

Abstract
Background
The viral transactivator Tat protein is a key modulator of HIV-1 replication, as it regulates transcriptional elongation from the integrated proviral genome. Tat recruits the human transcription elongation factor b, and other host proteins, such as the super elongation complex, to activate the cellular RNA polymerase II, normally stalled shortly after transcription initiation at the HIV promoter. By means of a complex set of interactions with host cellular factors, Tat determines the fate of viral activity within the infected cell. The virus will either actively replicate to promote dissemination in blood and tissues, or become dormant mostly in memory CD4+ T cells, as part of a small but long-living latent reservoir, the main obstacle for HIV eradication.
Objective
In this review, we summarize recent advances in the understanding of the multi-step mechanism that regulates Tat-mediated HIV-1 transcription and RNA polymerase II release, to promote viral transcription elongation. Early events of the human transcription elongation factor b release from the inhibitory 7SK small nuclear ribonucleoprotein complex and its recruitment to the HIV promoter will be discussed. Specific roles of the super elongation complex subunits during transcription elongation, and insight on recently identified cellular factors and mechanisms regulating HIV latency will be detailed.
Conclusion
Understanding the complexity of HIV transcriptional regulation by host factors may open the door for development of novel strategies to eradicate the resilient latent reservoir.
Keywords: Tat, Tat-dependent HIV transcription, HIV latency, P-TEFb, SEC, 7SK snRNP, host factors
1. INTRODUCTION
Incredible progress has been made during the last 30 years of research on the human immunodeficiency virus 1 (HIV-1). The development of novel antiretroviral therapies (ART) has significantly improved the quality and the life expectancy of infected individuals. Unfortunately, this treatment does not eradicate the virus and patients must remain on antiretroviral drugs for life. HIV persists in a latent state in so-called latent reservoirs mainly composed of resting memory CD4+ T lymphocytes and macrophages [1, 2]. This long-lived reservoir is established early on after infection and can re-seed viral infection upon ART interruption.
Soon after HIV-1 entry into the host cells, the viral genomic RNA is reverse transcribed, viral DNA integrates into the host cell chromosomes, and the provirus becomes a cellular transcription unit. HIV transcription balances between two states both beneficial to the virus, active viral production of new infectious particles or a latent dormancy. After infection, active transcription from the integrated provirus is critical for viral dissemination into blood and lymphoid tissues, while entrance into latency allows viral escape from host immunological responses, a wait and see strategy punctuated by moments of stochastic transcriptional reactivation, which promotes replenishment of the latent viral reservoir granting a lifetime host infection [3]. The dogma is that HIV latency is an epiphenomenon resulting from an accidental return of newly infected activated CD4+ T cells to a resting state [2]. However, some believe that latency may confer evolutionary advantage to enhance viral transmission [4]. In any case, the entrance into latency and its maintenance is a complicated phenomenon, not yet fully understood, that is controlled and regulated by various mechanisms involving cellular host factors, discussed below.
1.1 The viral transactivator Tat
To support its own transcription, HIV-1 hijacks the host RNA polymerase II (RNAPII) machinery with the help of the viral transcriptional activator Tat, critical for robust and efficient viral expression [5]. In the absence of Tat, cellular transcription factors such as NF-κB, Sp1 and the TATA-box binding protein promote initiation of basal viral transcription at the 5′ long terminal repeat (LTR) promoter. However, RNAPII stalls and pauses just after transcribing a short transcript encoding an RNA stem loop structure termed TAR [5, 6]. Tat is initially expressed from splicing of rare two-exon transcripts, generally as a 101-amino acid protein, however a shorter and fully functional 86-amino acid version is also observed in some laboratory adapted strains [7]. The first 72-amino acids encoded from the first exon are the minimum module required for Tat-mediated transcription (TMT) and is composed of five domains including an acidic/proline-rich motif (amino acids 1–20), a zinc-finger/cysteine-rich motif (22–37), a core domain (41–48), a basic arginine-rich motif (ARM) (49–57) and the glutamic acid-rich motif (59–72) (Fig. 1) [7, 8]. The first three domains (1–48) constitute the minimum activation domain, while ARM is the RNA-binding domain which also encompasses the nuclear localization signal [7]. Tat’s main function is to potentiate HIV provirus transcription elongation by RNAPII by recruiting to the 5′LTR all the necessary transcriptional players, a process mediated by the interaction of Tat with TAR [9, 10].
Fig. 1. Schematic representation of HIV-1 Tat domains.
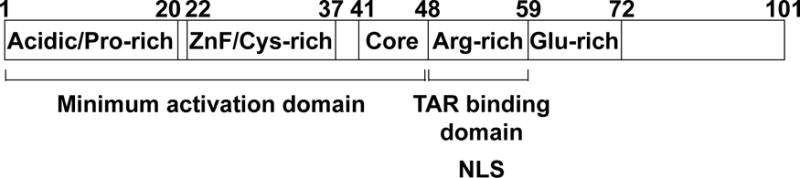
The minimum activation domain, the nuclear localization signal (NLS) as well as the basic region responsible for the interaction with TAR are represented. The numbering corresponds to the amino acids. ZnF, Zinc-finger.
1.2 Tat-regulated transcription elongation
A milestone in the field of HIV transcription was the discovery in the late nineties of the positive transcription elongation factor b (P-TEFb) as a necessary cofactor in TMT [11–13]. Human P-TEFb, composed of Cyclin-dependent kinase 9 (CDK9) and the Cyclin T (CycT) 1 or 2, is essential for RNAPII’s transition to productive elongation [14, 15]. Most cellular P-TEFb is found in an inactive form in complex with the 7SK small nuclear ribonucleoprotein (7SK snRNP) [16, 17]. However, P-TEFb can be recruited by Tat to the HIV-1 promoter, when Tat is acetylated at Lys28 by the p300/CBP-associated factor (PCAF) [18]. The activation domain of Tat folds upon interaction with CycT1 and with the T-loop of CDK9 [19]. These interactions stabilize P-TEFb and promote CDK9 autophosphorylation at a C-terminal Ser/Thr cluster, which is required for efficient TAR/Tat/P-TEFb complex formation at the vicinity of stalled RNAPII [20, 21]. Tat and P-TEFb post-translational modifications (PTMs) (acetylation, methylation, phosphorylation, polyubiquitination) play an important role in the regulation of TMT (reviewed in [3, 22, 23]). A recent study shows the importance of the lysine methyltransferase 7 (KMT7), encoded by the SET domain containing KMT7 gene, to facilitate TAR/Tat/P-TEFb complex formation and to increase TMT [24]. KMT7 binds TAR and monomethylates Tat at position Lys51 and Lys71, PTMs suggested to improve Tat’s affinity for TAR [24, 25]. The combination of Lys51Arg and Lys71Arg mutations almost completely abolish TMT [24]. Moreover, numerous studies have demonstrated the importance of the super elongation complex (SEC) to act in concert with the TAR/Tat/P-TEFb axis to promote TMT [26–28]. Mechanisms pertaining to SEC are detailed in section 3.
The transition of RNAPII from pausing at the HIV-1 genome start site, to transcription elongation, depends on CDK9 phosphorylation of several protein targets. CDK9 not only phosphorylates Ser2 of a heptapeptide repeat at RNAPII’s C-terminal domain (CTD), but also RNAPII-bound factors impairing transcription, such as the negative elongation factor complex (NELF) and the DRB sensitivity-inducing factor (DSIF) (reviewed in [29, 30]). Phosphorylation of the NELF-E subunit, promotes release of NELF from the complex, increasing the rate of RNAPII clearance from the LTR [31]. Phosphorylation of DSIF’s Spt5 subunit, converts it into a positive elongation factor, which then recruits Tat-SF1 and the human polymerase-associated factor complex 1 (PAF1) to the polymerase to promote full RNAPII activity [30]. Upon elongation, Tat becomes acetylated at Lys50 by p300/CBP and hGCN5 promoting its release from TAR and P-TEFb [32]. Tat can then recruit PCAF and the chromatin remodeling factor SWI/SNF to further facilitate the path of the polymerase by promoting an “open” chromatin environment [23, 33–35]. It has been shown that Tat and P-TEFb could stay associated with RNAPII during the elongation process and sustain several association/dissociation cycles [36, 37]. The highly processive RNAPII elongation complex can then increase transcription of the full-length viral mRNA by more than two orders of magnitude [38].
2. EARLY EVENTS IN TAT-MEDIATED HIV-1 TRANSCRIPTION
2.1. The 7SK snRNP complex
P-TEFb exists in all cells mostly in an inactive state as part of the 7SK snRNP complex. The latter includes the 7SK snRNA, a tridimensional 4 stem-loop structure that serves as a RNA scaffold for the La ribonucleoprotein domain family member 7 (LARP7), the methylphosphatase capping enzyme (MePCE), as well as the homodimer of the CDK9-inhibitory protein hexamethylene bisacetamide inducible 1 or 2 (HEXIM1/2) [39–43]. The primal function of LARP7 is to protect 7SK snRNA from nucleolytic degradation by binding the stem-loop 4 and the 3′ extremity of 7SK snRNA, and to promote the P-TEFb/7SK snRNP interaction by directly associating with CDK9 [44–47]. MePCE also stabilizes 7SK snRNA by binding at the base of the stem loop 1 and adding a methylphosphate cap at the 5′ end [41, 46, 48]. HEXIM1 directly binds to CycT1 and is responsible for the inactivation of the kinase activity of P-TEFb [49, 50]. The phosphorylation of the key residue Thr186, in the T-loop of CDK9, triggers binding of P-TEFb to HEXIM1 [49, 50]. A recent review thoroughly details the regulatory functions of the 7SK snRNP complex and surrounding factors, in the context of general RNAPII transcriptional elongation, with pertinent examples on HIV-1 Tat-dependent transcription [51]. In this section, we will report the latest developments regarding the role of cellular factors on the recruitment of P-TEFb/7SK snRNP to the chromatin, and the subsequent liberation of active P-TEFb from the inhibitory complex in the context of TMT.
2.2. Recruitment of P-TEFb/7SK snRNP to the HIV promoter
The 2010 study by the laboratories of D’Orso and Frankel revolutionized the thinking of how and when P-TEFb and Tat are mobilized to the HIV promoter, departing from the well-established idea of a sole TAR-dependent mechanism [52]. While their proposal is still not unanimously accepted, their successive studies demonstrated that Tat can be recruited to the HIV core promoter in association with the P-TEFb/7SK snRNP complex, even prior to TAR formation, in a Sp1-dependent manner [52–54]. Upon transcription initiation, Tat/P-TEFb can be rapidly mobilized to TAR appearing on nascent mRNA. Several studies have followed in an attempt to pinpoint the exact mechanism of P-TEFb/7SK snRNP recruitment to the HIV promoter.
2.2.1. KAP1
One of the most revealing studies came from the D’Orso laboratory which uncovered a role of the Krüppel-associated box (KRAB)-interacting protein 1 (KAP1), also called TRIM28 or TIF1β, in the P-TEFb/7SK snRNP recruitment to promoters with transcriptionally engaged but paused RNAPII where the transcription preinitiation complex (PIC) has been mobilized and the transcription initiated [55]. KAP1 interacts directly with the 7SK snRNP subunit LARP7 to pre-load the inactive P-TEFb to the promoter-proximal region, which upon stimulation facilitates a rapid transition of the paused RNAPII to productive transcription elongation [55]. This phenomenon was absent from both fully transcriptionally active genes and totally inactive genes [55, 56]. This mechanism, estimated to be specific to up to 70% of genes with promoter-proximal paused RNAPII, was first demonstrated at the HIV promoter [55]. Following KAP1 knock-down, P-TEFb/7SK snRNP localization at the 5′LTR decreased by 3 to 5-fold promoting a 2-fold RNAPII accumulation at the promoter most likely due to a decrease of its release to the open reading frame, without affecting PIC assembly and transcription initiation [55]. The authors demonstrated that upon TNF-α stimulation P-TEFb/7SK snRNP/KAP1 is recruited continuously as a whole to the promoter-proximal region to fuel the transcription elongation machinery with P-TEFb [55]. The study also points out that P-TEFb/7SK snRNP/KAP1 is recruited only at the promoter with ongoing transcription initiation and in absence of occluding nucleosomes, explaining the absence of P-TEFb/7SK snRNP/KAP1 in truly latently infected cell lines [55]. This study done in a Tat-free environment shows that the KAP1 recruiting function is Tat-independent. However, it could be hypothesized that the pre-loading of P-TEFb/7SK snRNP by KAP1 would facilitate P-TEFb release by Tat to initiate transcription elongation.
While the D’Orso study does not indicate how KAP1 is recruited to the promoter-proximal region, KAP1 has been described to bind to the KRAB motif of the Krüppel C2H2-type ZNF family and could therefore be recruited by a KRAB-ZNF to positively regulate transcription elongation [57, 58]. Two previous studies that contradict the D’Orso discovery have shown that KAP1, involved in the repression of HIV-1 LTR transcription, implicates two KRAB zinc finger proteins (ZNF), ZNF10 and ZNF350 [59, 60]. Alternatively, KAP1 recruitment could be mediated by a direct interaction with DNA or by binding through the intermediary of another protein, i.e. histones, subunit of the PIC or of the RNAPII transcription machinery. Future studies could clarify this question.
2.2.2. ZASC1
An interesting study proposed a mechanism in which Tat would be recruited along with P-TEFb/7SK snRNP to the HIV promoter, by the intermediary of the nine Krüppel-like ZNF 639, commonly called ZASC1 [61]. This cellular transcription factor binds both Tat and P-TEFb, recruiting them to the 5′LTR in complex with 7SK snRNP, in a TAR-independent manner (Fig. 2). To mediate DNA/protein interactions, ZASC1 was shown to bind two overlapping antiparallel binding sites immediately downstream of the TATA-box in the 5′LTR U3 promoter region [61]. This study also points out a possible cooperative role with Sp1 in the recruitment process. ZASC1 is found in complex with both the active and inactive forms of P-TEFb, suggesting a role beyond sole docking activity. While ZASC1 has no impact on HIV-1 basal transcription, the recruitment by ZASC1 of the Tat/P-TEFb/7SK snRNP complex promotes transcription elongation in cell lines and primary T-cells.
Fig. 2. Regulation of Tat-dependent HIV-1 transcription. 1) ZASC1 recruits the 7SK snRNP to the 5′LTR in a TAR-independent manner.
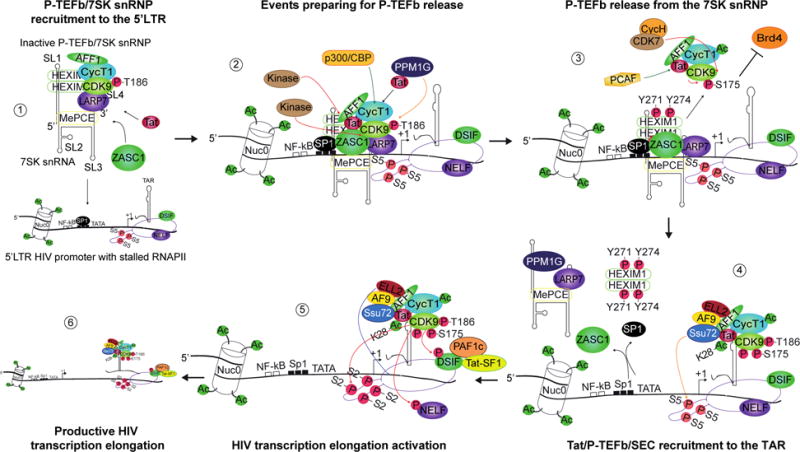
Tat binds to P-TEFb within the 7SK snRNP. ZASC1 binds to Tat and recruits the 7SK snRNP to the 5′LTR by binding two overlapping antiparallel binding sites immediately downstream of the TATA-box with a possible cooperative role of Sp1. 2) Events facilitating P-TEFb release from the 7SK snRNP. Tat competes HEXIM1 to bind P-TEFb and release it from the inhibitory 7SK snRNP complex, assisted by the scaffolding protein AFF1, which mediates an increase in Tat/CycT1 affinity for P-TEFb and by several coordinated PTMs of Tat, P-TEFb and HEXIM1. p300/CBP acetylates several residues within CycT1, a necessary step for P-TEFb release. A still uncharacterized kinase phosphorylates Tyr271 and Tyr274 on HEXIM1 facilitating 7SK snRNP dissociation. Phosphorylation of CDK9 Ser175 is also detrimental to the HEXIM1/P-TEFb interaction. Moreover, Tat recruits the phosphatase PPM1G to 7SK snRNP, which dephosphorylates CDK9 Thr186 leading to HEXIM1/P-TEFb dissociation and P-TEFb/Tat exit from the 7SK snRNP complex. 3) Tat/Brd4 competition for P-TEFb release from 7SK snRNP. Phosphorylated CDK9 at Ser175 favors Tat competition with Brd4 for P-TEFb binding, by increasing or decreasing their CDK9 affinity respectively and hence favors the formation of AFF1/Tat/P-TEFb over AFF1/Brd4/P-TEFb. Acetylation of Tat Lys28 is also important for TMT. Tat may be acetylated by PCAF within the 7SK snRNP complex or after Tat/P-TEFb release, a critical step in Tat-mediated P-TEFb recruitment to the TAR. The required CDK9 phosphorylation by CDK7/CycH at Thr186 is potentiated by Tat in complex with P-TEFb and the SEC subunit AFF1. Autophosphorylation of several Ser/Thr in the CDK9 C-terminus is required for Tat/P-TEFb association to TAR 4) SEC/Tat/TAR/P-TEFb association and role of Ssu72 in RNAPII-mediated transcription elongation. Following phosphorylation, HEXIM1 dissociates from 7SK snRNP and both release the 5′LTR. Active P-TEFb, is recruited by Tat to TAR, in complex with the complete AFF1/ELL2/AF9 SEC near the stalled RNAPII. Ssu72 is recruited by Tat and dephosphorylates RNAPII CTD Ser5. 5) CDK9 phosphorylates RNAPII and the negative elongation factors. CDK9 mediates Ser2 phosphorylation of the heptapeptide of the RNAPII CTD to activate transcription elongation. The negative factor DSIF is also phosphorylated by CDK9 becoming a positive elongation factor. PAF1c and Tat-SF1 are recruited to phosphorylated DSIF to facilitate productive elongation. NELF phosphorylation leads to its release and an increase of RNAPII clearance from the 5′LTR promoter. The SEC subunit ELL2 synergizes with Tat/P-TEFb to promote transcription elongation by preventing RNAPII backtracking. 6) RNAPII-mediated transcription elongation. The SEC/Tat/P-TEFb complex might stay associated to the elongating RNAPII. SL = stem-loop; P = phosphor; Ac = acetyl; T = Thr; Y = Tyr; S = Ser. Green arrow = acetylation; Red arrow = phosphorylation; Orange arrow = dephosphorylation; Blue arrow = ELL2 action on RNAPII.
2.2.3. HMGA1 and BCL11B
Another mechanism of 7SK snRNP recruitment to the HIV promoter, demonstrated in microglial cells, depends on the non-histone chromatin protein high mobility group AT-Hook 1 (HMGA1) [62]. HMAG1 recruits the B-cell CLL/lymphoma 11 B (BCL11B), also called CTIP2, found in complex with P-TEFb/7SK snRNP, by binding to both the 7SK stem-loop 2 and BCL11B in an RNA-dependent manner [62]. BCL11B, a C2H2-type ZNF, is exclusively expressed in microglial cells, and has been shown to inactivate the kinase activity of CDK9 in P-TEFb-containing 7SK snRNP [63]. Contrary to ZASC1, BCL11B and HMGA1 synergize to repress HIV-1 promoter activity inhibiting early Tat-independent transcription. The double protein knock-down of BCL11B and HMGA1 relieves the block, suggesting a role in the maintenance of latency in microglial cells, an important latent reservoir in the brain.
It is worth noting that BCL11B and ZASC1 which have a role in anchoring the 7SK snRNP to the HIV promoter share a C2H2-type ZNF motif, a feature commonly seen in mammalian transcription factors. Moreover, KAP1 is known to bind to KRAB-ZNF also presenting this motif. While the BCL11B/HMGA1 anchor function is only observed in microglial cells, ZASC1 and KAP1 have a broader distribution and are also expressed in primary T-cells, the main HIV-1 latent reservoir. However, as mentioned above, these proteins present opposite activities on HIV transcription, ZASC1 or KAP1 promotes transcription while BCL11B/HMGA1 promote latency. Questions remain such as cell-specificity, redundancy, or alternative mechanisms to recruit P-TEFb/7SK snRNP to the HIV promoter. Cell-specificity has already been observed with the KAP1 highly homologous TIF1γ (belonging to the same family), which can regulate transcription elongation in a small subset of erythroid specific genes. TIF1γ may interact and recruit P-TEFb in a yet unspecified manner, since the role of 7SK snRNP was not investigated in this study [64]. Moreover, KAP1 and ZASC1 may have redundant functions in CD4+ T cells or act sequentially depending on the degree of activation of the HIV promoter. KAP1 may be involved when Tat expression is low and most of the RNAPII is paused on the HIV promoter-proximal region, while ZASC1 could step up later to recruit both Tat and P-TEFb in complex with the 7SK snRNP. Further studies should address these multiple questions and help understand why HIV has hijacked, in different cell types, alternative anchoring mechanisms to regulate TMT.
2.3. Switching to active P-TEFb
2.3.1. P-TEFb release: Tat competes with HEXIM1
One proposed mechanism for P-TEFb release from 7SK snRNP is the direct competition between Tat and HEXIM1 for binding CycT1 and/or 7SK snRNA (Fig. 2) (reviewed in [51, 65]). In vitro studies show that Tat and HEXIM1 can both compete to recruit P-TEFb through the same N-terminal CycT1-binding site, with Tat/CycT1 affinity measured to be 10-fold greater, thus giving an advantage to Tat over HEXIM1 [16, 39, 66]. Moreover, AF4/FMR2 family member 1 (AFF1) part of the SEC supports Tat competition with HEXIM1 for P-TEFb release, by increasing the affinity of Tat for CycT1 [67]. Given the high homology between Tat ARM and HEXIM1 7SK snRNA-binding domain, Tat ARM could also interfere with HEXIM1 association with 7SK snRNA by binding to a TAR-like motif within the stem-loop 1 of 7SK snRNA [17, 68, 69]. However, this model backing a mechanic Tat/HEXIM1 competition for P-TEFb release done primarily in vitro using protein overexpression and non-stoichiometric condition might not fully reflect what is happening in vivo and not completely explain how Tat can access the CycT1 binding site already occupied by HEXIM1. Therefore, other studies point to the necessity of PTMs such as Tat and CycT1 acetylation, CDK9 and HEXIM1 phosphorylation/dephosphorylation, to trigger P-TEFb association/dissociation from the inhibitory 7SK snRNP complex (Fig. 2) [18, 54, 70–77].
2.3.2. P-TEFb release: influence of the P-TEFb/7SK snRNP PTMs
HEXIM1 phosphorylation is one proposed step in the release of inactivated P-TEFb from the 7SK snRNP complex. Previous studies showed that hexamethylene bisacetamide (HMBA) can activate the PI3K/Akt kinase pathway resulting in the HEXIM1 CycT1-binding domain phosphorylation at Thr270 and Ser278 and the subsequent P-TEFb release from HEXIM1 [72, 73]. Additionally, a positive role of the Jun N-terminal protein kinase (JNK) in HIV-1 reactivation from latency was discovered, by studying the activity of the JNK inhibitor AS601245, an event possibly mediated by the P-TEFb release from the inactive complex with HEXIM1 [74]. A recent study analyzing HEXIM1 PTMs upon PMA activation, using protein affinity purification coupled to mass spectrometry, revealed the importance of the phosphorylation of two Tyr at position 271 and 274 to release P-TEFb from 7SK snRNP (Fig. 2) [71]. The phosphomimetic double mutation Tyr271Glu/Tyr274Glu completely inhibits HEXIM1/P-TEFb formation, while a mutant hindering phosphorylation at position 271/274 completely blocks the release of P-TEFb [71]. Further studies are however needed to identify the kinase responsible for HEXIM1 phosphorylation and its general regulation.
The D’Orso laboratory reported the protein phosphatase, Mg2+/Mn2+ dependent 1G (PPM1G) as essential for the release of P-TEFb from the 7SK snRNP complex in a two-step mechanism (Fig. 2) [54]. As mentioned previously, the CDK9 phosphorylation at Thr186 is required to sequester P-TEFb into 7SK snRNP [49, 50]. However, the same Thr186 phosphorylation is required during Tat-dependent transactivation [78, 79]. This led to the postulate that P-TEFb phosphorylated at Thr186, while inhibited by HEXIM1 on 7SK snRNP complex, is in a pre-activated state to facilitate a rapid transition to transcription elongation upon activation [49, 51, 65]. In the D’Orso study, Tat recruits the PPM1G phosphatase to the LTR-bound 7SK snRNP complex to facilitate dephosphorylation of CDK9 at Thr186, abrogating P-TEFb/HEXIM1 interaction, resulting in the subsequent release of the transcription elongation factor [54]. PPM1A, a phosphatase of the same family, is also known to bind and dephosphorylate CDK9 at Thr186 in resting memory CD4+ T cells, but was unable to bind either Tat or the 7SK snRNP complex in the D’Orso study [54, 80, 81]. More studies are needed to fully understand the relevance of PPM1A phosphatase activity in the P-TEFb release. After dephosphorylation by PPM1G, P-TEFb is recognized by Tat, which potentiates CDK9 autophosphorylation and TAR/Tat/P-TEFb assembly. This study resulted in a better understanding of the Tat-dependent P-TEFb recruitment to the paused RNAPII to induce rapid gene activation and switch to efficient transcription elongation [54]. Of note, PPM1G can also initiate P-TEFb release from 7SK snRNP complex upon TNF-α activation in a Tat-independent manner, but only on specific cellular genes of the inflammatory pathway [54]. It is not the first time a phosphatase has been implicated in P-TEFb release from 7SK snRNP by dephosphorylating CDK9 Thr186. Indeed, Tat can also bind the protein phosphatase-1 (PP1), promoting its translocation to the nucleus where upon CDK9 dephosphorylation, releases P-TEFb, an essential step in the activation of TMT [75–77].
2.3.3. P-TEFb release: Tat competes with Brd4
Besides Tat, the bromodomain protein 4 (Brd4) can also recruit an active P-TEFb and release HEXIM1 from the 7SK snRNP [82]. On that account, P-TEFb is mainly found in complex with Brd4, when not inactivated by the 7SK snRNP complex [83]. Brd4 belongs to the large family of BRD-containing proteins which are a diverse set of transcriptional regulators that bind acetyl-lysine residues found on histones and transcription factors, and are readily targeted by small molecules [84]. Indeed, both Brd2 and Brd4 are known to regulate HIV latency, and a well-known BRD-inhibitor (JQ1) which targets these proteins, induces potent HIV reactivation [85–89]. Given the importance of acetyltransferases (p300/CBP) in regulating HIV-transcription, multiple BRD-containing proteins might regulate HIV-1 transcription. The inhibition of Brd4 function is therefore considered as a viable option to reactivate the virus from the latent reservoir, the first step in the Kick and Kill eradication strategy [27, 90]. This approach combines a reactivation “kick” event using latency-reversing agents (LRAs) with strategies to induce a robust anti-HIV immune response to “kill” the reactivated latently infected cells [90–92].
Brd4 binds acetylated H3/H4 histones near cellular promoters, enabling P-TEFb to contact the Mediator complex and promote P-TEFb-dependent transcription [93, 94]. To be noted, the Mediator complex, but not the Mediator CDK8 module, can interact with Tat and be recruited by the intermediate of the MED14 subunit to induce TMT [95]. Similar recruitment mechanism seems to take place on the HIV promoter with early data supporting a positive role of Brd4 on Tat-independent basal HIV-1 transcription [83, 94]. Other studies, mostly based on Brd4 overexpression, pointed out to a possible inhibitory effect of Brd4 on Tat-dependent transcription by directly competing with Tat for P-TEFb [87, 89, 94, 96]. Brd4/Tat competition was also supported by studies showing that BRD-inhibitors can reactivate HIV-1 from latency in an apparent Tat-dependent manner [87, 89]. However, JQ1 and other BRD-inhibitors were also able to reactivate latent HIV-1 from cell line defective in the TAR/Tat axis (ACH-2, U1) or lacking Tat (J-Lat A72) supporting at the least the existence of a Tat-independent mechanism [85, 88]. Nonetheless, if biologically relevant, the competition for P-TEFb between Brd4 and Tat would be particularly visible in primary latent memory CD4+ T cells where Tat expression is low [97–99]. Limited amounts of Tat and Brd4 expression are both factors influencing HIV latency. An important discovery by Jonathan Karn and colleagues demonstrates the determining role of the CDK9 Ser175 phosphorylation in regulating TMT by balancing the interaction ratio between Brd4/CDK9 and Tat/CDK9 (Fig. 2) [100]. Ser175 is found in the activation loop of CDK9 and is important for the interaction with Tat, but it seems critical for the interaction with Brd4, since an alanine mutation abolishes their association [100]. Moreover, CDK9 Ser175 phosphorylation reduces the binding to Brd4 and slightly enhances the interaction with Tat, stabilizing the Tat/CDK9 complex and excluding P-TEFb from the 7SK snRNP complex. CDK9 Ser175 phosphorylation is even more relevant in latent memory CD4+ T cells where residual amounts of Tat now become more competitive for P-TEFb recruitment to promote transcription elongation [100].
2.3.4. P-TEFb release: role of the SR-splicing factors 1 and 2
Both SRSF1 and SRSF2 associate with the 7SK snRNA and hence are recruited to endogenous active promoters through the 7SK snRNP to regulate cellular transcription pause release [101]. A study focusing mainly on SRSF2 demonstrated that this SR protein may act similarly to the TAR/Tat activation mechanism, by releasing together with P-TEFb from the 7SK snRNP complex, through binding of SRSF2 to the exonic splicing enhancer (ESE) binding-site appearing on nascent promoter-proximal RNA. Thereby, ESE are used as transcriptional elongation signals to trigger transcription, and participate in a chain of events that recruit the SEC, eventually leading to the release of RNAPII pausing at the gene promoter [101]. A study focusing on HIV transcription shows that early during infection the splicing factor SRSF1 plays a role in basal transcription by recruiting P-TEFb directly to the TAR sequence from the 7SK snRNP complex. SRSF1 downregulates Tat-dependent transactivation by competing with Tat for the binding to TAR on a partly overlapping sequence located on the bulge and apical loop of TAR, and by binding to sequences in the 7SK snRNA [102]. These results suggest that SR proteins have a role in both cellular and HIV transcriptional regulation to release and recruit P-TEFb, by directly binding to the viral or gene promoters.
2.3.5. SART3
Finally, the squamous cell carcinoma antigen recognized by T-cells 3 (SART3) has also been found to associate with the 7SK snRNP, but its exact role within the complex is still unknown [41, 54]. Several studies have reported a direct and specific contact of SART3 with both unphosphorylated RNAPII and Tat, promoting P-TEFb recruitment directly to the TAR/Tat/P-TEFb complex, leading to Ser2 phosphorylation of the RNAPII CTD and transcription elongation [103, 104]. The role of SART3 on HIV transcription has been recently reviewed by Whitmill et al. [105].
To summarize this section, we could hypothesize a model for the release of active P-TEFb from the 7SK snRNP to promoter-proximal stalled RNAPII, unifying the above-proposed mechanisms. In a Tat-free environment such as in latent cells, HIV-1 basal transcription elongation could be undertaken by several non-mutually exclusive mechanisms, for example a TAR-dependent pathway involving SRSF1, and a TAR-independent pathway mediated by Brd4 recruited through acetylated histones and the Mediator complex [83, 94, 102]. Once Tat is produced a new pathway is probably set in motion to replace less efficient and possibly competitive mechanisms such as the one mediated by Brd4. Tat would actively recruit P-TEFb from 7SK snRNP by competing directly HEXIM1 binding to an overlapping site within CycT1, a process facilitated by an AFF1-mediated increase in Tat/CycT1 affinity [16, 66, 67]. The important role of AFF1 and the SEC in TMT will be discussed below. A series of PTMs changes on the HEXIM1/P-TEFb complex appearing in a yet unknown sequence would promote the recruitment of P-TEFb by Tat. Phosphorylation of the HEXIM1 CycT1-binding domain either by a kinase from the PI3K/Akt pathway at Thr270 and Ser278 and/or by an unknown kinase at Tyr271 and Tyr274, would decrease the affinity of HEXIM1 for CycT1 [71–73]. Concomitantly, acetylation of CycT1 by p300/CBP would further decrease this interaction [70]. In parallel, Tat would most likely recruit one of the apparently redundant phosphatase, PPM1G or PP1, to dephosphorylate CDK9 Thr186, a necessary PTM for P-TEFb/7SK snRNP interaction [54, 75–77]. The switch in P-TEFb affinity from HEXIM1 to Tat would lead to P-TEFb release and its recruitment by Tat to the TAR region to phosphorylate the RNAPII CTD and promote efficient viral transcription elongation. P-TEFb/7SK snRNP and P-TEFb/Brd4 are the two-main sources of P-TEFb in vivo. Tat would compete Brd4 for P-TEFb release from 7SK snRNP facilitated by a change in affinity triggered by phosphorylation of CDK9 Ser175 [100]. It is still unclear whether the mechanisms described above are all relevant in vivo, since much of the supporting data came from in vitro studies. Additional research efforts should be directed at the understanding of the complex set of molecular events during basal transcription, TMT and latency.
3. THE SUPER ELONGATION COMPLEX
In 2010, two breakthrough studies from the Zhou and Benkirane’s laboratories showed Tat recruitment of P-TEFb to the viral promoter as part of a larger entity, the SEC [26, 28]. Several types of SEC exist in vivo, a family known to regulate transcription elongation of various cellular genes [106]. In addition to P-TEFb, a typical SEC is composed of MLL-fusion partners involved in leukemia, including the human transcription factors/coactivators AFF1/AFF4, the eleven-nineteen leukemia protein/ALL1-fused gene from chromosome 9 protein (ENL/AF9) and the elongation factor eleven-nineteen lysine-rich leukemia protein 1 or 2 (ELL1/ELL2) [106]. The Tat/P-TEFb/SEC complex play a crucial role in TMT by relieving promoter-proximal pausing of RNAPII resulting in a robust HIV transcription elongation. In this section, we will highlight the role of SEC subunits and host factors in the regulation of Tat-dependent transcription.
3.1. The scaffolding protein AFF1/4
The structurally disordered AFF1/4 serves as a flexible scaffold to bind other transcriptional elongation factors. AFF1/4 has separate binding sites for CycT1, ELL1/ELL2 and ENL/AF9 critical to bridge together the P-TEFb/SEC components [107, 108]. Insights into the overall architecture of the complex was given by Chou et al., which revealed short hydrophobic binding modules on AFF4 used to bind its different partners, with CycT1, ELL2 and ENL/AF9 binding respectively to AFF42–73, AFF4318–337 and AFF4710–729 [107]. A series of structural and biochemical studies helped clarify the role of SEC subunits especially AFF1/AFF4 in TMT. The tripartite 2.9-Å crystal structure between P-TEFb and the intrinsically disordered AFF42–73 fragment was first resolved revealing an interaction between AFF434–67, which fold on CycT1 opposite to the CycT1 CDK9-binding site [109]. The position and orientation taken by AFF4 in contact with CycT1 predicted a direct and adjacent interaction with Tat, later confirmed by the resolution of two independent quaternary structures of Tat/CycT1/CDK9/AFF4 at 2.9-Å and 3.0-Å [110, 111]. The Tat/CycT1 interaction is mediated by two Zn2+, with a special role of the second Tat zinc-finger located in the Tat Cys-rich motif, which interacts with the Cys261 residue within the CycT1249–261 Tat/TAR recognition motif (TRM) [112]. AFF4 increases the order of the flexible TRM, which by interacting tightly with Tat, exposes residues essential for Tat transcriptional activity such as the acetylated Lys28 and the Tat ARM, favoring an interaction with TAR [53, 110, 111]. Several key amino acids relevant for the Tat/CycT1/AFF1/4 interaction were revealed by these crystal structures. Modeling based on the quaternary crystal structure indicates that the TRM of CycT1 will be stabilized by the formation of a hydrogen bond between the acetylated Tat Lys28 and the CycT1 Asn257, thereby enhancing and facilitating the interaction with TAR [18, 110, 113]. The CycT1 binding site of AFF4 also makes multiple contacts with Tat, enhancing P-TEFb affinity for Tat by 11-fold and promoting SEC recruitment [109]. It also increases Tat-P-TEFb affinity for TAR by 30-fold favoring TAR recognition and binding [111]. Finally, the structure of TAR in complex with Tat/P-TEFb/AFF4 was recently solved albeit a 5.9-Å low-resolution [114]. Backed-up by various structural approaches such as small angle X-ray scattering, hydrogen-deuterium exchange, and 2′-hydroxyl acylation analyzed by primer (SHAPE), the structural study revealed a direct interaction of the TAR central loop with CycT1 TRM and the Tat Cys-rich motif containing the Zn2+ coordinating loop (Tat24–29), but detected no direct interaction with AFF4 [114]. Instead, AFF4 indirectly enhances TAR binding to the SEC by stabilizing the Tat/CycT1 interaction. While not directly observed due to poor resolution, modeling allowed positioning of the amino acid main chain of the Tat ARM within the major groove of TAR, in contact with both the TAR bulge and the RNA phosphate backbone [114].
3.2. AFF1-containing SEC as a true partner to HIV TMT
Recent studies indicate that the AFF1-containing SEC, but not AFF4, is the specific partner in the TMT pathway [67, 115, 116]. Lu et al. first demonstrated that AFF1 is found in all major P-TEFb-containing complexes that regulate cellular and HIV transcription (the 7SK snRNP, SEC and Brd4) and that P-TEFb/AFF1 is transferred as a single unit between those complexes during activation phases [67]. AFF1 increases the affinity of Tat for CycT1, thereby boosting the ability of Tat to compete with HEXIM1, extract P-TEFb from the 7SK snRNP complex, and integrate within SEC to stimulate transcriptional elongation [67]. When Tat-AFF1 interface was mutated, both their association and transactivation of the LTR were decreased. AFF1 rescues the lack of interaction between the CycT1 non-binding mutant Tat Cys22Gly and P-TEFb and boosts the transcriptional activity of this Tat mutant from 1.9 to 89-fold, showing the critical importance of AFF1 in TMT upregulation [67, 111]. It appears that full Tat/P-TEFb transcriptional activity is reached upon recruitment and formation of a complete SEC, containing all subunits, to the HIV promoter. A follow-up study showed that AFF1 and AFF4 are mutually exclusive and that AFF1-containing SEC seems to be more efficient at triggering Tat-dependent transcription elongation than the AFF4 SEC counterpart [116]. These differences lie in the differential ability of AFF1/4 to increase Tat affinity for P-TEFb. Structures of the Tat/P-TEFb/AFF4 complex reveal AFF4 Met62 and Phe65 as the main Tat-interacting residues, corresponding to Val67 and Phe70 in AFF1 [67, 111]. It is interesting to note that AFF1/Tat specificity lies within a single amino acid, Val67 in the cycT1-binding site of Tat [115, 116]. Indeed, the pocket containing the AFF1 Val67 is larger and can accommodate the acetylated Lys28 side chain of Tat, while the one with the bulkier AFF4 Met62 cannot [116]. This single difference makes AFF1 more prone to support Tat-dependent transcription while AFF4 is preferentially used during activation of the heat shock-response pathway [116].
The latest work from Zhou and colleagues, allied SEC-subunits knock-out and reactivation with LRAs, to study the role of AFF1/4 and ELL1/2 in the process of latency reversal in Jurkat-T cell line model of HIV latency [115]. They took advantage of the specificity of reactivation of two different types of LRAs, PMA and Prostratin to activate the PKC/NF-κB-mediated initiation pathway, and the Brd4-inhibitor JQ1, known to induce the Tat/SEC-stimulated elongation pathway [87, 89, 117–120]. They first showed that in HeLa and Jurkat T-lymphocytic cell lines, AFF4 is the predominant SEC-scaffolding protein, while AFF1 represents only a minor subset [115]. Both have separate roles in transcription elongation and reversal of latency. AFF4-SEC recruitment to the 5′LTR strongly impacts spontaneous Tat-independent reversal from latency, probably initiated by cellular complexes, such as the PAF complex (PAFc) or the Mediator complex. In contrast, AFF1-SEC is Tat-dependent, since reductions in AFF1 expression specifically repress Tat/SEC-dependent HIV-1 transcriptional elongation, as shown by experiments using the JQ1 Brd4-inhibitor believed to specifically affect this pathway [115]. Moreover, overexpression of AFF1 in Jurkat T cell lines, but not AFF4, could reverse latency without any reactivation agent. The same experimental setup showed the elongation factor ELL2, but not ELL1, as critical as AFF1 to reverse latency in a Tat-dependent manner [115]. This study also notes that the expression level of AFF1 and ELL2 in Jurkat T cells is suboptimal, representing a limiting factor for efficient reactivation from latency. Altogether, the small subset of AFF1/ELL2-containing SEC appears as a specific and fundamental complex in Tat-dependent HIV transcription (Fig. 2).
3.3. The elongation factor ELL2
The general role of the ELL1/ELL2 subunit is to prevent RNAPII backtracking to insure successful transcription elongation [121, 122]. As mentioned above, while ELL1 and ELL2 have redundant functions during cellular transcription elongation, only ELL2 has the ability to reverse HIV-1 from latency, attributed to differences in their N-terminal RNAPII binding domains [115]. In the presence of Tat, ELL2 can synergistically stimulate P-TEFb-dependent HIV transcription elongation [26]. This potent activation is due to a direct and close interaction of ELL2 with active P-TEFb mediated by AFF4, combined with a protective effect of Tat against ELL2 proteasomal degradation [26]. ELL2 is also able to connect the SEC complex to PAFc through binding to PAF1, a subunit previously found to affinity purify with the SEC [28, 107].
3.4. The co-activators ENL/AF9
Besides ELL2, ENL/AF9 can also directly bind to the PAF1 subunit, allowing PAFc to connect SEC to the RNAPII transcriptional unit in a Tat-independent manner [108]. Sohbian et al. demonstrated that AF9 is required for CDK9 to reach optimal kinase activity and to boost Tat-dependent transcription elongation [28]. AF9 and its homolog, ENL, compete for binding to the AFF4 scaffold and for direct interaction with P-TEFb. As such, these are not simultaneously present in the same SEC, but have an interchangeable role on Tat-dependent transcription elongation [108]. AF9 is however the preferred subunit in the SEC for optimal HIV transcription.
3.5. Role of the phosphatase Ssu72 within the SEC
Other factors revolving around the SEC may also help regulate TMT. Chen et al. show that the RNA polymerase II subunit A C-terminal domain phosphatase Ssu72 (Ssu72), a specific RNAPII Ser5 CTD phosphatase, is recruited by Tat, P-TEFb and AFF4 to the 5′ end of the HIV-1 promoter and associates with RNAPII until just before the termination site [123]. Tat seems to regulate the Ssu72 phosphatase/CDK9 kinase activity required for potent viral transcription elongation. Tat stimulates the phosphatase activity of Ssu72 in vitro in a dose-dependent manner, through direct binding to Ssu72 C-terminal coiled-coil domain, resulting in the removal of Ser5 and Ser7 phosphorylation, but not Ser2, from the RNAPII CTD heptatpeptide repeat [123]. Full-length Tat is required to bind Ssu72, with Arg49 and Arg53 of the Tat ARM being particularly important for the association. Moreover, the phosphatase activity of Ssu72 is necessary in vitro to decrease the CTD Ser5 phosphorylation to grant P-TEFb access to the CTD, a requirement possibly more nuanced and less pronounced in vivo. Altogether, this study shows that in the newly formed SEC, Tat in concert with the catalytic activity of Ssu72 and CDK9 promotes transition from Ser5 to Ser2 RNAPII CTD hyperhosphorylation and activates transcription elongation in a TAR- and CTD phosphatase-dependent manner (Fig. 2) [123].
In summary, in the last few years our knowledge of how the SEC assists Tat-dependent transcription has improved. The SEC has been found central and crucial in TMT, notably with the scaffolding protein AFF1/4 playing an early role in P-TEFb release from the 7SK snRNP inhibitory complex, increasing Tat/CycT1 affinity, by inducing an advantageous conformation to the newly formed Tat/P-TEFb/AFF1 complex. This mechanism supported by AFF1/4 seems in line with the unifying model presented earlier, with several modifying enzymes playing important roles to weaken/strengthen the interaction with P-TEFb, leading to recruitment by Tat (Fig. 2). The Tat/P-TEFb/AFF1 can then form a ternary complex with TAR, a process again facilitated by the SEC scaffolding proteins. AFF1 and AFF4 appear to have a redundant role, but AFF1 seems to be favored during TMT. Additional studies are needed to understand their relative roles in the formation of a competent elongation complex. Moreover, while AFF1 seems to be necessary for latency-reversal in a Tat-dependent fashion, AFF4 represents a likely general, alternate and complementary Tat-independent mechanism to reactivate HIV-1 from latency by the intermediate of cellular pathways. The discovery that AFF1/ELL2-SEC is found only as a minor complex in latently infected T cells, and that is the specific SEC complex required to initiate latency reversal, defines a potential target for the design of specific LRAs for the “Kick and Kill” strategy, reducing off target effects on AFF4-SEC regulated cellular pathways. An alternative would be to target specifically the subset Tat/P-TEFb/AFF1 with inhibitors to permanently suppress viral transcription and reactivation. This concept was demonstrated in our laboratory using the Tat inhibitor, didehydro-Cortistatin A (dCA) [124, 125]. Over time dCA drives HIV-1 gene expression into a state of persistent latency, or deep-latency, refractory to viral reactivation by the usual panel of LRAs in primary CD4+ T cells isolated from infected individuals [124]. Discontinuation of dCA treatment does not result in immediate viral rebound, as seen by measuring p24 capsid production and viral mRNA expression, and a lack of recruitment of RNAPII to the promoter. We postulated that the lack of occasional low-grade transcription caused by dCA may induce epigenetic changes that lock the HIV promoter in a sustainable latent state. Based on dCA’s mode of action, we have postulated a strategy of HIV-1 cure termed “Block-and-Lock”, where a specific HIV-1 transcriptional inhibitor would promote a durable state of latency, less susceptible to spontaneous reactivation during ART and when ART is discontinued [126–128].
4. HOST FACTORS INFLUENCING HIV-1 LATENCY
4.1 Generalities on HIV-1 latency
Multiple molecular mechanisms are responsible for the progressive silencing of HIV transcription in resting memory CD4+ T cells, and for maintenance of a latent state. These include transcriptional interference events, low levels or absence of important viral or cellular regulators of HIV transcription such as Tat, CycT1, NF-κB and NFAT, as well as the presence of repressors such as CBF-1 and YY1 (reviewed in [3]). As mentioned above, other complexes have a role in the induction/maintenance of latency such as the low level of the Tat-specific AFF1/ELL2-SEC, the competition of Tat/Brd4 for P-TEFb, and the HMGA1/BCL11B repressive role in microglial cells. The epigenetic regulation at nucleosomes surrounding the HIV promoter is also determinant for latency/active transcription balance. For instance, repressive marks including deacetylation and methylation of histone N-terminal tails are deposited by enzymes such as histone deacetylase 1 (HDAC1) and Suv39H1 [3]. Importantly, Tat acts as the master switch in the transition between HIV-1 latency and transcriptional activity [129]. Tat can recruit SWI/SNF a chromatin-remodeling factor, as well as histone acetyl transferases, such as p300/CBP to activate transcription from the integrated provirus [3, 130, 131]. It is therefore not surprising that host factors interact directly or indirectly with Tat to activate/suppress TMT and regulate viral transcription. The section below summarizes recent host factors and mechanisms influencing Tat transcriptional regulation.
4.2 7SK snRNA pseudouridylation
HIV latency seems to be linked to the pseudouridylation state of 7SK snRNA [132]. Most cellular 7SK snRNP is found pseudouridylated exclusively at uridine U250 by the cellular dyskerin pseudouridine synthase 1 (DKC1) [132]. Suppression of the catalytic subunit of DKC1, or mutations around U250, trigger 7SK snRNP disruption and P-TEFb release. This results in an increase of other P-TEFb-containing complexes, such as the SEC- and Brd4-P-TEFb consequently promoting cellular and HIV transcription. Interestingly, the DKC1 knock-down seems to preferentially increase the AFF1/ELL2-SEC formation favoring Tat-dependent transcription. Tat can then recruit P-TEFb-containing SEC to the 5′LTR, promoting transcription elongation and escape from latency. 7SK snRNA pseudouridylation seems to be an important intrinsic mechanism of maintenance of HIV latency by promoting a large inhibitory pool of P-TEFb/7SK snRNP.
4.3. The long non-coding RNA (lncRNA) NRON
Several studies have demonstrated the role of microRNA (miRNA) and lncRNA in regulating HIV replication and latency mostly by targeting cellular or viral factors [133, 134]. For example, a cluster of miRNAs targeting the 3′end of the viral mRNA have been found to potently promote latency in resting CD4+ T cells, by limiting general viral protein expression [135]. A recent study discovered the lncRNA NRON, enriched in resting CD4+ T cells, decreases viral transcription and induces latency by limiting the amounts of Tat [136]. NRON mediates specific Tat proteasomal degradation by linking Tat to the ubiquitin/proteasome subunits CUL4B and PSMD11. Moreover, it was shown in a primary CD4+ T cell model of HIV latency that NRON knocking-down alone could reactivate transcription from baseline by 12-fold, and in combination with the HDAC inhibitor SAHA, by 26-fold [136]. However, in resting CD4+ T lymphocytes isolated from three infected individuals, NRON knock-down alone was not sufficient but helped SAHA reactivation by 2 to 3-fold. Thus, by decreasing Tat abundance, this non-protein factor NRON, can help support latency in infected primary resting CD4+ T lymphocytes.
4.4. The facilitates chromatin transcription (FACT) complex
FACT complex, a heterodimer between SUPT16H and SSRP1, is a known histone chaperone that assembles/disassembles the nucleosomes on cellular promoters by interacting specifically with H2A/H2B dimer to promote transcription elongation by RNAPII [137]. However, Huang et al., using RNAi functional genomic screens, demonstrated that individual proteins from the FACT complex act as transcriptional repressors of HIV [138]. In this study, Tat was shown to recruit the FACT complex to the HIV-1 LTR promoter by interacting with SUPT16H [138]. By binding to Tat, SUPT16H decreases the Tat/CycT1 interaction interfering with transcriptional elongation in latent cell line models. Both SUPT16H and SSRP1 seem to have a role in maintaining latency since the depletion of either protein reverses latency in CD4+ T cell lines. However, the exact mechanism of inhibition by FACT is not known and could be different from a simple block between Tat and CycT1. Indeed, depletion of FACT in the U1 latent cell line, which contains a Tat-defective H13L mutant impairing P-TEFb binding, also lead to the activation of both transcription initiation and elongation, suggesting another unknown mechanism [138, 139]. Lastly, FACT seems to have a general role on retrovirus silencing since HTLV-1 Tax-dependent transcription was also affected [138]. Further studies will be required to understand the exact role of the subunits of FACT in TMT and HIV-1 latency in general.
4.5. The protein hypermethylated in cancer 1 (HIC1)
A recent study indicates that HIC1 represses Tat-dependent transcription in microglial cells [140]. The transcriptional repressor activity of HIC1 in tumor cell is regulated by post-translational modifications of its amino acid Lys314. This residue can be acetylated by p300/CBP, deacetylated by SIRT1, a class III NAD-dependent deacetylase, or SUMOylated by a pathway involving HDAC4 [141]. While SUMOylation is not necessary, deacetylation of Lys314 after SIRT1 recruitment by HIC1 is required for the transcriptional repressor to bind Tat and inhibit HIV-1 Tat-dependent transcription. HIC1 also co-immunoprecipitates with the chromatin regulator HMGA1, a protein known to bind TAR and to compete with Tat/TAR formation in vitro [142]. The authors suggested a coordinated role of HMGA1 recruiting HIC1/SIRT1 to the TAR vicinity, to suppress Tat-dependent transcription, a possible mechanism regulating latency maintenance [140].
4.6. The nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 (NUCKS1)
Low levels of NUCKS1 in latently infected cell line models such as ACH2 and J1.1 as compared to the parental cell lines, may explain HIV entry into latency [143]. NUCKS1 binds directly to Tat and seems important for the recruitment of Tat to TAR, since its depletion decreases Tat interaction with TAR. NUCKS1 appears as an important co-factor of Tat to promote transcriptional elongation in an NF-κB-independent manner. Altogether, the low abundance of the Tat-cofactor NUCKS1 in latently infected cells might be one more mechanism promoting HIV transcriptional dormancy.
4.7. The mTOR complex
Finally, Verdin and colleagues, using a pooled shRNA screen on a J-Lat based latent cell line model, identified host proteins regulating latency. The Ser-Thr kinase mTOR complexes 1 and 2 were reported as a possible pathway for latency reversal [126]. Other well-known complexes implicated in the latency process were identified such as P-TEFb and the silencing complex PRC2 (EED, EZH2 and YY1 subunits), validating the shRNA screen approach [23]. The authors used CRISPR interference to mTOR subunits, and specific small molecule inhibitors, to demonstrate the role of mTOR complexes in latency reversal [126]. Decrease in mTOR expression or reduced kinase activity impaired LRA-mediated or antigenic HIV-1 transcriptional activation, in latent cell line models and in primary CD4+ T lymphocytes isolated from ART-treated infected individuals. Tat-independent and Tat-dependent pathways were both moderately inhibited by mTOR inhibitors, with a 2-fold decrease of basal transcription and 2 to 3-fold dose-dependent inhibition of Tat-mediated LTR transactivation. This study suggests that inhibition of the mTOR signaling mediates a general downregulation of CDK9 phosphorylation subsequently impacting TMT and latency reversal [126].
CONCLUSION
The viral protein Tat, acting in concert with P-TEFb, plays a central role in the HIV replication cycle to promote strong transcriptional stimulation. In the last few years a great deal was uncovered regarding RNAPII promoter-proximal release in a Tat-dependent manner. The roles of major P-TEFb-containing complexes in TMT were clarified. Significant insight was gained on P-TEFb recruitment to the HIV promoter and release from the inhibitory 7SK snRNP complex, on Tat/Brd4 competition for P-TEFb to escape latency in primary CD4+ T lymphocytes, and finally on the crucial and specific role of AFF1/ELL2 SEC to initiate Tat-dependent transcription elongation. These key steps are all regulated by PTMs affecting several proteins involved in TMT. Additional studies are still needed to completely understand the Tat-dependent transcriptional regulation, e.g., uncovering all PTMs involved in regulating Tat/P-TEFb complex formation and the exact sequence of events. Much could be understood from higher-resolution structures of Tat/P-TEFb/SEC with TAR, as well as from additional structures of P-TEFb-containing complexes implicated in TMT. Future studies should focus on determining in vivo relevant events leading to the establishment of HIV latency reservoirs, and to unify found discrepancies, notably the order of events leading to P-TEFb recruitment to the HIV promoter. Fully understanding the mechanisms of TMT and latency establishment, will be incredibly helpful in designing specific strategies for HIV latency eradication, either through “Kick and Kill” or “Block and Lock” type of approaches.
Acknowledgments
Declared none.
G.M. is funded by the CFAR FY16 Pilot Award.
S.T.V. is funded by R01 AI097012-01A1, R01 AI118432-01A1.
This review was made possible by support for the Miami Center for AIDS Research (CFAR) at the University of Miami Miller School of Medicine funded by a grant (P30AI073961) from the National Institutes of Health (NIH), which is supported by the following NIH co-funding and participating institutes and centers: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, NIDDK, NIGMS, FIC and OAR. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
LIST OF ABBREVIATIONS
- 7SK snRNP
7SK small nuclear ribonucleoprotein
- AFF1/4
AF4/FMR2 family member 1 or 4
- ARM
Arginine-rich motif
- ART
Antiretroviral therapies
- BCL11B
B-cell CLL/lymphoma 11 B
- Brd4
Bromodomain protein 4
- CDK9
Cyclin-dependent kinase 9
- CTD
C-terminal domain
- CycT
Cyclin T
- dCA
Didehydro-Cortistatin A
- DKC1
Dyskerin pseudouridine synthase 1
- DSIF
DRB sensitivity-inducing factor
- ELL1/ELL2
Elongation factor eleven-nineteen lysine-rich leukemia protein 1 or 2
- ENL/AF9
Eleven-nineteen leukemia protein/ALL1-fused gene from chromosome 9 protein
- ESE
Exonic splicing enhancer
- FACT
Facilitates chromatin transcription complex
- HDAC
Histone deacetylase
- HEXIM1/2
Hexamethylene bisacetamide inducible 1 or 2
- HIC1
Protein hypermethylated in cancer 1
- HIV-1
Human immunodeficiency virus 1
- HMBA
Hexamethylene bisacetamide
- HMGA1
Non-histone chromatin protein high mobility group AT-Hook 1
- JNK
Jun N-terminal protein kinase
- KAP1
Krüppel-associated box (KRAB)-interacting protein 1
- KMT7
Lysine methyltransferase 7
- LARP7
La ribonucleoprotein domain family member 7
- lncRNA
Long non-coding RNA
- LRAs
Latency-reversing agents
- LTR
Long terminal repeat
- MePCE
Methylphosphatase capping enzyme
- mRNA
Messenger RNA
- miRNA
MicroRNA
- NELF
Negative elongation factor
- NUCKS1
Nuclear ubiquitous casein and cyclin-dependent kinase substrate 1
- PAF1
Polymerase-associated factor complex 1
- PAFc
PAF complex
- PCAF
p300/CBP-associated factor
- PIC
Transcription preinitiation complex
- PP1
Protein phosphatase-1
- PPM1A/G
Protein phosphatase, Mg2+/Mn2+ dependent 1A or G
- P-TEFb
Positive transcription elongation factor b
- PTMs
Post-translational modifications
- RNAPII
RNA polymerase II
- TAR
Transactivation response element
- TMT
Tat-mediated transcription
- TRM
Tat/TAR recognition motif
- SART3
Squamous cell carcinoma antigen recognized by T-cells 3
- SEC
Super elongation complex
- snRNA
Small nuclear RNA
- SRSF1/2
SR-splicing factors 1 and 2
- Ssu72
RNA polymerase II subunit A C-terminal domain phosphatase Ssu72
- ZASC1
Nine Krüppel-like zinc finger protein 639
- ZNF
Zinc finger protein.
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
G.M. and S.T.V. declare no conflict of interest.
References
- 1.Abbas W, Tariq M, Iqbal M, Kumar A, Herbein G. Eradication of HIV-1 from the macrophage reservoir: an uncertain goal? Viruses. 2015;7(4):1578–98. doi: 10.3390/v7041578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37(3):377–88. doi: 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:67. doi: 10.1186/1742-4690-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouzine IM, Weinberger AD, Weinberger LS. An evolutionary role for HIV latency in enhancing viral transmission. Cell. 2015;160(5):1002–12. doi: 10.1016/j.cell.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330(6147):489–93. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 6.Toohey MG, Jones KA. In vitro formation of short RNA polymerase II transcripts that terminate within the HIV-1 and HIV-2 promoter-proximal downstream regions. Genes Dev. 1989;3(3):265–82. doi: 10.1101/gad.3.3.265. [DOI] [PubMed] [Google Scholar]
- 7.Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem. 1999;274(41):28837–40. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 8.Jeang KT, Shank PR, Rabson AB, Kumar A. Synthesis of functional human immunodeficiency virus tat protein in baculovirus as determined by a cell-cell fusion assay. J Virol. 1988;62(10):3874–8. doi: 10.1128/jvi.62.10.3874-3878.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dingwall C, Ernberg I, Gait MJ, et al. HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. Embo j. 1990;9(12):4145–53. doi: 10.1002/j.1460-2075.1990.tb07637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dingwall C, Ernberg I, Gait MJ, et al. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc Natl Acad Sci U S A. 1989;86(18):6925–9. doi: 10.1073/pnas.86.18.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancebo HS, Lee G, Flygare J, et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11(20):2633–44. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92(4):451–62. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhu Y, Pe’ery T, Peng J, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11(20):2622–32. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall NF, Price DH. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992;12(5):2078–90. doi: 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270(21):12335–8. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 16.Barboric M, Yik JH, Czudnochowski N, et al. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007;35(6):2003–12. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sedore SC, Byers SA, Biglione S, et al. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007;35(13):4347–58. doi: 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Orso I, Frankel AD. Tat acetylation modulates assembly of a viral-host RNA-protein transcription complex. Proc Natl Acad Sci U S A. 2009;106(9):3101–6. doi: 10.1073/pnas.0900012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tahirov TH, Babayeva ND, Varzavand K, et al. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465(7299):747–51. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garber ME, Mayall TP, Suess EM, et al. CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat-P-TEFb complex to TAR RNA. Mol Cell Biol. 2000;20(18):6958–69. doi: 10.1128/mcb.20.18.6958-6969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baumli S, Lolli G, Lowe ED, et al. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. Embo j. 2008;27(13):1907–18. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho S, Schroeder S, Ott M. CYCLINg through transcription: posttranslational modifications of P-TEFb regulate transcription elongation. Cell Cycle. 2010;9(9):1697–705. doi: 10.4161/cc.9.9.11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ott M, Geyer M, Zhou Q. The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe. 2011;10(5):426–35. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali I, Ramage H, Boehm D, et al. The HIV-1 Tat Protein Is Monomethylated at Lysine 71 by the Lysine Methyltransferase KMT7. J Biol Chem. 2016;291(31):16240–8. doi: 10.1074/jbc.M116.735415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagans S, Kauder SE, Kaehlcke K, et al. The Cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription. Cell Host Microbe. 2010;7(3):234–44. doi: 10.1016/j.chom.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He N, Liu M, Hsu J, et al. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38(3):428–38. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu RD, Wu J, Shao R, Xue YH. Mechanism and factors that control HIV-1 transcription and latency activation. J Zhejiang Univ Sci B. 2014;15(5):455–65. doi: 10.1631/jzus.B1400059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sobhian B, Laguette N, Yatim A, et al. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38(3):439–51. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23(3):297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi Y, Shibata H, Handa H. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim Biophys Acta. 2013;1829(1):98–104. doi: 10.1016/j.bbagrm.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 31.Jadlowsky JK, Wong JY, Graham AC, et al. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol Cell Biol. 2014;34(11):1911–28. doi: 10.1128/MCB.01013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaehlcke K, Dorr A, Hetzer-Egger C, et al. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol Cell. 2003;12(1):167–76. doi: 10.1016/s1097-2765(03)00245-4. [DOI] [PubMed] [Google Scholar]
- 33.Agbottah E, Deng L, Dannenberg LO, Pumfery A, Kashanchi F. Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology. 2006;3:48. doi: 10.1186/1742-4690-3-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahmoudi T, Parra M, Vries RG, et al. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J Biol Chem. 2006;281(29):19960–8. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- 35.Treand C, du Chene I, Bres V, et al. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. Embo j. 2006;25(8):1690–9. doi: 10.1038/sj.emboj.7601074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bres V, Gomes N, Pickle L, Jones KA. A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev. 2005;19(10):1211–26. doi: 10.1101/gad.1291705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molle D, Maiuri P, Boireau S, et al. A real-time view of the TAR:Tat:P-TEFb complex at HIV-1 transcription sites. Retrovirology. 2007;4:36. doi: 10.1186/1742-4690-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sodroski J, Rosen C, Wong-Staal F, et al. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science. 1985;227(4683):171–3. doi: 10.1126/science.2981427. [DOI] [PubMed] [Google Scholar]
- 39.Michels AA, Nguyen VT, Fraldi A, et al. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol. 2003;23(14):4859–69. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yik JH, Chen R, Nishimura R, et al. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003;12(4):971–82. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 41.Jeronimo C, Forget D, Bouchard A, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27(2):262–74. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krueger BJ, Jeronimo C, Roy BB, et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008;36(7):2219–29. doi: 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markert A, Grimm M, Martinez J, et al. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 2008;9(6):569–75. doi: 10.1038/embor.2008.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barboric M, Lenasi T, Chen H, et al. 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc Natl Acad Sci U S A. 2009;106(19):7798–803. doi: 10.1073/pnas.0903188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He N, Jahchan NS, Hong E, et al. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell. 2008;29(5):588–99. doi: 10.1016/j.molcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muniz L, Egloff S, Kiss T. RNA elements directing in vivo assembly of the 7SK/MePCE/Larp7 transcriptional regulatory snRNP. Nucleic Acids Res. 2013;41(8):4686–98. doi: 10.1093/nar/gkt159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uchikawa E, Natchiar KS, Han X, et al. Structural insight into the mechanism of stabilization of the 7SK small nuclear RNA by LARP7. Nucleic Acids Res. 2015;43(6):3373–88. doi: 10.1093/nar/gkv173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue Y, Yang Z, Chen R, Zhou Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010;38(2):360–9. doi: 10.1093/nar/gkp977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen R, Yang Z, Zhou Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J Biol Chem. 2004;279(6):4153–60. doi: 10.1074/jbc.M310044200. [DOI] [PubMed] [Google Scholar]
- 50.Li Q, Price JP, Byers SA, et al. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem. 2005;280(31):28819–26. doi: 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- 51.Quaresma AJC, Bugai A, Barboric M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016;44(16):7527–39. doi: 10.1093/nar/gkw585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Orso I, Frankel AD. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol. 2010;17(7):815–21. doi: 10.1038/nsmb.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D’Orso I, Jang GM, Pastuszak AW, et al. Transition step during assembly of HIV Tat:P-TEFb transcription complexes and transfer to TAR RNA. Mol Cell Biol. 2012;32(23):4780–93. doi: 10.1128/MCB.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McNamara RP, McCann JL, Gudipaty SA, D’Orso I. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 2013;5(5):1256–68. doi: 10.1016/j.celrep.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McNamara RP, Reeder JE, McMillan EA, et al. KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol Cell. 2016;61(1):39–53. doi: 10.1016/j.molcel.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McNamara RP, Guzman C, Reeder JE, D’Orso I. Genome-wide analysis of KAP1, the 7SK snRNP complex, and RNA polymerase II. Genom Data. 2016;7:250–5. doi: 10.1016/j.gdata.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedman JR, Fredericks WJ, Jensen DE, et al. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996;10(16):2067–78. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 58.Iyengar S, Ivanov AV, Jin VX, Rauscher FJ, 3rd, Farnham PJ. Functional analysis of KAP1 genomic recruitment. Mol Cell Biol. 2011;31(9):1833–47. doi: 10.1128/MCB.01331-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishitsuji H, Abe M, Sawada R, Takaku H. ZBRK1 represses HIV-1 LTR-mediated transcription. FEBS Lett. 2012;586(20):3562–8. doi: 10.1016/j.febslet.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 60.Nishitsuji H, Sawada L, Sugiyama R, Takaku H. ZNF10 inhibits HIV-1 LTR activity through interaction with NF-kappaB and Sp1 binding motifs. FEBS Lett. 2015;589(15):2019–25. doi: 10.1016/j.febslet.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 61.Bruce JW, Reddington R, Mathieu E, et al. ZASC1 stimulates HIV-1 transcription elongation by recruiting P-TEFb and TAT to the LTR promoter. PLoS Pathog. 2013;9(10):e1003712. doi: 10.1371/journal.ppat.1003712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eilebrecht S, Le Douce V, Riclet R, et al. HMGA1 recruits CTIP2-repressed P-TEFb to the HIV-1 and cellular target promoters. Nucleic Acids Res. 2014;42(8):4962–71. doi: 10.1093/nar/gku168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cherrier T, Le Douce V, Eilebrecht S, et al. CTIP2 is a negative regulator of P-TEFb. Proc Natl Acad Sci U S A. 2013;110(31):12655–60. doi: 10.1073/pnas.1220136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bai X, Kim J, Yang Z, et al. TIF1gamma controls erythroid cell fate by regulating transcription elongation. Cell. 2010;142(1):133–43. doi: 10.1016/j.cell.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu H, Li Z, Xue Y, Zhou Q. Viral-host interactions that control HIV-1 transcriptional elongation. Chem Rev. 2013;113(11):8567–82. doi: 10.1021/cr400120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schulte A, Czudnochowski N, Barboric M, et al. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem. 2005;280(26):24968–77. doi: 10.1074/jbc.M501431200. [DOI] [PubMed] [Google Scholar]
- 67.Lu H, Li Z, Xue Y, et al. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Proc Natl Acad Sci U S A. 2014;111(1):E15–24. doi: 10.1073/pnas.1318503111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muniz L, Egloff S, Ughy B, Jady BE, Kiss T. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010;6(10):e1001152. doi: 10.1371/journal.ppat.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yik JH, Chen R, Pezda AC, Samford CS, Zhou Q. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol Cell Biol. 2004;24(12):5094–105. doi: 10.1128/MCB.24.12.5094-5105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cho S, Schroeder S, Kaehlcke K, et al. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. Embo j. 2009;28(10):1407–17. doi: 10.1038/emboj.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mbonye UR, Wang B, Gokulrangan G, Chance MR, Karn J. Phosphorylation of HEXIM1 at Tyr271 and Tyr274 Promotes Release of P-TEFb from the 7SK snRNP Complex and Enhances Proviral HIV Gene Expression. Proteomics. 2015;15(12):2078–86. doi: 10.1002/pmic.201500038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choudhary SK, Archin NM, Margolis DM. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J Infect Dis. 2008;197(8):1162–70. doi: 10.1086/529525. [DOI] [PubMed] [Google Scholar]
- 73.Contreras X, Barboric M, Lenasi T, Peterlin BM. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007;3(10):1459–69. doi: 10.1371/journal.ppat.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wolschendorf F, Bosque A, Shishido T, et al. Kinase control prevents HIV-1 reactivation in spite of high levels of induced NF-kappaB activity. J Virol. 2012;86(8):4548–58. doi: 10.1128/JVI.06726-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ammosova T, Jerebtsova M, Beullens M, et al. Nuclear targeting of protein phosphatase-1 by HIV-1 Tat protein. J Biol Chem. 2005;280(43):36364–71. doi: 10.1074/jbc.M503673200. [DOI] [PubMed] [Google Scholar]
- 76.Ammosova T, Yedavalli VR, Niu X, et al. Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J Biol Chem. 2011;286(5):3798–804. doi: 10.1074/jbc.M110.196493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen R, Liu M, Li H, et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008;22(10):1356–68. doi: 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramakrishnan R, Dow EC, Rice AP. Characterization of Cdk9 T-loop phosphorylation in resting and activated CD4(+) T lymphocytes. J Leukoc Biol. 2009;86(6):1345–50. doi: 10.1189/jlb.0509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramakrishnan R, Rice AP. Cdk9 T-loop phosphorylation is regulated by the calcium signaling pathway. J Cell Physiol. 2012;227(2):609–17. doi: 10.1002/jcp.22760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Budhiraja S, Ramakrishnan R, Rice AP. Phosphatase PPM1A negatively regulates P-TEFb function in resting CD4(+) T cells and inhibits HIV-1 gene expression. Retrovirology. 2012;9:52. doi: 10.1186/1742-4690-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y, Dow EC, Liang YY, et al. Phosphatase PPM1A regulates phosphorylation of Thr-186 in the Cdk9 T-loop. J Biol Chem. 2008;283(48):33578–84. doi: 10.1074/jbc.M807495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krueger BJ, Varzavand K, Cooper JJ, Price DH. The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS One. 2010;5(8):e12335. doi: 10.1371/journal.pone.0012335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jang MK, Mochizuki K, Zhou M, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–34. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 84.Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel. 2009;12(5):659–65. [PMC free article] [PubMed] [Google Scholar]
- 85.Banerjee C, Archin N, Michaels D, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol. 2012;92(6):1147–54. doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem. 2012;287(43):36609–16. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu J, Gaiha GD, John SP, et al. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012;2(4):807–16. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boehm D, Calvanese V, Dar RD, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013;12(3):452–62. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013;41(1):277–87. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Archin NM, Margolis DM. Emerging strategies to deplete the HIV reservoir. Curr Opin Infect Dis. 2014;27(1):29–35. doi: 10.1097/QCO.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cillo AR, Mellors JW. Which therapeutic strategy will achieve a cure for HIV-1? Curr Opin Virol. 2016;18:14–9. doi: 10.1016/j.coviro.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 92.Margolis DM, Garcia JV, Hazuda DJ, Haynes BF. Latency reversal and viral clearance to cure HIV-1. Science. 2016;353(6297):aaf6517. doi: 10.1126/science.aaf6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lu X, Zhu X, Li Y, et al. Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res. 2016;44(14):6853–67. doi: 10.1093/nar/gkw571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang Z, Yik JH, Chen R, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19(4):535–45. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 95.Ruiz A, Pauls E, Badia R, et al. Characterization of the influence of mediator complex in HIV-1 transcription. J Biol Chem. 2014;289(40):27665–76. doi: 10.1074/jbc.M114.570341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. 2007;104(34):13690–5. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Donahue DA, Kuhl BD, Sloan RD, Wainberg MA. The viral protein Tat can inhibit the establishment of HIV-1 latency. J Virol. 2012;86(6):3253–63. doi: 10.1128/JVI.06648-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122(2):169–82. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 99.Yukl S, Pillai S, Li P, et al. Latently-infected CD4+ T cells are enriched for HIV-1 Tat variants with impaired transactivation activity. Virology. 2009;387(1):98–108. doi: 10.1016/j.virol.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mbonye UR, Gokulrangan G, Datt M, et al. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog. 2013;9(5):e1003338. doi: 10.1371/journal.ppat.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ji X, Zhou Y, Pandit S, et al. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;153(4):855–68. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Paz S, Krainer AR, Caputi M. HIV-1 transcription is regulated by splicing factor SRSF1. Nucleic Acids Res. 2014;42(22):13812–23. doi: 10.1093/nar/gku1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu Y, Li J, Kim BO, Pace BS, He JJ. HIV-1 Tat protein-mediated transactivation of the HIV-1 long terminal repeat promoter is potentiated by a novel nuclear Tat-interacting protein of 110 kDa, Tip110. J Biol Chem. 2002;277(26):23854–63. doi: 10.1074/jbc.M200773200. [DOI] [PubMed] [Google Scholar]
- 104.Zhao W, Liu Y, Timani KA, He JJ. Tip110 protein binds to unphosphorylated RNA polymerase II and promotes its phosphorylation and HIV-1 long terminal repeat transcription. J Biol Chem. 2014;289(1):190–202. doi: 10.1074/jbc.M113.529784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Whitmill A, Timani KA, Liu Y, He JJ. Tip110: Physical properties, primary structure, and biological functions. Life Sci. 2016;149:79–95. doi: 10.1016/j.lfs.2016.02.062. [DOI] [PubMed] [Google Scholar]
- 106.Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13(9):543–7. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- 107.Chou S, Upton H, Bao K, et al. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc Natl Acad Sci U S A. 2013;110(2):E123–31. doi: 10.1073/pnas.1216971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.He N, Chan CK, Sobhian B, et al. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108(36):E636–45. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schulze-Gahmen U, Upton H, Birnberg A, et al. The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife. 2013;2:e00327. doi: 10.7554/eLife.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gu J, Babayeva ND, Suwa Y, et al. Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell Cycle. 2014;13(11):1788–97. doi: 10.4161/cc.28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schulze-Gahmen U, Lu H, Zhou Q, Alber T. AFF4 binding to Tat-P-TEFb indirectly stimulates TAR recognition of super elongation complexes at the HIV promoter. Elife. 2014;3:e02375. doi: 10.7554/eLife.02375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garber ME, Wei P, KewalRamani VN, et al. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12(22):3512–27. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kiernan RE, Vanhulle C, Schiltz L, et al. HIV-1 tat transcriptional activity is regulated by acetylation. Embo j. 1999;18(21):6106–18. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schulze-Gahmen U, Echeverria I, Stjepanovic G, et al. Insights into HIV-1 proviral transcription from integrative structure and dynamics of the Tat:AFF4:P-TEFb:TAR complex. Elife. 2016;5 doi: 10.7554/eLife.15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li Z, Lu H, Zhou Q. A Minor Subset of Super Elongation Complexes Plays a Predominant Role in Reversing HIV-1 Latency. Mol Cell Biol. 2016;36(7):1194–205. doi: 10.1128/MCB.00994-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lu H, Li Z, Zhang W, et al. Gene target specificity of the Super Elongation Complex (SEC) family: how HIV-1 Tat employs selected SEC members to activate viral transcription. Nucleic Acids Res. 2015;43(12):5868–79. doi: 10.1093/nar/gkv541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chatila T, Silverman L, Miller R, Geha R. Mechanisms of T cell activation by the calcium ionophore ionomycin. J Immunol. 1989;143(4):1283–9. [PubMed] [Google Scholar]
- 118.McKernan LN, Momjian D, Kulkosky J. Protein Kinase C: One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv Virol. 2012;2012:805347. doi: 10.1155/2012/805347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Siekevitz M, Josephs SF, Dukovich M, et al. Activation of the HIV-1 LTR by T cell mitogens and the trans-activator protein of HTLV-I. Science. 1987;238(4833):1575–8. doi: 10.1126/science.2825351. [DOI] [PubMed] [Google Scholar]
- 120.Williams SA, Chen LF, Kwon H, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem. 2004;279(40):42008–17. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- 121.Shilatifard A, Duan DR, Haque D, et al. ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc Natl Acad Sci U S A. 1997;94(8):3639–43. doi: 10.1073/pnas.94.8.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. An RNA polymerase II elongation factor encoded by the human ELL gene. Science. 1996;271(5257):1873–6. doi: 10.1126/science.271.5257.1873. [DOI] [PubMed] [Google Scholar]
- 123.Chen Y, Zhang L, Estaras C, et al. A gene-specific role for the Ssu72 RNAPII CTD phosphatase in HIV-1 Tat transactivation. Genes Dev. 2014;28(20):2261–75. doi: 10.1101/gad.250449.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mousseau G, Kessing CF, Fromentin R, et al. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio. 2015;6(4):e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mousseau G, Clementz MA, Bakeman WN, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe. 2012;12(1):97–108. doi: 10.1016/j.chom.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Besnard E, Hakre S, Kampmann M, et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe. 2016;20(6):785–97. doi: 10.1016/j.chom.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mousseau G, Mediouni S, Valente ST. Targeting HIV transcription: the quest for a functional cure. Curr Top Microbiol Immunol. 2015;389:121–45. doi: 10.1007/82_2015_435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Darcis G, Van Driessche B, Van Lint C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017;38(3):217–28. doi: 10.1016/j.it.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 129.Razooky BS, Pai A, Aull K, Rouzine IM, Weinberger LS. A hardwired HIV latency program. Cell. 2015;160(5):990–1001. doi: 10.1016/j.cell.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Easley R, Carpio L, Dannenberg L, et al. Transcription through the HIV-1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology. 2010;405(2):322–33. doi: 10.1016/j.virol.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mahmoudi T. The BAF complex and HIV latency. Transcription. 2012;3(4):171–6. doi: 10.4161/trns.20541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhao Y, Karijolich J, Glaunsinger B, Zhou Q. Pseudouridylation of 7SK snRNA promotes 7SK snRNP formation to suppress HIV-1 transcription and escape from latency. EMBO Rep. 2016;17(10):1441–51. doi: 10.15252/embr.201642682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rice AP. Roles of microRNAs and long-noncoding RNAs in human immunodeficiency virus replication. Wiley Interdiscip Rev RNA. 2015;6(6):661–70. doi: 10.1002/wrna.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun B, Yang R, Mallardo M. Roles of microRNAs in HIV-1replication and latency. Microrna. 2016 doi: 10.2174/2211536605666160829123118. [DOI] [PubMed] [Google Scholar]
- 135.Huang J, Wang F, Argyris E, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13(10):1241–7. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 136.Li J, Chen C, Ma X, et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat Commun. 2016;7:11730. doi: 10.1038/ncomms11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Formosa T. The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta. 2013;1819(3–4):247–55. [PubMed] [Google Scholar]
- 138.Huang H, Santoso N, Power D, et al. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J Biol Chem. 2015;290(45):27297–310. doi: 10.1074/jbc.M115.652339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reza SM, Rosetti M, Mathews MB, Pe’ery T. Differential activation of Tat variants in mitogen-stimulated cells: implications for HIV-1 postintegration latency. Virology. 2003;310(1):141–56. doi: 10.1016/s0042-6822(03)00106-5. [DOI] [PubMed] [Google Scholar]
- 140.Le Douce V, Forouzanfar F, Eilebrecht S, et al. HIC1 controls cellular- and HIV-1- gene transcription via interactions with CTIP2 and HMGA1. Sci Rep. 2016;6:34920. doi: 10.1038/srep34920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stankovic-Valentin N, Deltour S, Seeler J, et al. An acetylation/deacetylation-SUMOylation switch through a phylogenetically conserved psiKXEP motif in the tumor suppressor HIC1 regulates transcriptional repression activity. Mol Cell Biol. 2007;27(7):2661–75. doi: 10.1128/MCB.01098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Eilebrecht S, Wilhelm E, Benecke BJ, Bell B, Benecke AG. HMGA1 directly interacts with TAR to modulate basal and Tat-dependent HIV transcription. RNA Biol. 2013;10(3):436–44. doi: 10.4161/rna.23686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim HY, Choi BS, Kim SS, et al. NUCKS1, a novel Tat coactivator, plays a crucial role in HIV-1 replication by increasing Tat-mediated viral transcription on the HIV-1 LTR promoter. Retrovirology. 2014;11:67. doi: 10.1186/s12977-014-0067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]