

Abstract
The Rosetta molecular modeling software package provides a large number of experimentally validated tools for modeling and designing proteins, nucleic acids, and other biopolymers, with new protocols being added continually. While freely available to academic users, external usage is limited by the need for expertise in the Unix command line environment. To make Rosetta protocols available to a wider audience, we previously created a web server called Rosetta Online Server that Includes Everyone (ROSIE), which provides a common environment for hosting web‐accessible Rosetta protocols. Here we describe a simplification of the ROSIE protocol specification format, one that permits easier implementation of Rosetta protocols. Whereas the previous format required creating multiple separate files in different locations, the new format allows specification of the protocol in a single file. This new, simplified protocol specification has more than doubled the number of Rosetta protocols available under ROSIE. These new applications include pK a determination, lipid accessibility calculation, ribonucleic acid redesign, protein‐protein docking, protein‐small molecule docking, symmetric docking, antibody docking, cyclic toxin docking, critical binding peptide determination, and mapping small molecule binding sites. ROSIE is freely available to academic users at http://rosie.rosettacommons.org.
Keywords: web server, molecular modeling, design, prediction
Introduction
Rosetta is a molecular modeling suite which provides a wide array of tools for prediction and design of biological macromolecules. Rosetta has been used in a number of diverse publications, from prediction of protein structure,1 ribonucleic acid (RNA) structure,2 protein–protein interactions,3, 4 protein–peptide interactions,5, 6 and protein–small molecule interactions,7, 8 to the use of nuclear magnetic resonance9, 10 and electron density information11, 12 in structure prediction and refinement, to the design of novel protein folds,13, 14 protein–protein interactions,15, 16 protein–small molecule interactions,17, 18 enzymes,19, 20 macromolecular cages,21, 22 and protein–interacting peptides.23
Rosetta is maintained by the RosettaCommons, a collaborative association of more than 45 principal investigators and collaborators at 55 institutions. With over 350 active developers and a modular architecture,24 new protocols and functionality are continually being added. While recent efforts have expanded the usability of Rosetta with interfaces to Python (PyRosetta25), and XML (RosettaScripts26), and the publication of introductory tutorials,27, 28, 29 most of Rosetta's functionality still requires familiarity with the Unix command line environment, limiting use by nonexperts.
Web‐accessible servers are one way to lower the barrier for nonspecialist users to access Rosetta protocols. Indeed, many groups creating Rosetta protocols have independently implemented servers making their protocols available to anyone with an internet connection and a web browser.30, 31, 32, 33, 34 Unfortunately, setting up a new scientific web server is a laborious process, one which introduces a significant “barrier to entry” for exposing new protocols. To reduce the complexity of setting up a new web server, we have previously implemented Rosetta Online Server that Includes Everyone (ROSIE), a single server framework which can accommodate a range of Rosetta protocols.35
Here, we describe an improvement to the application program interface (API) for the ROSIE framework. This “meta” API simplifies the process of taking a protocol implemented in the Unix command line and converting it to a web‐accessible server. This simplified API has permitted Rosetta developers to more than double the number of protocols exposed through ROSIE since the previous publication. It should also facilitate user requests for additional Rosetta protocols to be exposed through the ROSIE interface.
Results
Our previous paper35 has already described the general ROSIE server architecture, including the structure of the database and servers. The primary improvement over the previous approach is a simplified method for specifying new protocols. This new meta API acts as a domain‐specific mini‐language, where input and output facilities are specified through object specification (“widgets”) and the execution is specified through a list of “triggers.” An example of the meta application specification format can be found in the Supporting Information.
Overview of simplified protocol definition
In contrast to the previously published method of implementing a ROSIE protocol, which required multiple different files in defined locations,35 ROSIE protocols can now be implemented within a single Python file located in the rosie.front/rosie/meta/directory of the ROSIE server. This file contains declarations of all the relevant contents of the submission, results and documentation pages on the ROSIE website, as well as the specification of how to run the protocol. These contents are contained within a “named tuple” (NT) object in the Python file.
Submission form definition
The format of the submission form (Fig. 1) is described by the “input_” data member of the NT. This member contains a list of the various input elements from which the form can be constructed. The input elements are represented as Python objects, with different input types being a different subclass of the “Input” widget class (Table 1). Each different input widget contains the html and javascript code needed for proper display of the input field on the submission page. A protocol developer need only to specify the widget type and relevant parameters: for example,
Figure 1.
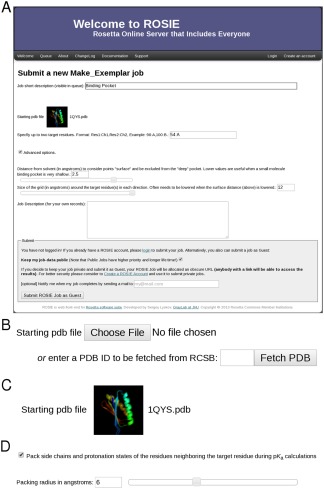
ROSIE input elements. A: Example of a submission form. B: Example of the FileInput widget, customized for structure file loading. C: Example of the FileInput widget, after file selection. Icons of the submitted structures are shown. D: Example of the CheckboxInput and FloatInput widgets.
Table 1.
Input Widgets
| Input widget | Description |
|---|---|
| FileInput | Upload an arbitrary input file |
| StringInput | An arbitrary text string |
| IntInput | An integer in a specified range |
| FloatInput | A real number in a specified range |
| CheckboxInput | True/False option input |
| SelectInput | Pick one of several specified options, in dropdown format |
| RadioButtonListInput | Pick one of several specified options, in selection list format |
| HRuleInput | No input, but add a separator to the input page |
[FloatInput(name=‘pocket_width’, min=0.0, max=7.0, default=5.0, description=‘Maximum radius to search (in Angstroms) from starting coordinate’, optional=True)]
From the list of input elements, the submission form is built automatically, placing the desired input elements one after each other on the page. Additional input elements common to all protocols, such as job descriptions and account information, are handled by the ROSIE server framework and do not need to be explicitly included by the protocol writer.
Validation of input
As the input parameters are entered by potentially untrusted users on the internet, it is critical that any values that are entered are checked and validated to make sure that they are in the proper form and do not contain any malicious content. Misplaced punctuation or deliberately crafted input could compromise the integrity of the server. Additionally, as the aim of the web server is to provide access to protocols to people with limited modeling experience, it is important to check the range of the input parameters, to make sure that they will not produce anything that is compromised scientifically.
To that end, all input fields in ROSIE contain a validation component. While certain fields (such as numeric entry) can be checked automatically by the input widget to ensure they are correctly formatted and are in the appropriate range, other fields (such as structure file uploads) require special attention to ensure they are correctly formatted. To allow for custom validation, each input widget accepts a list of validators, which are Python functions. These validators can signal an improper input value by raising a Python exception.
As combinations of input values may also be inappropriate, rather than input values in isolation, the ROSIE framework also provides an “input_validator” entry, which is a Python function that will be passed all of the values in the input form. This validator may perform any comparisons the protocol author deems necessary and signal an improper value by returning an error message string.
Execution of the protocol
Protocol execution is specified in the “commands” section of the NT. This is a list of decorated Python functions (triggers) which are executed by the ROSIE server backend when the submitted job is run. The Python decorators allow the protocol writer to specify which of the input fields are passed to the trigger functions.
These trigger functions are then responsible for setting up the input files for the runs (typically by using Python string formatting functions on a template string that is included in the one Python file), and then launching the execution of the appropriate backend job. These backend jobs can be arbitrary programs, but most typically are Rosetta command‐line programs, RosettaScripts extensible markup language (XML) runs, or PyRosetta scripts. As ROSIE can interface with several cluster backends, the actual launching of the backend job is done indirectly, through an hpc_driver object. This object is set up by the ROSIE server prior to trigger function execution, based on the high‐performance computing (HPC) cluster in use at the time.
The trigger function is also responsible for output validation, checking to ensure that the backend run completed successfully and raising a Python exception if not. As a final step, the trigger function specifies the filenames for any produced structures and any output report files, which the ROSIE server architecture will then store in its database.
Presentation of results
The formatting of the results page (Fig. 2) is specified by the “output” section of the NT. As with the input section, we have created different output widgets (Table 2), which the ROSIE framework will assemble into a results page. Standard widgets are available to display the input parameters (including a structural rendering of any input protein, nucleic acid, or small molecule structures), as well as renderings of output structures, graphs of various scoring parameters, and tables of scoring and structural evaluation results.
Figure 2.
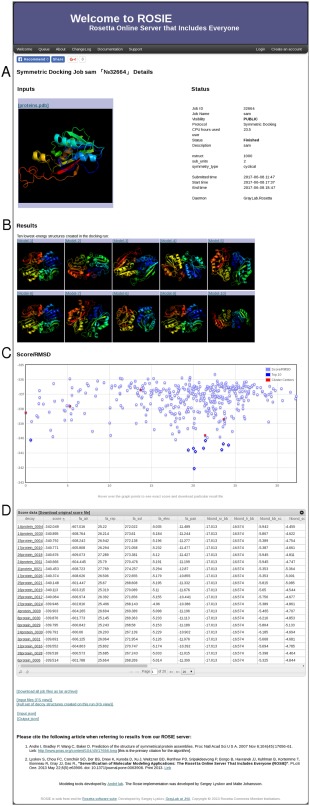
A ROSIE result page, showing output elements. A: The JobHeader widget provides details about the protocol run. B: The TopModels widget shows the selected result structures. Each structure can be downloaded by clicking on it. C: The ScorePlot widget displays a plot of two selected scores. Additional information for each point can be obtained by hovering over it. D: The ScoreTable widget. The results can be sorted by each column, and any particular structure can be downloaded by clicking its name.
Table 2.
Output Widgets
| Output widget | Description |
|---|---|
| JobHeader | An overview of the inputs and running statistics |
| TopModels | Display and permit downloading the structures of the best results of the run |
| ScoreTable | Display a Rosetta scorefile in sortable tabular form |
| ScorePlot | Display a scatter plot of data from a Rosetta scorefile |
| Plot | Produce a scatter plot of arbitrary data |
| File | Display the text contents of an output file |
In addition to the customizable rendering of output results, each results page contains links which allow the download of all produced results. Logging information is also available under the full output link, allowing users of failed runs to debug why their runs may have failed.
Documentation
As the intent of the ROSIE server is to make Rosetta protocols accessible to people with limited experience in computational biology, extensive documentation is needed to explain the usage and to describe how the adjustable parameters will affect the results. For this purpose, a “documentation” slot is provided in the NT protocol specification, which allows the protocol writer to provide an html‐formatted string which will be included on the documentation page for the protocol. Additionally, there are “citations” and “developed by” slots provided, which are included not only on the documentation page, but also on other pages of the ROSIE server. This information allows users to find the relevant papers describing the underlying protocol and the contact information for the protocol maintainer, respectively.
Protocols available through ROSIE
Brief summaries of the currently available protocols are given in Table 3. For more extensive discussion, as well as details of their operation, we refer the reader to the documentation on the ROSIE website (http://rosie.rosettacommons.org/documentation) and the cited papers.
Table 3.
Protocols Available Through ROSIE
| Application | Developer | API | Introduced | Jobsa | References |
|---|---|---|---|---|---|
| Docking2 | Gray at JHU | Meta | January 2012b | 17,054b | 50 and 51 |
| Symmetric docking | André at Lund | Meta | August 2013 | 1081 | 52 |
| RNA redesign | Das at Stanford | Meta | October 2013 | 76c | 36 |
| Ligand docking | Meiler at Vanderbilt | Meta | January 2014 | 3722 | 53 and 54 |
| pK a | Gray at JHU | Meta | January 2014 | 1078 | 55 |
| Peptiderive | Furman at HUJ | Meta | May 2015 | 2057 | 56 and 57 |
| Make exemplar | Karanicolas at FHCC | Meta | September 2015 | 167 | 58 |
| Snug dock | Gray at JHU | Meta | May 2016 | 363 | 51 and 59 |
| Tox dock | Bonneau at NYU | Meta | May 2016 | 50 | |
| Lipid accessibility | Bonneau at NYU | Meta | August 2016 | 141 | 60 |
| FARFAR | Das at Stanford | Original | February 2012 | 1706 | 36 |
| ERRASER | Das at Stanford | Original | October 2012 | 232 | 37 |
| Beta peptide design | Das at Stanford | Original | November 2012 | 19 | 38 |
| Supercharge | Kuhlman at UNC | Original | November 2012 | 1013 | 43 |
| Antibody | Gray at JHU | Original | December 2012 | 5133 | 44 and 45 |
| NCBB design | Bonneau at NYU | Original | December 2012 | 43 | 46 |
| Sequence tolerance | Kortemme at UCSF | Original | January 2013 | 741 | 47 and 48 |
| VIP | Havranek at WUSTL | Original | March 2013 | 412 | 49 |
As of September 2017.
Includes jobs run with the original API.
RNA Redesign runs on a separate backend, and not all jobs may be adequately counted.
Protocols using the original framework
In addition to the new simplified framework, ROSIE still supports the original protocol specification framework. All eight of these protocols were also mentioned in the original ROSIE paper.35
Fragment assembly of RNA with full atom refinement—De novo RNA structure prediction
The Fragment Assembly of RNA with Full Atom Refinement (FARFAR) application models RNA structure de novo by combining short (1–3 nucleotide) fragments from existing RNA crystal structures.36 These fragment assembly models are then further refined with a full‐atom relaxation, resulting in predicted structures for RNA molecules.
Enumerative Real‐space Refinement ASsisted by Electron‐density under Rosetta
The Enumerative Real‐space Refinement ASsisted by Electron‐density under Rosetta (ERRASER) application is an RNA structure refinement protocol which uses electron density information to inform and guide the refinement.37
Beta peptide design
Beta peptides contain noncanonical backbones, with each residue containing an extra backbone carbon. The ROSIE Beta peptide design application takes such a backbone structure and predicts which sidechains (from the standard 20) would best support that backbone structure.38
Supercharge
Increasing the net charge on the surface of a protein can prevent aggregation,39, 40 increase expression and protein lifetime, alter cell entry,41 and affect kidney filtration.42 The ROSIE Supercharge application takes a protein structure and attempts either to maximize the surface charge (positive or negative, as specified), or to obtain a specific net charge, all while attempting to maintain the protein stability.43
Antibody
Antibody structure prediction is aided by the consideration of antibody‐specific structural features, which can be reliably predicted from their sequence. The ROSIE Antibody application uses this domain specific knowledge to model the predicted structure of an antibody given its primary sequence.44, 45 Due to the increased complexity, modeling the challenging HCDR3 loop is provided as a separate option.
Noncanonical backbones design
Noncanonical backbones (NCBB) can be more thermodynamically stable than standard alpha amino acids, and they are more resistant to native peptidases. The NCBB Design application redesigns existing peptide‐protein complexes by replacing the peptide's backbone residues with oligooxopiperazines, hydrogen bond surrogates and peptoids, while maintaining peptide–protein binding.46
Sequence tolerance
The Sequence Tolerance application examines a defined set of positions in a given protein–protein interface, predicting which mutations may be tolerated, and which may destabilize either the monomers or the protein–protein interaction.47, 48
VIP—Core redesign to eliminate voids
Voids in the hydrophobic core of a protein are correlated with reduced stability. The ROSIE Void Identification and Packing (VIP) application can examine the structure of a protein, locate voids in the core, and suggest mutations that could be made to improve core packing without destabilizing the protein.49
Protocols using the new meta API
The simplified meta API framework for protocol specification has allowed a number of additional Rosetta protocols to be exposed through ROSIE.
Docking2
The ROSIE Docking2 application is a rewrite of the previous protein‐protein docking protocol to use the new meta API. This application does a local docking search of a protein–protein interface, given a close starting conformation.50, 51
Symmetric docking
Assembly of a monomer into a symmetric oligomer is assisted by knowledge of that symmetry. The Symmetric Docking application uses knowledge of the desired symmetry to convert a structure of a monomer into a symmetric oligomer.52
RNA redesign
Given a three dimensional (3D) structure of a folded RNA, the RNA Redesign application will attempt to find an RNA sequence (A/U/C/G) which best stabilizes that particular backbone conformation.36
Ligand docking
The ROSIE Ligand Docking application allows users with a given small molecule to predict the binding conformation of that small molecule to a designated pocket on a 3D structure of a protein.53, 54
pKa
The pKa of charged amino acids in a protein can be substantially perturbed due to their surrounding environment and the charge state of surrounding residues. The ROSIE pKa application predicts the pKa of various residues in the protein based on their three‐dimensional context.55
Peptiderive
While many protein–protein interfaces are large, it has been found that much of the energy of interaction is localized to specific regions in the interface. The Peptiderive application examines a protein‐protein interface and attempts to locate the short peptide sequences which contribute the most to the interaction energy.56, 57 These peptides can then potentially be used as inhibitors of the protein–protein interaction.
Make Exemplar—Map small molecule binding pockets
Evaluating potential small molecule binders (such as in high throughput screening) is assisted by having a map of the “ideal” configuration of a ligand binding pocket. The Make Exemplar application creates such a map from the 3D structure of the protein, by placing atoms at the appropriate locations for hydrogen bond donors, hydrogen bond acceptors, and locations of hydrophobic atoms.58
Snug dock—Antibody/antigen docking
Standard protein–protein docking techniques do not necessarily sample all the relevant degrees of freedom needed for antibody/antigen docking. In particular, the flexibility of the antibody loops and the heavy/light chain interface are typically neglected in typical protein‐protein docking techniques. The Snug Dock application is tailored toward antibody‐antigen docking, with explicit additional sampling of the relevant internal degrees of freedom of the antibody.51, 59
Tox dock
Disulfide‐cyclized peptides are one major class of ion channel inhibitors. While these peptides are found natively as neurotoxins, their inhibitory effects may have therapeutic uses. The Tox Dock application docks these cyclized peptides into structures and homology models of ion channels.
Lipid accessibility
When examining membrane proteins, it is useful to know where the lipid bilayer contacts the protein. The mp_lipid_acc application examines structures of membrane proteins, and encodes the lipid accessibility of each atom in the B‐factor column of a Protein Data Bank (PDB) file.60 This annotated PDB can then be viewed in standard molecular viewers.
Server usage to date
From March 2013 (when the statistics in the previous ROSIE paper were gathered) to the time of writing (September 2017), over 4000 new users have registered with ROSIE. Along with an unknown number of anonymous users, they have submitted over 31,000 different jobs to the server. Through that time, the growth in the number of registered users as well as the number of new jobs has been approximately linear, adding approximately 80 new registered users per month, and serving approximately 875 new jobs per month. The computational demand in the same time period has been over 3.5 million CPU hours, or the equivalent of just under 100 CPUs in continuous use for the past 4 years. Much of this computational power was supplied on the Stampede cluster at the Texas Advanced Computing Center, through a grant from the Extreme Science and Engineering Discovery Environment (XSEDE).
Discussion
We have improved the method by which ROSIE server protocols are specified, which has greatly simplified the method by which new protocols are implemented. As such, the number of protocols freely available through the ROSIE website at http://rosie.rosettacommons.org has more than doubled since the last paper describing it.
Our long‐term goal continues to be to provide free web versions of all core Rosetta protocols. While there are 18 protocols currently on the ROSIE web server, additional protocols are not yet available through the server. The reduced complexity of the meta API described in this paper means that compatible protocols can be converted to run on the ROSIE server in a few weeks of work by a knowledgeable Rosetta developer. We plan to continue to add additional protocols to ROSIE, and encourage users interested in protocols not yet available on ROSIE to contact the ROSIE help forum at http://www.rosettacommons.org/forums/rosie/rosie-general to help guide which protocols get exposed as a web server.
Materials and Methods
ROSIE server infrastructure and meta‐API
ROSIE is implemented in the TurboGears web server framework, using a PostgresSQL database. The server is freely available to the public at http://rosie.rosettacommons.org.
Protocol for creation of new ROSIE applications
The following is a summary of the steps required for a developer to create a new ROSIE server with the new meta protocols specification format. ROSIE development tools and source code are available to registered developers through the RosettaCommons.
Install a local ROSIE test server
To facilitate rapid testing of the protocol, the ROSIE server environment is made available within a VirtualBox (http://www.virtualbox.org) virtual machine (VM). This VM can be downloaded, with the appropriate user name and password, from http://graylab.jhu.edu/ROSIE
All the software needed to run a ROSIE server locally is provided, including Rosetta (under ∼/rosetta/) and the ROSIE server software itself (under ∼/rosie/). As these may be out of date, you may wish to update one or both before continuing development, by using standard git revision control commands. (Note that any “push” of changes should be to a personal GitHub fork of the ROSIE repository, rather than the main ROSIE repository.)
Create a python script with your meta protocols
Your new meta protocol will live in a single Python script file in the rosie.front/rosie/meta/. (The other applications in this directory can be used as a guide when formatting and structuring your protocol.) The main specification of the protocol is the NT object, which contains entries for name, display name, input, input validator, commands, output, developed_by, citations, and documentation. Name, display name, developed_by, citations, and documentation entries are strings which provide textual information about the protocol for various locations in the server.
The input_ entry contains a Python list of input widget objects, and controls how the submission form is constructed. A list of currently available input widgets is given in Table 1. Each widget will be placed one after the other on the submission page. As the server is accepting potentially untrusted data over the internet, each widget accepts input validation parameters to raise an error if nonsensical or potentially malicious entries are given. In addition, the input_validator entry accepts a Python function which will be passed all the input values at the time of job submission to identify problematic interdependencies between entries.
The commands entry contains a list of triggers which will be run in order by the ROSIE backend. These triggers are specified using specially decorated Python functions which will be passed the values from the input widgets. These functions can execute arbitrary Python code to set up input files for external Rosetta runs, which are launched using a specialized HPC driver object to potentially run on a remote machine. To avoid resource starvation for other users, settings should be limited such that a single ROSIE job never takes more than 2000 CPU hours, with the average run preferably staying below 500 CPU hours. The trigger functions are also responsible for postprocessing the runs and passing back the processed results through annotated parameters. In this way the results of one trigger command can be used as input for the next.
The output entry controls the display of the results page for finished (and in progress) runs. This is a Python list of output widget objects, which will be placed one after the other on the results page. A list of currently available output widgets is given in Table 2.
Enable your new protocol in the ROSIE test environment
To enable your protocols on the website, import your protocol's Python module rosie.front/rosie/meta/__init__.py and add it to the list of protocols in that file.
Add a 1024 × 1024 sized image to rosie.front/rosie/public/images/<protocol name>_icon.png
To enable running your protocol on the local backend, add your protocol to rosie.back/rosie/rosie‐daemon.ini.template (and rosie‐daemon.ini, if present)
Go to rosie.front/. Run “source ∼/prefix/TurboGears‐2.2/bin/activate” then “python update_protocol_schema.py” to update the database
Test your new protocol server locally
In one terminal window, run the ./run‐rosie‐server.sh script in ∼/rosie/ to launch the webserver
In another terminal, run the ./run_rosie‐daemon.sh script in ∼/rosie/rosie.back/ to launch the ROSIE backend.
Open “localhost:8080” in your web browser to visit the local version of your web server
Test your new protocol in your browser to make sure it runs appropriately, updating your meta protocol file as needed.
When finished, push the results to your personal fork of the ROSIE GitHub repository, and inform the ROSIE system administrators for code review and integration into the central server.
Supporting information
Supporting Information
Acknowledgments
We thank the numerous members of the Rosetta community who have implemented and debugged ROSIE applications. Computational power for ROSIE has been provided by XSEDE (“Stampede Power for the ROSIE Gateway” to J.J.G., S.L., and R.D.).
Importance/Impact: Rosetta, a comprehensive program for investigating biological macromolecules, can be difficult for nonexperts to use. The Rosetta Online Server that Includes Everyone (ROSIE) web server, available at http://rosie.rosettacommons.org, provides a web interface to a number of Rosetta protocol, allowing more users to leverage Rosetta. The simplified protocol specification format described in this paper makes it easy to add even more Rosetta protocols on the web server, allowing wider access to these techniques.
References
- 1. Ovchinnikov S, Park H, Varghese N, Huang PS, Pavlopoulos GA, Kim DE, Kamisetty H, Kyrpides NC, Baker D (2017) Protein structure determination using metagenome sequence data. Science 355:294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miao Z, Adamiak RW, Antczak M, Batey RT, Becka AJ, Biesiada M, Boniecki MJ, Bujnicki JM, Chen SJ, Cheng CY, Chou F‐C, Ferre‐D'Amare AR, Das R, Dawson WK, Ding F, Dokholyan NV, Dunin‐Horkawicz S, Geniesse C, Kappel K, Kladwang W, Krokhotin A, Lach GE, Major F, Mann TH, Magnus M, Pachulska‐Wieczorek K, Patel DJ, Piccirilli JA, Popenda M, Purzycka KJ, Ren A, Rice GM, Santalucia J, Jr , Sarzynska J, Szachniuk M, Tandon A, Trausch JJ, Tian S, Wang J, Weeks KM, Williams BII, Xiao Y, Xu X, Zhang D, Zok T, Westhof E (2017) RNA‐Puzzles Round III: 3D RNA structure prediction of five riboswitches and one ribozyme. RNA 23:655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weitzner BD, Jeliazkov JR, Lyskov S, Marze N, Kuroda D, Frick R, Adolf‐Bryfogle J, Biswas N, Dunbrack RL, Jr , Gray JJ (2017) Modeling and docking of antibody structures with Rosetta. Nat Protoc 12:401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marze NA, Jeliazkov JR, Roy Burman SS, Boyken SE, DiMaio F, Gray JJ (2017) Modeling oblong proteins and water‐mediated interfaces with RosettaDock in CAPRI rounds 28–35. Proteins 85:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alam N, Schueler‐Furman O (2017) Modeling peptide‐protein structure and binding using Monte Carlo sampling approaches: Rosetta FlexPepDock and FlexPepBind. Methods Mol Biol 1561:139–169. [DOI] [PubMed] [Google Scholar]
- 6. Marcu O, Dodson EJ, Alam N, Sperber M, Kozakov D, Lensink MF, Schueler‐Furman O (2017) FlexPepDock lessons from CAPRI peptide‐protein rounds and suggested new criteria for assessment of model quality and utility. Proteins 85:445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lemmon G, Kaufmann K, Meiler J (2012) Prediction of HIV‐1 protease/inhibitor affinity using RosettaLigand. Chem Biol Drug Des 79:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis IW, Raha K, Head MS, Baker D (2009) Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci 18:1998–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Z, Porter J, Tripsianes K, Lange OF (2014) Robust and highly accurate automatic NOESY assignment and structure determination with Rosetta. J Biomol NMR 59:135–145. [DOI] [PubMed] [Google Scholar]
- 10. Lange OF, Rossi P, Sgourakis NG, Song Y, Lee HW, Aramini JM, Ertekin A, Xiao R, Acton TB, Montelione GT, Baker D (2012) Determination of solution structures of proteins up to 40 kDa using CS‐Rosetta with sparse NMR data from deuterated samples. Proc Natl Acad Sci USA 109:10873–10878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DiMaio F (2017) Rosetta structure prediction as a tool for solving difficult molecular replacement problems. Methods Mol Biol 1607:455–466. [DOI] [PubMed] [Google Scholar]
- 12. Wang RY, Kudryashev M, Li X, Egelman EH, Basler M, Cheng Y, Baker D, DiMaio F (2015) De novo protein structure determination from near‐atomic‐resolution cryo‐EM maps. Nat Methods 12:335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D (2003) Design of a novel globular protein fold with atomic‐level accuracy. Science 302:1364–1368. [DOI] [PubMed] [Google Scholar]
- 14. Koga N, Tatsumi‐Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D (2012) Principles for designing ideal protein structures. Nature 491:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D (2011) Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strauch EM, Bernard SM, La D, Bohn AJ, Lee PS, Anderson CE, Nieusma T, Holstein CA, Garcia NK, Hooper KA, Ravichandran R, Nelson JW, Sheffler W, Bloom JD, Lee KK, Ward AB, Yager P, Fuller DH, Wilson IA, Baker D (2017) Computational design of trimeric influenza‐neutralizing proteins targeting the hemagglutinin receptor binding site. Nat Biotechnol 35:667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, Baker D (2013) Computational design of ligand‐binding proteins with high affinity and selectivity. Nature 501:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allison B, Combs S, DeLuca S, Lemmon G, Mizoue L, Meiler J (2014) Computational design of protein‐small molecule interfaces. J Struct Biol 185:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF III, Hilvert D, Houk KN, Stoddard BL, Baker D (2008) De novo computational design of retro‐aldol enzymes. Science 319:1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D (2008) Kemp elimination catalysts by computational enzyme design. Nature 453:190–195. [DOI] [PubMed] [Google Scholar]
- 21. King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO, Baker D (2014) Accurate design of co‐assembling multi‐component protein nanomaterials. Nature 510:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, Gonen T, Yeates TO, Baker D (2012) Computational design of self‐assembling protein nanomaterials with atomic level accuracy. Science 336:1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SV, Kaas Q, Eletsky A, Huang P‐S, Johnsen WA, Greisen PJ, Rocklin GJ, Song Y, Linsky TW, Watkins A, Rettie SA, Xu X, Carter LP, Bonneau R, Olson JM, Coutsias E, Correnti CE, Szyperski T, Craik DJ, Baker D (2016) Accurate de novo design of hyperstable constrained peptides. Nature 538:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leaver‐Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban Y‐EA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P (2011) ROSETTA3: an object‐oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaudhury S, Lyskov S, Gray JJ (2010) PyRosetta: a script‐based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26:689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fleishman SJ, Leaver‐Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D (2011) RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One 6:e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J (2010) Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry 49:2987–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bender BJ, Cisneros A III, Duran AM, Finn JA, Fu D, Lokits AD, Mueller BK, Sangha AK, Sauer MF, Sevy AM, Sliwoski G, Sheehan JH, DiMaio F, Meiler J, Moretti R (2016) Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry 55:4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gray JJ (2017) The PyRosetta interactive platform for protein structure prediction and design: a set of educational modules. Mountain View, CA: Createspace, p 98. [Google Scholar]
- 30. Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y, Kuhlman B (2006) Rosetta Design server for protein design. Nucleic Acids Res 34:W235–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. London N, Schueler‐Furman O (2008) Funnel hunting in a rough terrain: learning and discriminating native energy funnels. Structure 16:269–279. [DOI] [PubMed] [Google Scholar]
- 33. London N, Raveh B, Cohen E, Fathi G, Schueler‐Furman O (2011) Rosetta FlexPepDock web server–high resolution modeling of peptide‐protein interactions. Nucleic Acids Res 39:W249–W253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lauck F, Smith CA, Friedland GF, Humphris EL, Kortemme T (2010) RosettaBackrub–a web server for flexible backbone protein structure modeling and design. Nucleic Acids Res 38:W569–W575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lyskov S, Chou FC, Conchuir SO, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, Borgo B, Havranek JJ, Kuhlman B, Kortemme T, Bonneau R, Gray JJ, Das R (2013) Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8:e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Das R, Karanicolas J, Baker D (2010) Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 7:291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R (2013) Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10:74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Molski MA, Goodman JL, Chou FC, Baker D, Das R, Schepartz A (2013) Remodeling a beta‐peptide bundle. Chem Sci 4:319–324. [Google Scholar]
- 39. Fields GB, Alonso DOV, Stigter D, Dill KA (1992) Theory for the aggregation of proteins and copolymers. J Phys Chem 96:3974–3981. [Google Scholar]
- 40. Fink AL (1998) Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des 3:R9–R23. [DOI] [PubMed] [Google Scholar]
- 41. Cronican JJ, Beier KT, Davis TN, Tseng JC, Li W, Thompson DB, Shih AF, May EM, Cepko CL, Kung AL, Zhou Q, Liu DR (2011) A class of human proteins that deliver functional proteins into mammalian cells in vitro and in vivo. Chem Biol 18:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lund U, Rippe A, Venturoli D, Tenstad O, Grubb A, Rippe B (2003) Glomerular filtration rate dependence of sieving of albumin and some neutral proteins in rat kidneys. Am J Physiol Renal Physiol 284:F1226–F1234. [DOI] [PubMed] [Google Scholar]
- 43. Der BS, Kluwe C, Miklos AE, Jacak R, Lyskov S, Gray JJ, Georgiou G, Ellington AD, Kuhlman B (2013) Alternative computational protocols for supercharging protein surfaces for reversible unfolding and retention of stability. PLoS One 8:e64363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sivasubramanian A, Sircar A, Chaudhury S, Gray JJ (2009) Toward high‐resolution homology modeling of antibody Fv regions and application to antibody‐antigen docking. Proteins 74:497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marze NA, Lyskov S, Gray JJ (2016) Improved prediction of antibody VL‐VH orientation. Protein Eng Des Sel 29:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Drew K, Renfrew PD, Craven TW, Butterfoss GL, Chou FC, Lyskov S, Bullock BN, Watkins A, Labonte JW, Pacella M, Kilambi KP, Leaver‐Fay A, Kuhlman B, Gray JJ, Bradley P, Kirshenbaum K, Arora PS, Das R, Bonneau R (2013) Adding diverse noncanonical backbones to Rosetta: enabling peptidomimetic design. PLoS One 8:e67051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith CA, Kortemme T (2010) Structure‐based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol 402:460–474. [DOI] [PubMed] [Google Scholar]
- 48. Smith CA, Kortemme T (2011) Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PLoS One 6:e20451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Borgo B, Havranek JJ (2012) Automated selection of stabilizing mutations in designed and natural proteins. Proc Natl Acad Sci USA 109:1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, Gray JJ (2011) Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 6:e22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lyskov S, Gray JJ (2008) The RosettaDock server for local protein‐protein docking. Nucleic Acids Res 36:W233–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andre I, Bradley P, Wang C, Baker D (2007) Prediction of the structure of symmetrical protein assemblies. Proc Natl Acad Sci USA 104:17656–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, Meiler J (2013) Small‐molecule ligand docking into comparative models with Rosetta. Nat Protoc 8:1277–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DeLuca S, Khar K, Meiler J (2015) Fully flexible docking of medium sized ligand libraries with RosettaLigand. PLoS One 10:e0132508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kilambi KP, Gray JJ (2012) Rapid calculation of protein pKa values using Rosetta. Biophys J 103:587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. London N, Raveh B, Movshovitz‐Attias D, Schueler‐Furman O (2010) Can self‐inhibitory peptides be derived from the interfaces of globular protein‐protein interactions?. Proteins 78:3140–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sedan Y, Marcu O, Lyskov S, Schueler‐Furman O (2016) Peptiderive server: derive peptide inhibitors from protein‐protein interactions. Nucleic Acids Res 44:W536–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johnson DK, Karanicolas J (2016) Ultra‐high‐throughput structure‐based virtual screening for small‐molecule inhibitors of protein‐protein interactions. J Chem Inf Model 56:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sircar A, Gray JJ (2010) SnugDock: paratope structural optimization during antibody‐antigen docking compensates for errors in antibody homology models. PLoS Comput Biol 6:e1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koehler Leman J, Lyskov S, Bonneau R (2017) Computing structure‐based lipid accessibility of membrane proteins with mp_lipid_acc in RosettaMP. BMC Bioinform 18:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information