

Abstract
Purpose of review
This review will critically highlight the role of leukotrienes as mediators of renal diseases and drug nephrotoxicity. It will also discuss the recently identified mechanism of cysteinyl leukotrienes induction and action, and will propose clinical implementation of these findings.
Recent findings
Since last reviewed in 1994, leukotrienes were shown to mediate drug-associated nephrotoxicity, transplant rejection and morbidity in several models of renal diseases. Although leukotrienes may be released by various infiltrating leukocytes, a recent study demonstrated that cytotoxic agents trigger production of leukotriene C4 (LTC4) in mouse kidney cells by activating a biosynthetic pathway based on microsomal glutathione-S-transferase 2 (MGST2). LTC4 then elicits nuclear accumulation of hydrogen peroxide-generating NADPH oxidase 4, leading to oxidative DNA damage and cell death. LTC4 inhibitors, commonly used as systemic asthma drugs, alleviated drug-associated damage to proximal tubular cells and attenuated mouse morbidity.
Summary
Cysteinyl leukotrienes released by mast cells trigger the symptoms of asthma, including bronchoconstriction and vasoconstriction. Therefore, effective leukotriene inhibitors were approved as orally administered asthma drugs. The findings that leukotrienes mediate the cytotoxicity of nephrotoxic drugs, and are involved in numerous renal diseases, suggest that such asthma drugs may ameliorate drug-induced nephrotoxicity, as well as some renal diseases.
Keywords: drug-associated toxicity, leukotriene C4, microsomal glutathione-S-transferase 2, NADPH oxidase 4
INTRODUCTION
Leukotrienes are members of the eicosanoid family of bioactive oxygenated fatty acids, containing 20 carbon atoms, and generated by a range of highly regulated biosynthetic pathways. An initial event, common to most of these pathways, is the activation of cytosolic phospholipase A2α (cPLA2α, officially termed PLA2G4A), resulting in its translocation to cellular membranes, where it hydrolyzes phospholipids to generate arachidonic acid. Free arachidonic acid may then be oxidized by the cyclooxygenases COX-1 and COX-2, followed by additional enzymatic transformations, to generate the families of prostaglandins and thromboxanes. Alternatively, arachidonic acid may be oxidized by 5-lipoxygenase (5LO, officially termed ALOX5), followed by additional enzymes, to form leukotrienes, lipoxins and resolvins. Eicosanoids act by binding to specific G-protein-coupled receptors (GPCRs), triggering a broad range of physiological and immunological activities. Several comprehensive reviews provide detailed description of these transformations, the structure and regulation of the enzymes involved, the eicosanoid receptors, and available drugs and other agents that inhibit biosynthesis of specific eicosanoids, or act as receptor antagonists [1–6,7▪].
Two types of leukotrienes, leukotriene B4 (LTB4) and leukotriene C4 (LTC4) are generated in mast cells and other immune cells by colocalization at the nuclear envelope of four components, thereby forming an active biosynthetic machinery: cPLA2α, 5LO, 5LO-activating protein (FLAP, officially termed ALOX5AP) and leukotriene C4 synthase (LTC4S). Arachidonic acid released by cPLA2α is oxidized by FLAP-activated 5LO to yield leukotriene A4 (LTA4), a short-lived intermediate that rapidly undergoes two alternative transformations. Its epoxide ring is either hydrolyzed by cytoplasmic LTA4 hydrolase (LTA4H), a fifth component of leukotriene biosynthetic machinery, to yield LTB4. Alternatively, LTC4S conjugates LTA4 with glutathione to form LTC4. LTB4 and LTC4 are actively effluxed from cells by the transporters MRP4 and MRP1, respectively. Following efflux, the glutathione residue of LTC4 may undergo partial and consecutive proteolysis by two outer cytoplasmic membrane-anchored proteases: γ-glutamyl transpeptidase and dipeptidase, generating LTD4 and LTE4. LTC4, D4 and E4 are collectively termed cysteinyl leukotrienes [2,3].
Circulating leukotrienes B4, C4, D4 and E4 bind to their GPCRs on target cells. LTB4 binds to two receptors termed BLT1 and BLT2. BLT1 is expressed on lymphocytes and mast cells, whereas the less specific BLT2 is ubiquitously expressed. LTB4 is a potent chemoattractant, triggering inflammatory responses. Two GPCRs, CysLT1 and CysLT2, bind the various cysteinyl leukotrienes with different affinities. CysLT1 binds LTD4 with the highest affinity, followed by LTC4 and LTE4 in decreasing order. CysLT2 binds LTC4 and LTD4 with equally high affinity and LTE4 with a lower affinity. Binding of these ligands to CysLT1 expressed by smooth muscle cells of the lungs triggers bronchoconstriction and vasoconstriction, the two symptoms of asthma. Binding to CysLT2, which is expressed in various organs, but not in lung cells, increases vascular permeability of small blood vessels.
The role of leukotrienes in various nephropathies was last reviewed in 1994. It covered studies showing that cysteinyl leukotrienes reduce renal blood flow and glomerular filtration rate (GFR) by triggering vasoconstriction. It also included studies showing the role of LTB4 in recruiting infiltrating polymorphonuclear (PMN) cells, thereby exacerbating immune-mediated damage to kidney functions [8]. Since then, many studies appeared, elucidating the role of leukotrienes in renal diseases, and further revealing the biochemistry and physiology of leukotrienes in general. The present review will focus on these more recent findings.
Box 1.
no caption available
ROLE OF LEUKOTRIENES IN MODELS OF RENAL DISEASES
The development of specific CysLT1 and CysLT2 receptor antagonists, as well as inhibitors of leukotriene biosynthesis, provided simple and powerful tools for studying the role of leukotrienes in renal diseases and drug-associated nephrotoxicity. Spurney et al. who studied the role of leukotrienes in renal allograft rejection, employing the FLAP inhibitor MK886, provided the first example. This agent attenuated the decline in GFR and renal plasma flow, and prolonged the survival of rats following allograft transplantation, [9]. FLAP activity is common to the biosynthesis of all leukotrienes, lipoxins and resolvins. Acting in a more specific manner, the cysteinyl leukotriene receptor antagonist SKF106203 was less effective than MK886, indicating that both cysteinyl leukotrienes and LTB4 promoted allograft rejection [9]. In another study, glomerular nephritis was initiated in rats by administrating rabbit anti-rat glomerular basement membrane (GBM) antibodies. Urinary excretion of LTC4, LTD4 and acetylated LTE4 was greatly increased following antibody administration, concomitantly with increased renal LTC4 synthase activity [10]. Burn-induced injury affects remote organs in a complex manner, involving oxidative stress and immune responses. In a rat model of burn injury, montelukast, a specific CysLT1 receptor antagonist, reduced kidney malondialdehyde (MDA), a marker of oxidative damage. It also reduced myeloperoxidase levels and kidney hemorrhages, and attenuated glomerular degeneration [11]. Similar results were seen in rats undergoing unilateral nephrectomy followed by ischemia–reperfusion triggered by transient ligation of the remaining renal pedicle. In addition, montelukast attenuated the treatment-associated increase in plasma LTB4 and pro-inflammatory cytokines [12], suggesting a cross talk between cysteinyl leukotrienes and LTB4. In another study by the same group, chronic renal failure was established in rats by 5/6 resection of the left kidney, followed by right kidney nephrectomy. Here too, montelukast attenuated the rise in kidney MDA, myeloperoxidase, LTB4 and cytokine levels, the drop in GSH and the damage to glomeruli structure [13]. In a mouse model of renal ischemia–reperfusion, MK886 attenuated oxidative stress, histopathological markers of tissue damage, cytokine release and damage to renal function [14]. Montelukast also reduced renal injury in a model of lipopolysaccharide-induced sepsis in rats, as determined by the levels of inflammatory and oxidative stress markers and by preservation of tissue morphology [15]. Furthermore, montelukast protected rats against acute kidney injury triggered by remote muscle rhabdomyolysis and by intestinal ischemia–reperfusion [16,17]. In all of these studies, no attempt was made to identify the cysteinyl leukotrienes producer cells.
Several models of renal diseases are associated with elevated levels of LTB4. Following administration of rabbit anti-rat GBM antibodies to rats, the specific BLT1 receptor antagonist ONO-4057 effectively reduced proteinuria and hematuria, kidney necrotizing lesions, mononuclear cell infiltration and glomerular deformation, despite no effect on glomerular IgG deposition [18]. Experimental nephritis is established in rats by administration of a monoclonal antibody to rat Thy-1.1, expressed on mesangial cells. Treatment with ONO-4057 somewhat attenuated proteinuria, reduced glomeruli PMN infiltration and reduced mesangial cell proliferation [19]. Diet-induced hyperlipidemia leads to glomerular sclerosis, contributing to renal injury. ONO-4057 significantly attenuated both basal and cholesterol-induced proteinuria and glomerular macrophage infiltration in kidneys of spontaneously hypercholesterolemic rats fed with a cholesterol-rich diet. Unexpectedly, it also reduced urinary LTB4 secretion, despite increased availability of the LT4H substrate LTA4, again suggesting a regulatory cross talk between LTB4 and cysteinyl leukotrienes [20].
LEUKOTRIENES AS MEDIATORS OF DRUG-ASSOCIATED NEPHROTOXICITY
Arachidonic acid is the common substrate of cyclooxygenases and lipoxygenases. It is, therefore, likely that cyclooxygenase inhibitors may increase the biosynthesis of lipoxygenase-derived products and vice versa. This issue is of great importance in view of the extensive and unregulated use of COX inhibitors as pain relievers. An early study proposed that ‘COX to LOX’ shunting may contribute to the development of a nephrotic syndrome in patients taking COX inhibitors [21]. The findings that SC75416, a COX-2 inhibitor, reduce renal blood flow and GFR in dogs given low-sodium diet supports this hypothesis. PF-150, a 5LO inhibitor, reversed these effects of SC75416, suggesting that blockade of COX-2 may have shunted arachidonic acid towards increased production of 5LO end products [22]. Yet, the effect was rather limited, in line with the fact that 5LO is a highly regulated enzyme and not just by substrate availability.
Very few studies linked nephrotoxic agents with the induction of LTB4 biosynthesis. In one study, cisplatin induced the biosynthesis of LTB4 and the expression of its BLT1 receptor in the kidney. The BLT1 receptor antagonist U-75302, BLT1 deficiency and the LTA4H inhibitor SC-57461A reduced kidney structural and functional damage, inflammation and apoptosis [23]. It should be noticed, however, that inhibition of LTA4H might direct its substrate LTA4 towards increased biosynthesis of cysteinyl leukotrienes. In another study, dioxin was shown to induce the expression of 5LO in infiltrating neutrophils in several tissues, including the kidney, resulting in elevated production of LTB4[24].
Several other studies implicated the production of cysteinyl leukotrienes in drug-associated nephrotoxicity. The immunosuppressant cyclosporine A (CsA) exhibits significant nephrotoxicity. The role of leukotrienes in CsA-associated nephrotoxicity was studied in rats following nonrejecting isograft renal transplantation. CsA administration resulted in vacuolization of proximal tubule cells, significantly reducing the GFR and renal plasma flow of the implanted kidney. In parallel, CsA triggered a 10-fold increase in cysteinyl leukotriene biosynthesis. Administration of SKF106203, a CysLT receptor antagonist completely reversed all of the CsA-mediated damage to renal structure and functions. These findings indicated that CsA is a potent inducer of cysteinyl leukotrienes, which serve as major mediators of CsA-triggered nephrotoxicity. Acting as an immunosuppressant, CsA reduced the biosynthesis of the chemoattractant LTB4 in the kidney. Nevertheless, the authors suggested that infiltrating macrophages were the source of LTC4[25]. Similar attenuation of CsA toxicity was later reported using the specific CysLT1 receptor antagonist montelukast [26]. In another study, treatment of rats with the bisphosphonate alderonate increased the expression of kidney myeloperoxidase, causing oxidative stress. Kidney histology revealed severe hemorrhagia and damage to glomeruli. Coadministration of montelukast ameliorated all the adverse effects of alderonate. The authors suggested that neutrophil infiltration was the source of the damaging cysteinyl leukotrienes [27]. Similar results were observed in kidneys of rats treated with cisplatin [28,29], amikacin [30], gentamycin [31] and methotrexate [32]. In all of these cases subsequent administration of montelukast attenuated the drug-triggered nephrotoxicity. The agricultural fungicide NDPS [N-(3,5-dichlorophenyl)-succinimide] and its metabolites NDHS [N-(3,5-dichlorophenyl)-2-hydroxysuccinimide] and 2-NDHSA [N-(3,5-dichlorophenyl)-2-hydroxysuccinamic acid] were shown to be nephrotoxic. Inhibition of 5LO markedly reduced all aspects of their nephrotoxicity in rats [33]. Several chemotherapeutic and cytotoxic agents were recently shown to act by inducing LTC4. In particular, LTC4 activity was implicated in tunicamycin and 5-fluorouracil (5-FU)-triggered nephrotoxicity [34▪▪]. Details of these observations are discussed in the following.
RECENT ADVANCES IN UNDERSTANDING THE MECHANISM OF LEUKOTRIENE INDUCTION AND ACTION
The function of LTB4 as a chemoattractant of inflammatory cells is relatively well understood. In contrast, until recently, studies describing the involvement of cysteinyl leukotrienes in kidney diseases failed to identify the producer cells or to reveal the mechanism of leukotriene action. Most studies assumed that infiltrating immune cells, expressing LTC4S, are the source of these leukotrienes. Yet, several studies demonstrated that nonimmune cells might also produce leukotrienes. Thus, MGST2 expressed in endothelial cells effectively converted exogenously added LTA4 to LTC4[35–37]. Furthermore, production of LTC4 in the testis was not impaired in LTC4S-deficient mice [38]. These studies indicated that MGST2 or MGST3 might serve as LTC4-generating enzymes in nonimmune cells. In the kidney, trauma and hemorrhagic shock led to colocalization of 5LO and FLAP in the interstitium and the tubule lumen [39]. As kidney cells were shown to express LTC4S [35,40], a biosynthetic machinery of cysteinyl leukotrienes based on 5LO, FLAP and LTC4S could theoretically be assembled in these cells. A recent study, however, revealed that ER stress and a broad range of cytotoxic agents trigger MGST2-based rather than LTC4S-based biosynthesis of LTC4 in many types of nonimmune cells and in mouse kidneys. In analogy with the LTC4S based machinery, biosynthesis was initiated by stress-triggered translocation and colocalization at the nuclear envelope of cPLA2, 5LO, FLAP and MGST2, thereby generating LTC4[34▪▪].
The same study also revealed the downstream action of MGST2-generated LTC4. Under stress, the two LTC4 receptors, CysLT1 and CysLT2 translocated to the nuclear envelope. Acting in an intracrine manner, LTC4 then triggered nuclear translocation of NADPH oxidase 4 (NOX4), resulting in oxidative DNA damage, dsDNA breaks and subsequent cell death (Fig. 1). Thus, MGST2-generated LTC4 orchestrates a death-promoting pathway parallel to mitochondria-mediated apoptosis. Indeed, tunicamycin-triggered ER stress in wild type mice induced the expression of MGST2 in kidney cells, subsequent nuclear translocation of NOX4, extensive oxidative DNA damage and destruction of proximal tubular cells. Despite the presence of LTC4S in the kidney [35,40], Mgst2 deficiency prevented nuclear translocation of NOX4, thereby greatly attenuating oxidative DNA damage and apoptosis and preserving the proximal tubular cells (Fig. 2). Similarly, the CysLT1 receptor antagonist pranlukast prevented MGST2 induction, nuclear translocation of NOX4 and subsequent oxidative DNA damage in kidneys of wild type mice challenged with 5-FU [34▪▪]. This study revealed the physiological function of MGST2 as a mediator of stress-induced production of LTC4 and the role of the latter as a major mediator of oxidative stress and DNA damage in nonimmune cells.
FIGURE 1.
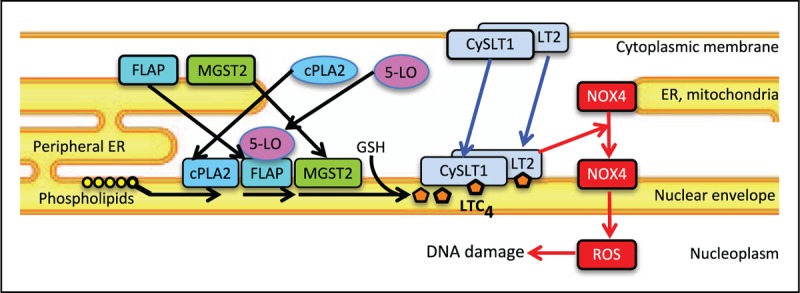
The mechanism of stress-triggered oxidative DNA damage. ER stress and cytotoxic agents trigger translocation and assembly at the nuclear envelope of MGST2-based biosynthetic machinery of LTC4 (black arrows). The two LTC4 receptors CysLT1 and CysLT2 translocate from the cytoplasmic membrane (and the ER) to the nuclear envelope as well (blue arrows). Binding of LTC4 to its receptors triggers translocation of NOX4 from the ER and mitochondria to the nucleus, resulting in nuclear accumulation of reactive oxygen species and subsequent oxidative DNA damage (red arrows).
FIGURE 2.
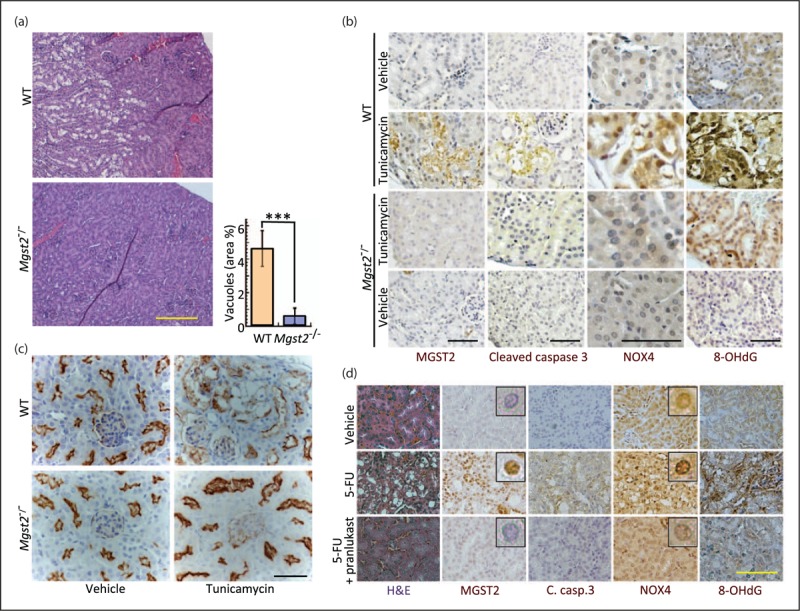
Mgst2 deficiency and pranlukast attenuate nephrotoxic drug-triggered DNA damage and apoptosis in mouse kidneys. (a) Hematoxylin–eosin stained kidney slices from wild type and Mgst2-deficient mice treated with tunicamycin (a single dose of 1.5 mg/ kg, i.p.). Kidneys were removed and processed on day 4. Bar = 200 μm. Percentage area of vacuoles represents the damage to kidney cells. n = 5, ∗∗∗P < 0.001. Values are means ± SD. No vacuoles were observed in kidneys of untreated mice. (b) Immunohistochemical staining of the indicated markers in kidney sections of mice treated as in (a). Notice the nuclear accumulation of NOX4. 8-OHdG is a marker of oxidative DNA damage. Bars = 50 μm. (c) Immunohistochemical stains of proximal tubules (brown) using antiaminopeptidase A in kidney sections from wild type and Mgst2-deficient mice treated with tunicamycin as in (a). Nuclei were counterstained with hematoxylin (grey-blue). Bar = 50 μm. Notice the damage to the tubular cells and its absence in the Mgst2-deficient mice. (d) Hematoxylin–eosin (H&E) stain and immunohistochemical staining of the indicated proteins and of 8-OHdG in kidney slices of wild type mice treated with 5-FU (300 mg/kg, i.p. at time = 0) followed by six administrations of PBS or pranlukast (Pran., 3 mg/kg, i.p., administered at days 0, 1, 2 and 5–7). Kidneys were processed at day 13. Bar = 50 μm. Insets: enlarged images showing immunostained nuclei. Reproduced from an open access article [34▪▪].
One study implicated MGST3 as the source of cysteinyl leukotriene biosynthesis in aristolochic acid I-triggered apoptosis of proximal tubular epithelial cells in culture [41]. That study, however, lacked a confirmatory evidence for MGST3 involvement, for example, by Mgst3 knockdown or by in-vivo studies.
CONCLUSION
In view of the relatively large number of promising preclinical studies and availability of leukotriene inhibitors as approved drugs, it is frustrating to find out that only one clinical study, evaluating the role of leukotrienes in a renal diseases, was reported, and reaching an inconclusive outcome. In that study, treatment of children having primary nephrotic syndrome with montelukast on top of steroids did not provide a clear benefit [42▪]. Properly designed clinical trials using leukotriene inhibitors are required for evaluating their potential use in attenuating nephrotoxicity of drugs such as cyclosporine, gentamycin, alderonate, methotrexate and amikacin. The MGST2–LTC4 pathway was shown to mediate part of the cytotoxic action of doxorubicin, 5-FU, vincristine and bortezomib, but not in tumor cells of hematopoietic lineage [34▪▪]. Therefore, leukotriene inhibitors should be evaluated for their potential to reduce the side effects of chemotherapy, including nephrotoxicity, in patients treated for hematopoietic malignancies. Leukotriene inhibitors may also alleviate the damaging effect of transient hypoxia–reperfusion during kidney transplantation and following burn-induced injury. Measurement of urinary leukotriene metabolites such as N-acetyl LTE4[43] may help identify patients with these and other diseases that will benefit from leukotriene inhibitors. Similarly, clinical development of LTB4 inhibitors may provide tools for managing inflammatory nephropathies and possibly allograft rejection.
Acknowledgements
The author's study was supported by Grant No. 425/12 from the Israel Science Foundation.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Haeggström JZ, Rinaldo-Matthis A, Wheelock CE, Wetterholm A. Advances in eicosanoid research, novel therapeutic implications. Biochem Biophys Res Commun 2010; 396:135–139. [DOI] [PubMed] [Google Scholar]
- 2.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev 2011; 111:5866–5898. [DOI] [PubMed] [Google Scholar]
- 3.Di Gennaro A, Haeggstrom JZ. The leukotrienes: immune-modulating lipid mediators of disease. Adv Immunol 2012; 116:51–92. [DOI] [PubMed] [Google Scholar]
- 4.Singh RK, Tandon R, Dastidar SG, Ray A. A review on leukotrienes and their receptors with reference to asthma. J Asthma 2013; 50:922–931. [DOI] [PubMed] [Google Scholar]
- 5.Back M, Powell WS, Dahlen SE, et al. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR review 7. Br J Pharmacol 2014; 171:3551–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol 2015; 6:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7▪.Werz O, Gerstmeier J, Garscha U. Novel leukotriene biosynthesis inhibitors (2012–2016) as anti-inflammatory agents. Expert Opin Ther Pat 2017; 27:607–620. [DOI] [PubMed] [Google Scholar]; Review on past and future prospectives of the development of leukotriene inhibitors.
- 8.Badr KF, Lakkis FG. Lipoxygenase products in normal and diseased glomerulia. Ann N Y Acad Sci 1994; 744:216–228. [DOI] [PubMed] [Google Scholar]
- 9.Spurney RF, Ibrahim S, Butterly D, et al. Leukotrienes in renal transplant rejection in rats. Distinct roles for leukotriene B4 and peptidoleukotrienes in the pathogenesis of allograft injury. J Immunol 1994; 152:867–876. [PubMed] [Google Scholar]
- 10.Petric R, Ford-Hutchinson AW. Elevated cysteinyl leukotriene excretion in experimental glomerulonephritis. Kidney Int 1994; 46:1322–1329. [DOI] [PubMed] [Google Scholar]
- 11.Şener G, Kabasakal L, Çetinel Ş, et al. Leukotriene receptor blocker montelukast protects against burn-induced oxidative injury of the skin and remote organs. Burns 2005; 31:587–596. [DOI] [PubMed] [Google Scholar]
- 12.Şener G, Şehirli Ö, Velioğlu-Öğünç A, et al. Montelukast protects against renal ischemia/reperfusion injury in rats. Pharmacol Res 2006; 54:65–71. [DOI] [PubMed] [Google Scholar]
- 13.Şener G, Sakarcan A, Şehirli Ö, et al. Chronic renal failure-induced multiple-organ injury in rats is alleviated by the selective CysLT1 receptor antagonist montelukast. Prostaglandins Other Lipid Mediat 2007; 83:257–267. [DOI] [PubMed] [Google Scholar]
- 14.Hadi NR, Al-amran FG, Hussein AA. Effects of thyroid hormone analogue and a leukotrienes pathway-blocker on renal ischemia/reperfusion injury in mice. BMC Nephrol 2011; 12:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khodir AE, Ghoneim HA, Rahim MA, Suddek GM. Montelukast reduces sepsis-induced lung and renal injury in rats. Can J Physiol Pharmacol 2014; 92:839–847. [DOI] [PubMed] [Google Scholar]
- 16.Helmy MM, El-Gowelli HM. Montelukast abrogates rhabdomyolysis-induced acute renal failure via rectifying detrimental changes in antioxidant profile and systemic cytokines and apoptotic factors production. Eur J Pharmacol 2012; 683:294–300. [DOI] [PubMed] [Google Scholar]
- 17.Wu S, Zhu X, Jin Z, et al. The protective role of montelukast against intestinal ischemia-reperfusion injury in rats. Sci Rep 2015; 5:15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki S, Kuroda T, Kazama J-I, et al. The leukotriene B4 receptor antagonist ONO-4057 inhibits nephrotoxic serum nephritis in WKY rats. J Am Soc Nephrol 1999; 10:264–270. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki Y, Tanji M, Takano K, et al. The leukotriene B4 receptor antagonist ONO-4057 inhibits mesangioproliferative changes in anti-Thy-1 nephritis. Nephrol Dial Transplant 2005; 20:2697–2703. [DOI] [PubMed] [Google Scholar]
- 20.Doi K, Hamasaki Y, Noiri E, et al. Role of leukotriene B4 in accelerated hyperlipidaemic renal injury. Nephrology 2011; 16:304–309. [DOI] [PubMed] [Google Scholar]
- 21.Gambaro G, Perazella MA. Adverse renal effects of anti-inflammatory agents: evaluation of selective and nonselective cyclooxygenase inhibitors. J Intern Med 2003; 253:643–652. [DOI] [PubMed] [Google Scholar]
- 22.Salazar F, Salazar FJ, Saez F, et al. Leukotrienes, but not angiotensin II, are involved in the renal effects elicited by the prolonged cyclooxygenase-2 inhibition when sodium intake is low. J Cardiovasc Pharmacol 2013; 61:329–336. [DOI] [PubMed] [Google Scholar]
- 23.Deng B, Lin Y, Ma S, et al. The leukotriene B4-leukotriene B4 receptor axis promotes cisplatin-induced acute kidney injury by modulating neutrophil recruitment. Kidney Int 2017; 92:89–100. [DOI] [PubMed] [Google Scholar]
- 24.Takeda T, Komiya Y, Koga T, et al. Dioxin-induced increase in leukotriene B4 biosynthesis through the aryl hydrocarbon receptor and its relevance to hepatotoxicity owing to neutrophil infiltration. J Biol Chem 2017; 292:10586–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butterly DW, Spurney RF, Ruiz P, et al. A role for leukotrienes in cyclosporine nephrotoxicity. Kidney Int 2000; 57:2586–2593. [DOI] [PubMed] [Google Scholar]
- 26.Atakan A, Arikan H, Macunluoglu B, et al. Renal protective effects of leukotriene receptor blockers in an experimental model of cyclosporine nephrotoxicity. Transplant Proc 2008; 40:279–284. [DOI] [PubMed] [Google Scholar]
- 27.Şener G, Kapucu C, Cetinel S, et al. Gastroprotective effect of leukotriene receptor blocker montelukast in alendronat-induced lesions of the rat gastric mucosa. Prostaglandins Leukot Essent Fatty Acids 2005; 72:1–11. [DOI] [PubMed] [Google Scholar]
- 28.Beytur A, Kose E, Sarihan ME, et al. Beneficial effects of montelukast against cisplatin-induced acute renal damage in rats. Ren Fail 2012; 34:343–349. [DOI] [PubMed] [Google Scholar]
- 29.Suddek GM. Montelukast ameliorates kidney function and urinary bladder sensitivity in experimentally induced renal dysfunction in rats. Fundam Clin Pharmacol 2013; 27:186–191. [DOI] [PubMed] [Google Scholar]
- 30.Kose E, Beytur A, Dogan Z, et al. The effects of montelukast against amikacin-induced acute renal damage. Eur Rev Med Pharmacol Sci 2012; 16:503–511. [PubMed] [Google Scholar]
- 31.Otunctemur A, Ozbek E, Cekmen M, et al. Protective effect of montelukast which is cysteinyl-leukotriene receptor antagonist on gentamicin-induced nephrotoxicity and oxidative damage in rat kidney. Ren Fail 2013; 35:403–410. [DOI] [PubMed] [Google Scholar]
- 32.Abdel-Raheem IT, Khedr NF. Renoprotective effects of montelukast, a cysteinyl leukotriene receptor antagonist, against methotrexate-induced kidney damage in rats. Naunyn Schmiedebergs Arch Pharmacol 2014; 387:341–353. [DOI] [PubMed] [Google Scholar]
- 33.Rankin GO, Hong SK, Anestis DK, et al. Role of leukotrienes in N-(3,5-dichlorophenyl)succinimide (NDPS) and NDPS metabolite nephrotoxicity in male Fischer 344 rats. Toxicology 2012; 300:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34▪▪.Dvash E, Har-Tal M, Barak S, et al. Leukotriene C4 is the major trigger of stress-induced oxidative DNA damage. Nat Commun 2015; 6:10112. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, discussed in detail in the text, elucidated the role of MGST2 in stress and chemotherapy-triggered biosynthesis of LTC4 in nonimmune cells, and the mechanism by which LTC4 triggers oxidative stress, DNA damage and cell death.
- 35.Scoggan KA, Jakobsson P-J, Ford-Hutchinson AW. Production of leukotriene C4 in different human tissues is attributable to distinct membrane bound biosynthetic enzymes. J Biol Chem 1997; 272:10182–10187. [DOI] [PubMed] [Google Scholar]
- 36.Sjostrom M, Jakobsson PJ, Heimburger M, et al. Human umbilical vein endothelial cells generate leukotriene C4 via microsomal glutathione S-transferase type 2 and express the CysLT(1) receptor. Eur J Biochem 2001; 268:2578–2586. [DOI] [PubMed] [Google Scholar]
- 37.Carnini C, Accomazzo MR, Borroni E, et al. Synthesis of cysteinyl leukotrienes in human endothelial cells: subcellular localization and autocrine signaling through the CysLT2 receptor. FASEB J 2011; 25:3519–3528. [DOI] [PubMed] [Google Scholar]
- 38.Kanaoka Y, Maekawa A, Penrose JF, et al. Attenuated zymosan-induced peritoneal vascular permeability and IgE-dependent passive cutaneous anaphylaxis in mice lacking leukotriene C4 synthase. J Biol Chem 2001; 276:22608–22613. [DOI] [PubMed] [Google Scholar]
- 39.Stringham JR, Moore EE, Gamboni F, et al. Mesenteric lymph diversion abrogates 5-lipoxygenase activation in the kidney following trauma and hemorrhagic shock. J Trauma Acute Care Surg 2014; 76:1214–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jakobsson PJ, Mancini JA, Ford-Hutchinson AW. Identification and characterization of a novel human microsomal glutathione S-transferase with leukotriene C4 synthase activity and significant sequence identity to 5-lipoxygenase-activating protein and leukotriene C4 synthase. J Biol Chem 1996; 271:22203–22210. [DOI] [PubMed] [Google Scholar]
- 41.Yang H, Dou Y, Zheng X, et al. Cysteinyl leukotrienes synthesis is involved in aristolochic acid I-induced apoptosis in renal proximal tubular epithelial cells. Toxicology 2011; 287:38–45. [DOI] [PubMed] [Google Scholar]
- 42▪.Zedan MM, El-Refaey A, Zaghloul H, et al. Montelukast as an add-on treatment in steroid dependant nephrotic syndrome, randomised-controlled trial. J Nephrol 2016; 29:585–592. [DOI] [PubMed] [Google Scholar]; The first and only published clinical study using a leukotriene inhibitor for renal disease. The results were inconclusive.
- 43.Reinhold SW, Scherl T, Stölcker B, et al. Lipoxygenase products in the urine correlate with renal function and body temperature but not with acute transplant rejection. Lipids 2013; 48:167–175. [DOI] [PubMed] [Google Scholar]