) in Assay Complete Cell Plating 5 Reagent (DiscoveRx) 16–20 hours prior to measuring the signal. Cells were treated for 90 minutes with increasing concentrations of test compounds at 37 °C and βarrestin2 recruitment was determined using the PathHunter® Detection Kit with the β-galactosidase substrate to detect functional β-galactosidas. The resulting increase in luminescence was measured using a SpectraMax M5e Microplate Reader (Molecular Devices). The average vehicle for the PathHunter U2OS OPRM1 βarrestin cells was 446 ± 25 RLU and the average fold over vehicle for DAMGO was 36 ± 1.
To determine βarrestin2 recruitment to the mMOR, an imaging-based assay as was used (Zhou et al., 2013). U2OS-βarrestin2-GFP-mMOR cells were plated at a density of 5,000 cells per well of a 384-well, black-walled, clear-bottom optical imaging microplate (Brooks) in normal media 16–20 hours prior to assaying. Cells were serum-starved for 1 hour and then treated with increasing concentrations of test compounds for 20 minutes at 37 °C. Cells were fixed with 4% paraformaldehyde (PFA) containing Hoechst nuclear stain at a dilution of 1:1000. βArrestin 2 translocation was measured using the 20X objective on a CellInsight CX5 High Content Screening Platform (ThermoFisher Scientific). Punctae (normalized to Hoechst stain) were quantified using the Cellomics’ Spot Detection BioApplication (ThermoFisher Scientific). The average punctae / Hoechst ratio for vehicle treated U2OS-βarrestin2-GFP-mMOR cells was 2.2 ± 0.54 and the average fold over vehicle for DAMGO was 61 ± 13.
To determine whether the compounds have activity at NOP, βarrestin2 recruitment to the receptor was determined in the U2OS-Tango-hOPRL1-bla cells. U2OS-Tango-hOPRL1-bla cells were plated at a density of 10,000 cells per well of a 384-well, black-walled, clear-bottom assay plate in 32 μl assay media (DMEM + 10% dialyzed FBS, 0.1 mM NEAA, 25 mM HEPES and 1% pen/strep) 16–20 hours prior to assaying. Cells were treated with increasing concentrations of test compounds for 5 hours at 37 °C. NOP activation was determined using the LiveBLAzer FRET-B/G loading kit with Solution D (ThermoFisher Scientific), according to the manufacturer’s protocol. FRET signal (excitation 409 nm, emissions at 460 nm and 530 nm) was measured using a SpectraMax M5e Microplate Reader (Molecular Devices). The average 460/530 ratio vehicle treated U2OS-Tango-hOPRL1-bla cells was 0.31 ± 0.03 and the average fold over vehicle for nociceptin was 7.6 ± 0.68.
Pharmacokinetics and plasma protein binding
Pharmacokinetic parameters were determined in the C57BL/6J mice by i.p. dosing. Plasma was generated by standard centrifugation techniques, resulting in ~10 μl of plasma that was immediately frozen. For brain collection, mice were sacrificed by cervical dislocation and brains were isolated and flash frozen in liquid nitrogen. Drug levels were determined using a LC (Shimadzu)-tandem mass spectrometry (AB Sciex) operated in positive-ion mode using multiple reaction monitoring methods (Brust et al., 2016). Plasma protein binding for fentanyl and morphine was determined using Rapid Equilibrium Dialysis (RED) devices (ThermoFisher). For the SR compounds, plasma samples (0.5 mL at 0.5 μM test compound) were prepared and 900 μl was transferred to a 2 mL polycarbonate ultracentrifuge tube. The sample was centrifuged at 400,000 x g for two hours using a Beckman Coulter Optima Max ultracentrifuge (130,000 RPM max) with a TLA 120.2 rotor held at 25°C. The centrifuged sample separates into three layers. The protein-rich bottom layer contains most of the albumin and is easily visualized. The top layer is not as easily discerned, but contains a high concentration of lipoproteins. The middle layer (1–2 mm below surface using the described conditions) has very low protein concentrations and can be used to determine the amount of unbound drug. The percent unbound compound was determined by LC-MS/MS by comparison of the compound concentration in the middle layer of the centrifuged sample to the concentration of a parallel sample that did not undergo centrifugation (Kieltyka et al., 2016).
Antinociception
Antinociceptive responses to thermal stimuli were determined according to previously published protocols (Bohn et al., 1999; Raehal, 2011). Basal nociceptive responses were determined by measuring the amount of time until a mouse rapidly flicked its tail when placed into a 49 °C water bath (tail flick test) or until it licked or flicked its fore- or hind-paws when placed on a to a 52 °C hot plate (hot plate test; Hotplate Analgesia Meter, Columbus Instruments). Baseline response latencies averaged 2.95 ± 0.07 seconds (tail flick) and 6.17 ± 0.06 seconds (hot plate) for C57BL/6J male mice, 2.34 ± 0.18 seconds (tail flick) and 6.78 ± 0.14 seconds (hot plate) for C57BL/6J female mice and 2.29 ± 0.12 seconds (tail flick) and 6.54 ± 0.17 seconds (hot plate) for MOR-KO male mice. Antinociceptive responses were determined at the indicated time points over the course of 6 hours immediately following injection. To minimize tissue damage, maximum response latencies were limited to 30 and 20 seconds for tail flick and hot plate assays, respectively. Data are presented as “% maximum possible effect” which was calculated by (response latency – baseline) / (maximal response cutoff latency – baseline) * 100.
Respiration
A MouseOx Plus® pulse oximeter (Starr Life Sciences Corporation) was used to monitor mouse vital signs following drug treatment. Two days prior to testing, mice were shaved around the neck and habituated to the oximeter collars and 50 ml conical tubes that had been modified to restrain mice during testing. Mice were again habituated to the collars and conical tubes one day prior to testing. On the testing day, mice were fit with collars and returned to the conical tubes and basal vital signs were monitored for 30 minutes; mice were then immediately injected with drug and vital signs were monitored for an additional hour. Raw data were averaged into 5 minute bins. The average baseline responses (average over first 30 minutes) for C57BL/6J male mice were 95.11 ± 0.12 % (% oxygen saturation) and 165.0 ± 0.2 bpm (breath rate). The average baseline responses for C57BL/6J female mice were 96.30 ± 0.32 % (% oxygen saturation) and 150.1 ± 1.6 bpm (breath rate). The average baseline responses for MOR-KO male mice were 94.14 ± 0.38 % (% oxygen saturation) and 156.6 ± 2.5 bpm (breath rate). Data are presented as “% maximum possible effect” which was calculated by (response – average baseline) / (maximal response cutoff – average baseline) * 100. The maximum responses cutoff for % oxygen saturation and breath rate were set at 70% O2 and 75 breaths per minute (brpm), respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Software and analysis
GraphPad Prism software (v. 7.0) was used for data and statistical analyses which are specifically described in the figure legends. All data are presented as mean ± S.E.M or 95% confidence intervals, as indicated. For the in vitro studies, the compounds were assayed in duplicate or triplicate, with at least 3 independent replicates. Concentration response curves are presented as % of DAMGO, as indicated in the figures, and were fit to a non-linear regression (three parameter) model to determine EC50 and EMAX, with the average of the values from each individual experiment reported. For brainstem GTPγS binding assays, all studies were performed on brainstem taken from individual mice (one mouse brainstem per n, n ≥ 3). For the in vivo experiments, the number of animals used in each of the assays is provided in Table S4.
Calculation of signaling bias
For the in vitro assays, DAMGO was used as the reference compound in every experiment for normalization. For the calculation of bias, each concentration response curve was fit to the operational model based on the model by Black and Leff (Black and Leff, 1983) and is provided in the Prism software:
where EMAX is the maximal response, A is the molar concentration of the drug, KA is the equilibrium dissociation constant, the τ parameter is defined as the agonist efficacy and the log(τ/KA) is the transduction coefficient. For each assay, the EMAX is constrained to be a shared value and the log(KA) is constrained so that it must be between zero and -15 M to permit convergence of the model. Within each individual experiment, the log(τ/KA) values were calculated for the reference agonist, DAMGO, and the test agonists and then Δlog(τ/KA) values were calculated with the constraint that they be an absolute less than 10 by the equation:
The Δlog(τKA) values were fit within each individual experiment and then the values were averaged for to generate a mean Δlog(τ/KA) and error (S.E.M.), which are provided in Table 2. (This method of determining Δlog(τ/KA) has been demonstrated to be normally distributed and, therefore, appropriately estimates the Δlog(τ/KA) of the test ligand in each population (Stahl et al., 2015).) ΔΔLog(τ/KA) values were derived by subtracting the mean Δlog(τ/KA) produced from multiple independent experiments for assay 2 (βarrestin assay) from the similarly calculated Δlog(τ/KA) for assay 1 (GTPγS binding or cAMP inhibition) and is presented as the mean with the 95% confidence intervals. Error is propagated by using Prism v. 7.0 and comparing the two Δlog(τ/KA) values via an unpaired t-test and acquiring the difference between the two values with 95% confidence intervals. Bias factors were calculated by taking the antilog of the ΔΔlog(τ/KA) (Griffin et al., 2007; Kenakin et al., 2012):
Determination of ED50 and Therapeutic Window
For the in vivo studies, raw data were converted to the % maximal possible effect, as described in each behavioral method section above. An area under the curve (AUC) analysis was performed using GraphPad Prism, on each individual animal, with the baseline defined as time zero in the antinociception assays, and the mean response during the 30-minute habituation phase in the respiratory assays. Peaks that go below baseline were also considered. For the calculation of the AUC in the antinociception assays, only the data from the first hour after drug treatment was used. The AUC was then normalized to the percent maximal possible response within each assay (75 for the antinociception assays and 5750 for the respiratory assays). The ED50 values and asymmetric 95% confidence intervals were then calculated by fitting the AUC %MAX to a hyperbolic fit, with the maximum constrained to 100%. The therapeutic window was calculated by:
Supplementary Material
Figure S1. Related to Table 1. Synthesis of MOR selective ligands
General synthesis scheme for novel piperidine MOR agonists. The versatile 5-step route includes a nucleophilic aromatic substitution reaction, nitro reduction, urea formation, Boc group deprotection, and finally a direct alkylation or a reductive amination reaction. Solvent or reagent abbreviations are as follows: dimethyl sulfoxide (DMSO), carbonyl diimidazole (CDI), tetrahydrofuran (THF) and trifluoroacetic acid (TFA).
See also: Table 1 for structure activity relationships in this series; Table S1 for binding affinities for MOR over other opioid receptors and Figure S2 for functional profiling at DOR, KOR and NOP.
Figure S2. Related to Table 1. Functional effects of MOR agonists at the human DOR, KOR and NOP.
The SR compounds are not potent agonists or antagonists in DOR, KOR or NOP functional assays. Data are presented as mean ± S.E.M. of 3 independent assays, assayed in duplicate. Agonism and antagonism (“antag”, right side of graph) of forskolin-stimulated cAMP accumulation were determined in (A) CHO-hDOR or (B) CHO-hKOR cells. The antagonist assays were performed in the presence of 100 nM SNC80 and compared to naltrindole for the DOR and in the presence of 100 nM U69,593 and compared to nor-Binaltorphimine (NorBNI) for the KOR. (C) Agonism and antagonism of βarrestin2 recruitment to NOP was determined by a FRET-based assay in U2OS-Tango-hOPRL1-bla cells. The antagonist assays were performed in the presence of 10 nM nociceptin, and compared to the NOP antagonist, [Nphe1]Nociceptin(1–13)NH2.
See also: Tables 1 and S1 for potency and binding affinities at the human opioid receptors.
Figure S3. Related to Figure 1 and Table 2. GTPγS binding at mouse MOR expressed in CHO cells and mouse brainstem compared to βarrestin2 recruitment to mouse MOR
MOR agonist stimulated 35S-GTPγS binding in membranes from (A) CHO-mMOR cells or from (B) brainstem membranes from C57BL/6 mice. (C) βarrestin2 recruitment to the mMOR as determined by an imaging based assay in U2OS-βarrestin2-GFP-mMOR cells. For A–C, the data were normalized to the % maximal response for DAMGO and are presented as mean ± S.E.M. of 3 or more assays (1 mouse/assay) performed in duplicate or triplicate. (D) The SR compounds do not stimulate 35S-GTPγS binding over vehicle levels in brainstem membranes isolated from MOR-KO mice. The data are presented as fold over vehicle, with the mean ± S.E.M. of 3 or more assays (1 mouse/assay) assayed in quadruplicate.
See also: Tables 2 and S2 for potency, efficacy and bias factors, and Figure 1E for graphical representation.
Figure S4. Related to Figure 3 and Table 3. Dose response for the fentanyl, morphine and the SR compounds in the antinociception and respiratory assays and efficacy in female mice.
Antinociception dose response studies (mg/kg, i.p. doses in legends) in C57BL/6J male mice were performed for (A) hot plate (52°C) and (B) warm-water (49°C) tail flick assays over time. Respiratory dose response studies (mg/kg, i.p. doses in legends) in C57BL/6J male mice fit with a pulse oximeter to detect (C) % arterial oxygen saturation and (D) breath rate changes over time. Basal responses were determined for 30 minutes prior to injection (at time 0). Female C57BL/6J mice have similar (E) antinociceptive and (F) respiratory responses as their male counterparts at the doses tested (mg/kg i.p., doses in legend), n=4. MOR-KO mice displayed no response to the doses of compounds that produced near maximal effects in C57 mice (within MOR-KO, Student’s t-test: p > 0.05). The data are presented as mean ± S. E. M. of the % maximal possible effect (%MPE), with baselines determined for each mouse and the maximums set at 20 seconds for hot plate, 30 seconds for tail flick, 70% for oxygen saturation and 75 breaths per minute for breath rate measures.
See also: Figure 3C and Table 3 for ED50 values and therapeutic windows and Table S4 for the number of male mice used in each assay.
Acknowledgments
This work was supported by NIH grant R01 DA033073 (to L.M.B and T.D.B.) and R01 DA 038694 (to L.M.B.). The NIDA Drug Supply Program provided the morphine sulfate pentahydrate and sufentanil used in this study. We thank Dr. Lawrence Barak and the Addiction Research GPCR Assay Bank at Duke University for supplying the βarr2-GFP U2OS cells. WE thank Dr. Edward Stahl for helpful discussions regarding error analysis and bias factor calculations and Dr. Travis Grim for helpful discussions regarding in vivo statistical analyses. The authors have filed for a patent on compounds described herein.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, L.M.B, C.L.S. and T.D.B. Methodology, L.M.B, T.D.B, M.D.C., C.L.S., N.M.K, N.C.R., K.M.L. and Z.Y. Validation, J.M., N.M.K and K.M.L. Formal analysis, L.M.B. and C.L.S. Investigation, C.L.S., N.M.K, N.C.R, K.M.L., Z.Y. and J.M. Writing—original draft, L.M.B., T.D.B., C.L.S. and N.M.K. Writing—reviewing and editing, N.C.R. and M.D.C. Visualization, L.M.B., C.L.S. and T.D.B. Supervision, L.M.B. and T.D.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ananthan S, Saini SK, Dersch CM, Xu H, McGlinchey N, Giuvelis D, Bilsky EJ, Rothman RB. 14-Alkoxy- and 14-acyloxypyridomorphinans: mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J Med Chem. 2012;55:8350–8363. doi: 10.1021/jm300686p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, Zhou L, Stahl EL, Cameron MD, Scarry SM, et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal. 2016;9:ra117. doi: 10.1126/scisignal.aai8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu H, Liu X, Tian X, Yang H, Gao F. Enhancement of morphine analgesia and prevention of morphine tolerance by downregulation of beta-arrestin 2 with antigene RNAs in mice. Int J Neurosci. 2015;125:56–65. doi: 10.3109/00207454.2014.896913. [DOI] [PubMed] [Google Scholar]
- Budzik B, Garzya V, Shi DC, Walker G, Woolley-Roberts M, Pardoe J, Lucas A, Tehan B, Rivero RA, Langmead CJ, et al. Novel N-Substituted Benzimidazolones as Potent, Selective, CNS-Penetrant, and Orally Active M-1 mAChR Agonists. Acs Med Chem Lett. 2010;1:244–248. doi: 10.1021/ml100105x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, Sarton E, Teppema L, Olievier C, Nieuwenhuijs D, Matthes HW, Kieffer BL. Anesthetic potency and influence of morphine and sevoflurane on respiration in mu-opioid receptor knockout mice. Anesthesiology. 2001;94:824–832. doi: 10.1097/00000542-200105000-00021. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. The Journal of pharmacology and experimental therapeutics. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Frank RG, Pollack HA. Addressing the Fentanyl Threat to Public Health. N Engl J Med. 2017;376:605–607. doi: 10.1056/NEJMp1615145. [DOI] [PubMed] [Google Scholar]
- Griffin MT, Figueroa KW, Liller S, Ehlert FJ. Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. The Journal of pharmacology and experimental therapeutics. 2007;321:1193–1207. doi: 10.1124/jpet.107.120857. [DOI] [PubMed] [Google Scholar]
- Groer CE, Schmid CL, Jaeger AM, Bohn LM. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem. 2011;286:31731–31741. doi: 10.1074/jbc.M111.248310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen PA, Niemegeers CJ, Schellekens KH, Marsboom RH, Herin VV, Amery WK, Admiraal PV, Bosker JT, Crul JF, Pearce C, et al. Bezitramide (R 4845), a new potent and orally long-acting analgesic compound. Arzneimittelforschung. 1971;21:862–867. [PubMed] [Google Scholar]
- Kalvass JC, Olson ER, Cassidy MP, Selley DE, Pollack GM. Pharmacokinetics and pharmacodynamics of seven opioids in P-glycoprotein-competent mice: assessment of unbound brain EC50,u and correlation of in vitro, preclinical, and clinical data. J Pharmacol Exp Ther. 2007;323:346–355. doi: 10.1124/jpet.107.119560. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieltyka K, McAuliffe B, Cianci C, Drexler DM, Shou W, Zhang J. Application of Cassette Ultracentrifugation Using Non-labeled Compounds and Liquid Chromatography-Tandem Mass Spectrometry Analysis for High-Throughput Protein Binding Determination. J Pharm Sci. 2016;105:1036–1042. doi: 10.1016/S0022-3549(15)00177-X. [DOI] [PubMed] [Google Scholar]
- Kruegel AC, Gassaway MM, Kapoor A, Varadi A, Majumdar S, Filizola M, Javitch JA, Sames D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J Am Chem Soc. 2016;138:6754–6764. doi: 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu X, Liu C, Kang J, Yang J, Pei G, Wu C. Improvement of morphine-mediated analgesia by inhibition of beta-arrestin2 expression in mice periaqueductal gray matter. Int J Mol Sci. 2009;10:954–963. doi: 10.3390/ijms10030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley CW, Zhao ZJ, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorganic & Medicinal Chemistry Letters. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Melnikova I. Pain market. Nat Rev Drug Discov. 2010;9:589–590. doi: 10.1038/nrd3226. [DOI] [PubMed] [Google Scholar]
- Obase H, Takai H, Teranishi M, Nakamizo N. Synthesis of (1-Substituted Piperidin-4-Yl)-1h-Benzimidazoles and (1-Substituted Piperidin-4-Yl)-3,4-Dihydroquinazolines as Possible Antihypertensive Agents. J Heterocyclic Chem. 1983;20:565–573. [Google Scholar]
- Patel V, Bhatt N, Bhatt P, Joshi HD. Synthesis and pharmacological evaluation of novel 1-(piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one derivatives as potential antimicrobial agents. Med Chem Res. 2014;23:2133–2139. [Google Scholar]
- Raehal K, Groer CE, Schmid CL, Bohn LM. Functional selectivity at the mu opioid receptor: Implications for understanding opiate analgesia and tolerance. Pharmacological Reviews. 2011;63:58–65. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. Mu opioid receptor regulation and opiate responsiveness. AAPS J. 2005;7:E587–591. doi: 10.1208/aapsj070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Rankovic Z, Brust TF, Bohn LM. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett. 2016;26:241–250. doi: 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd RA, Seth P, David F, Scholl L. Increases in Drug and Opioid-Involved Overdose Deaths - United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65:1445–1452. doi: 10.15585/mmwr.mm655051e1. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at kappa-opioid receptors in striatal neurons. J Biol Chem. 2013;288:22387–22398. doi: 10.1074/jbc.M113.476234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla N, Minkowitz H, Soergel D, Burt D, Skobieranda F. (432) Respiratory safety signal with oliceridine (TRV130), a novel mu receptor G protein pathway selective modulator (mu-GPS), vs morphine: a safety analysis of a Phase 2b randomized clinical trial. J Pain. 2016;17:S82. [Google Scholar]
- Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Stahl EL, Zhou L, Ehlert FJ, Bohn LM. A novel method for analyzing extremely biased agonism at G protein-coupled receptors. Mol Pharmacol. 2015;87:866–877. doi: 10.1124/mol.114.096503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. Functional selectivity and classical concepts of quantitative pharmacology. The Journal of pharmacology and experimental therapeutics. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Zhao ZJ, William HL, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME, et al. Discovery of 2,3,5-trisubstituted pyridine derivatives as potent AM Akt1 and Akt2 dual inhibitors. Bioorganic & Medicinal Chemistry Letters. 2005;15:905–909. doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, et al. Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem. 2013;288:36703–36716. doi: 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Table 1. Synthesis of MOR selective ligands
General synthesis scheme for novel piperidine MOR agonists. The versatile 5-step route includes a nucleophilic aromatic substitution reaction, nitro reduction, urea formation, Boc group deprotection, and finally a direct alkylation or a reductive amination reaction. Solvent or reagent abbreviations are as follows: dimethyl sulfoxide (DMSO), carbonyl diimidazole (CDI), tetrahydrofuran (THF) and trifluoroacetic acid (TFA).
See also: Table 1 for structure activity relationships in this series; Table S1 for binding affinities for MOR over other opioid receptors and Figure S2 for functional profiling at DOR, KOR and NOP.
Figure S2. Related to Table 1. Functional effects of MOR agonists at the human DOR, KOR and NOP.
The SR compounds are not potent agonists or antagonists in DOR, KOR or NOP functional assays. Data are presented as mean ± S.E.M. of 3 independent assays, assayed in duplicate. Agonism and antagonism (“antag”, right side of graph) of forskolin-stimulated cAMP accumulation were determined in (A) CHO-hDOR or (B) CHO-hKOR cells. The antagonist assays were performed in the presence of 100 nM SNC80 and compared to naltrindole for the DOR and in the presence of 100 nM U69,593 and compared to nor-Binaltorphimine (NorBNI) for the KOR. (C) Agonism and antagonism of βarrestin2 recruitment to NOP was determined by a FRET-based assay in U2OS-Tango-hOPRL1-bla cells. The antagonist assays were performed in the presence of 10 nM nociceptin, and compared to the NOP antagonist, [Nphe1]Nociceptin(1–13)NH2.
See also: Tables 1 and S1 for potency and binding affinities at the human opioid receptors.
Figure S3. Related to Figure 1 and Table 2. GTPγS binding at mouse MOR expressed in CHO cells and mouse brainstem compared to βarrestin2 recruitment to mouse MOR
MOR agonist stimulated 35S-GTPγS binding in membranes from (A) CHO-mMOR cells or from (B) brainstem membranes from C57BL/6 mice. (C) βarrestin2 recruitment to the mMOR as determined by an imaging based assay in U2OS-βarrestin2-GFP-mMOR cells. For A–C, the data were normalized to the % maximal response for DAMGO and are presented as mean ± S.E.M. of 3 or more assays (1 mouse/assay) performed in duplicate or triplicate. (D) The SR compounds do not stimulate 35S-GTPγS binding over vehicle levels in brainstem membranes isolated from MOR-KO mice. The data are presented as fold over vehicle, with the mean ± S.E.M. of 3 or more assays (1 mouse/assay) assayed in quadruplicate.
See also: Tables 2 and S2 for potency, efficacy and bias factors, and Figure 1E for graphical representation.
Figure S4. Related to Figure 3 and Table 3. Dose response for the fentanyl, morphine and the SR compounds in the antinociception and respiratory assays and efficacy in female mice.
Antinociception dose response studies (mg/kg, i.p. doses in legends) in C57BL/6J male mice were performed for (A) hot plate (52°C) and (B) warm-water (49°C) tail flick assays over time. Respiratory dose response studies (mg/kg, i.p. doses in legends) in C57BL/6J male mice fit with a pulse oximeter to detect (C) % arterial oxygen saturation and (D) breath rate changes over time. Basal responses were determined for 30 minutes prior to injection (at time 0). Female C57BL/6J mice have similar (E) antinociceptive and (F) respiratory responses as their male counterparts at the doses tested (mg/kg i.p., doses in legend), n=4. MOR-KO mice displayed no response to the doses of compounds that produced near maximal effects in C57 mice (within MOR-KO, Student’s t-test: p > 0.05). The data are presented as mean ± S. E. M. of the % maximal possible effect (%MPE), with baselines determined for each mouse and the maximums set at 20 seconds for hot plate, 30 seconds for tail flick, 70% for oxygen saturation and 75 breaths per minute for breath rate measures.
See also: Figure 3C and Table 3 for ED50 values and therapeutic windows and Table S4 for the number of male mice used in each assay.