

Abstract
The Protein Data Bank Japan (PDBj), a member of the worldwide Protein Data Bank (wwPDB), accepts and processes the deposited data of experimentally determined biological macromolecular structures. In addition to archiving the PDB data in collaboration with the other wwPDB partners, PDBj also provides a wide range of original and unique services and tools, which are continuously improved and updated. Here, we report the new RDB PDBj Mine 2, the WebGL molecular viewer Molmil, the ProMode‐Elastic server for normal mode analysis, a virtual reality system for the eF‐site protein electrostatic molecular surfaces, the extensions of the Omokage search for molecular shape similarity, and the integration of PDBj and BMRB searches.
Keywords: PDB, BMRB, protein structures, structural dynamics, semantic web, virtual reality
Abbreviations
- PDB
Protein Data Bank
- BMRB
BioMagResBank
- EMDB
Electron Microscopy Data Bank
- iDR
incremental Distance Rank
Introduction
The Protein Data Bank Japan (PDBj, https://pdbj.org/)1, 2 accepts and annotates biological macromolecular structure data in collaboration with the other worldwide Protein Data Bank (wwPDB, https://wwpdb.org/)3, 4 partners, PDBe,5 RCSB PDB6 and BMRB.7 All depositions from Asia and the Middle East are processed and annotated at PDBj, and the updated data are released simultaneously with the other partner sites, every Wednesday at midnight (UTC + 0). In addition to processing depositions and maintaining the archive, PDBj provides unique service tools and derived databases to facilitate structural biology and bioinformatics research. Here, we describe these services along with their new functions and updates.
Characteristic Tools and Functions at PDBj
First, we describe our updated system of the relational database (RDB), PDBj Mine 2 that processes many different kind of queries about the PDB metadata. In order to integrate the PDB data with other biological information, it includes the resource of SIFTS (Structure integration with function, taxonomy, and sequence),8 which has been developed by EMBL‐EBI. Second, we introduce the web‐based molecular viewer, Molmil,9 which is now implemented in many PDBj services. Third, we describe the server showing the protein dynamics using the all‐atom normal mode analysis (NMA), ProMode‐Elastic.10 Fourth, we show a virtual reality (VR) application for the database of the electrostatic molecular surfaces, eF‐site,11 for educational purposes. Fifth, we introduce the extension of molecular shape comparison server, Omokage search,12 where searching for the Small Angle Scattering Biological Data Bank (SASBDB)13 entries is now available in addition to PDB and Electron Microscope Data Bank (EMDB).14 Finally, we integrate the database of NMR experimental data, BMRB,7 with PDB information for an advanced search, and describe semantic web services.
PDBj mine 2
A biological researcher may have his/her own specific questions to PDB, such as “Are there any 3D structures of Homeobox superfamily with better than 2.0 Å resolution and <60 residues?”. To answer this type of questions, the PDBj provides the PDBj Mine service, which enables the user to input his/her own SQL (Structured Query Language) query in the web form. Although the knowledge of the SQL query is required, it allows users to ask wide ranges of queries to the PDBj database. PDBj Mine 2 RDB is the RDB for PDBj, and was updated from the previous PDBj Mine.15 It can be directly accessed via the interactive web interface at https://pdbj.org/mine/sql or via the REST API at https://pdbj.org/rest/mine2_sql (see https://pdbj.org/help/rest-interface for the details of the REST API). Furthermore, the complete database dump is available at ftp://ftp.pdbj.org/mine2/ for local installation, using PostgreSQL (https://www.postgresql.org/) version 9.3 or higher (see https://pdbj.org/help/mine2-rdb-local-install for instructions). Most of the tables in the PDBj Mine 2 RDB correspond to the categories defined in the PDBx/mmCIF dictionary (http://mmcif.wwpdb.org/). For a complete description of the database schema, see https://pdbj.org/mine-rdb-docs. We have also integrated the SIFTS resource (https://www.ebi.ac.uk/pdbe/docs/sifts/).8 Currently, only the “quick access” files of SIFTS are incorporated in the PDBj Mine 2 RDB, in table structures that reflect the tab‐separated format of the original SIFTS files (see https://pdbj.org/help/sifts for details).
A representative SQL query that integrates PDB data and SIFTS resources is shown in Figure 1. A more comprehensive list of examples is available at https://pdbj.org/help/mine2-sql.
Figure 1.

An example query in SQL for our system of the RDB, PDBj Mine 2, that integrates PDB data and SIFTS resources.8 An advanced and flexible search is available using the SQL for the RDB. This query retrieves PDB chain sequences matching to the Pfam16 accession “PF00046” (Homeobox) and having a resolution better than 2.0 Angstrom and a sequence length ≥ 58 (residues), ordered by the resolution, sequence length, and chain ID. Comments are in blue. The result of this query can be accessed at https://goo.gl/C12oPE.
Molmil and PDBx/mmJSON
The PDBj developed its own Web‐based molecular graphics program, Molmil.9 It provides fast and smooth graphics for all the platforms supporting Javascript and WebGL. The Molmil in the PDBj server can quickly download large 3D structural data, such as ribosome and virus, due to employing the PDBx/mmJSON binary format.
Molmil supports the current standard formats, PDBx/mmCIF17 and its XML version, PDBML,18 as well as the conventional PDB format. For displaying even the large structures with millions of atoms, as shown in Figure 2, the JSON (JavaScript Object Notation)‐based PDBx/mmJSON format, derived from the standard PDBx/mmCIF format, is employed. It is a web‐optimized version of the PDBx/mmCIF data, as an alternative to the recently introduced MMTF format19, 20 that lacks much of the metadata described in the PDBx/mmCIF, because of its special purpose to only draw molecular pictures. PDBx/mmJSON is used by many services in PDBj, including PDBj's Mine 2 PDB Explorer, ProMode‐Elastic, and eF‐site, which we will also describe in this paper. The mmJSON files of the PDB entries are available via a REST service described at https://pdbj.org/help/rest-interface.
Figure 2.
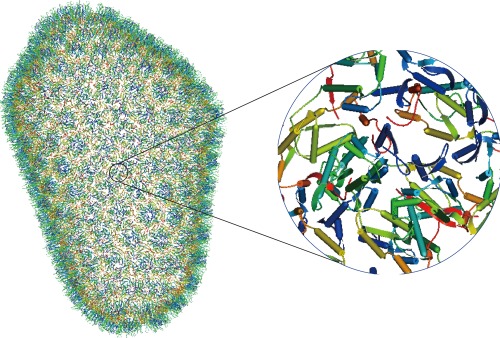
Molmil displays a large structure HIV‐1 Capsid (PDB ID, 3j3q) with 1,356 chains and more than 2.4 million atoms. The whole and zoomed pictures are shown.
ProMode‐Elastic
ProMode‐Elastic10 provides an elastic‐network‐model based NMA of PDB data (https://pdbj.org/promode-elastic/). The NMA results characterize the dynamic aspects of biomolecules quite well. In particular, it is known that some of the low‐frequency normal modes are strongly correlated with the large amplitude conformational changes in proteins observed upon ligand binding.21, 22, 23, 24
ProMode‐Elastic is characterized by the use of dihedral angles as independent variables and its application to the full‐atom system of the PDB data including protein, DNA, RNA, and ligand molecules without coarse‐graining them. It provides the results associated with the fluctuations of atoms and dihedral angles, and the correlations between fluctuating atoms. For oligomeric proteins and complexes with other molecules such as DNA and RNA, these properties are calculated for not only the whole system but also the individual components; that is, subunits, DNA, and RNA.
The fluctuations of atoms are represented by a fluctuation profile, displacement vectors on the 3D structure, and GIF animation. They are given for an average over all of the normal modes and time and for the 10 lowest‐frequency normal modes individually. In particular, the concerted motions of residues, such as opening‐and‐closing motions and sliding motions, around the interfaces of domains and subunits are worthy of attention. A fluctuation profile of dihedral angles furnishes information about internal fluctuations of local structures, such as secondary structures and hinge regions. The correlative movements of atoms are important to characterize protein dynamics, and they are shown in a triangle map that shows correlations between residues. Domains in a dynamic sense, that is, clusters of residues that move in a positively correlated manner with each other, and their mutual movement reveal structurally and functionally important aspects of a protein structure. The results are displayed on the web pages, and the raw data are also available there.
Figure 3 shows a snapshot of ProMode‐Elastic for the RNA polymerase elongation complex for an illustration. This is a core enzyme composed of five subunits, α2ββ’ω (A, B, C, D, and E chains in the PDB data, respectively), and two DNA and one RNA strands (G, I, and H chains, respectively) that form a fourteen‐bp double‐stranded DNA, a nine‐bp DNA/RNA hybrid, and a seven‐nucleotide single‐stranded RNA; a full‐atom system of 2880 amino acid residues and 52 nucleotides.
Figure 3.
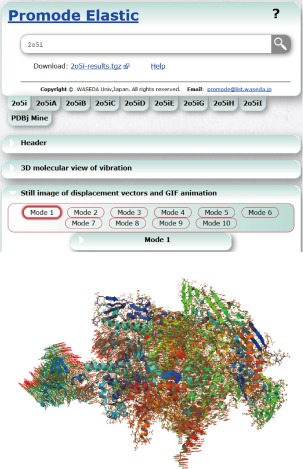
Snapshot of ProMode‐Elastic for RNA polymerase elongation complex (PDB ID, 2o5i).25 The displacement vectors of the atoms for the first lowest‐frequency normal mode are shown with the 3D structure. The results for the individual chains are also available on the web.
eF‐site with VR
The eF‐site, a database of electrostatic surfaces of protein functional sites (http://service.pdbj.org/eF-site/), was originally developed in 2004 and has been maintained for more than 10 years, by adding newly determined structures every week.11 The database has been widely used for various types of research, and is also useful for education, by providing concrete views of biomolecules. However, biomolecules such as proteins are too small to obtain a realistic view of the molecules.
To overcome this difficulty, we have extended the eF‐site by exporting our data for VR technology. We presently provide a VR experience with a VR interface for an Oculus VR LLC headset, and the application software was developed with the Unity framework. In this implementation, we paid special attention to show the flexibility of proteins, and thus used soft bodies to represent the molecular surfaces. Each vertex of a molecular surface fluctuates according to the B‐factor of the nearest atom to the vertex. In addition, we enabled the integrative manipulation of proteins with Oculus Touch. With the devices, users can easily and interactively observe protein molecular surfaces as if the protein were in their hand, like a molecular model in the real world (Fig. 4). The VR application and a data convert script for the application are freely available from the Tools page at the eF‐site.
Figure 4.
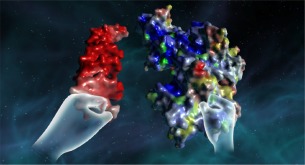
An example of a VR experience with a DNA molecule (left) and a protein (right).
Omokage search and Gmfit
Omokage search is a Web‐based service for structure searches focused on global shape similarities, for both three‐dimensional density maps and atomic models (https://pdbj.org/omokage).12 Although it is primarily for electron microscopy data, it is also useful to find some special similarity similarities among atomic models, such as molecular mimicry. The dataset contains more than 200,000 structures, consisting of EMDB maps,14 PDB atomic models, and their biological assemblies. We established a new method, incremental distance rank (iDR) profile comparison, to rapidly search through this large dataset. The iDR profile is a one‐dimensional profile of the distances of points, which are generated by the vector quantization method implemented in the Situs package.26 As a search query, the user can use a structure in the databanks or an original structure by uploading it to the server. In addition, the user can obtain the 3D fitting of the query with a found similar structure by gmfit,27 which uses a Gaussian mixture model (GMM). Since the recent improvements in cryo‐EM methodology and hybrid structure analysis are quite remarkable, we expect this tool to be used in wider fields, and thus we have improved on several points in the Omokage search.
The first improvement is the additional dataset from the structure models in the SASBDB, which is a databank for experimental data and structural models of small angle scattering (SAS).13 Some of data entries in the SASBDB have either one or multiple structural models. Each of the models is represented by one of three types of structures: atomic model, dummy atom model, and a mixture of them. The dummy atom model is also known as SAS bead model, which represents a molecular structure by a set of same spheres (dummy atoms or beads). Most of models employ a sphere corresponding to the amino acid size (radius = 1. 9 Å), but some use larger spheres. The iDR profile comparison and the GMM fitting are both compatible with all of the models. Now, the user can search the structure data in the SASBDB as well as a 3D map in the EMDB or an atomic model in the PDB. The user can also obtain the fitted view of the query structure onto found similar structures in any combination of data types, including the dummy atom model. For example, a search using the query of the exportin‐1 dummy atom model (model‐ID 68 in the SASBDB‐SASDAJ4) gave a result of similar structures including another dummy atom model of exportin‐1 (model‐ID 72 in the SASBDB‐SASDAK4) at rank 1, an atomic model of importin (PDB ID, 2xwu) at rank 3, and an atomic model of exportin‐1 (PDB ID, 4hzk) at rank 5. These models were fitted successfully by gmfit (Fig. 5).
Figure 5.
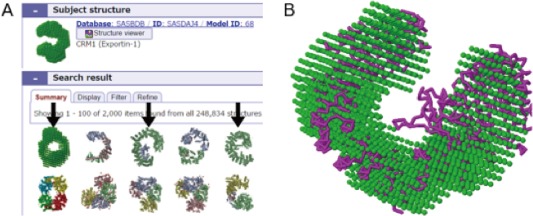
A. Result of Omokage search using SAS dummy atom model, SASBDB‐SASDAJ4, exportin‐1 as the search query. Found structures are ordered from left to right by the similarity scores. Arrows indicate exportin or importin structures. B. Superimposition of exportin‐1 and importin structures; query model with atomic model at rank 3 (PDB ID, 2xwu).
The second improvement is the filtering of the results. For instance, for users concerned with 3D map data only, the structures from the PDB and SASBDB in the search results are not necessary. Therefore, we implemented two types of filters, keyword and database. From the search results, the user can eliminate the data entry without matching particular keywords and the data entries that are not from a particular database: PDB, EMDB14 or SASBDB.13 For example, a search using a query of a low resolution map (38.5 Å) of Trypanosoma ribosome (EMD‐ID 8590) produces a list of similar structures containing many ribosomes of other species. Using the keyword “Trypanosoma”, the results were narrowed and all three Trypanosoma ribosome data were ranked in the top three.
The third improvement is the refinement of the search results. The GMM comparison in gmfit is more accurate than the iDR profile comparison.12 However, gmfit is not so fast as the iDR profile comparison, that it is not practical for a search against a large data set with more than 200,000 structures. This is why we have employed the iDR profile for the search and GMM for the fitting of the found structures. When the number of entries is in the range of hundreds, the computational time is on the order of minutes and is sufficiently practical. Now the user can re‐order the top 100 similar structures in the search results by the cross‐correlation coefficient value obtained by the GMM fitting. Using this functionality, within an additional several minutes, the user can obtain more accurate search results. For example, a search using the structure of the DNA gyrase complex (PDB ID, 2xct) gave results that included seven non‐related structures within the top 50. After the gmfit refinement, all of the structures that ranked in the top 50 were DNA gyrases or related molecular complexes.
The last improvement is about the gmfit comparison. To superimpose molecular structures, gmfit needs GMM for the target and reference structures. The server contains pre‐calculated GMMs for the three databanks, which are now calculated by the new algorithm, Gaussian‐input GMM. The new algorithm can consider the size of input atoms and grids, and yield a GMM with the same size (radius of gyration) as the input atomic model or map. The algorithm works especially well for the dummy atom model composing of a small number of spheres with large radius. And the algorithm is robust; it can convert thousands of maps without any failures caused by singularities. We also employ down‐sampled Gaussian‐input GMMs to convert a 3D density map with many voxels into a GMM, which require less computational time. The details of these algorithms will be published elsewhere. Additionally, the rapid molecular graphic program Molmil9 has now been implemented on the pairwise gmfit page, as well as JSmol and Jmol_S.
PDBj‐BMRB integrated search
PDBj‐BMRB (http://bmrbdep.pdbj.org) is a satellite BMRB7 repository for experimental and derived data gathered from nuclear magnetic resonance (NMR) spectroscopic studies. The primary repository consists of text files called NMR‐STAR, which are used for a wide variety of NMR researches and third‐party software applications. To enhance interoperability of NMR‐STAR data, we published common open representations in several Web standard formats, XML and RDF.28 To further encourage the reuse of NMR‐STAR data, we have launched PDBj‐BMRB integrated search service on our portal site that utilizes rich remote resources for annotating the original repository.
PDBj‐BMRB integrated search enables users to find biomolecules and biochemical information archived by BMRB, PDB, EMDB, UniProt, BMRB‐Metabolomics and Ligand Expo on the same screen and to associate obtained information. In addition to regular keyword search and sequence search, users can easily create highly structured queries by assistance of both an auto‐complete function being aware of dictionaries and a personalized query history storage function. (see http://bmrbdep.pdbj.org/search_help.html for instructions).
Search results are displayed as a list of entry panes by each information resource as shown in Figure 6. Each entry pane has interactive contents representing entry relationship between different databases via citation sharing, sequence homology, protein‐protein interaction and identity of chemical compound.
Figure 6.
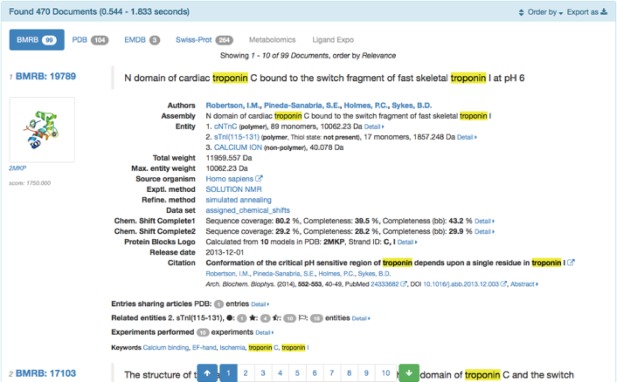
Search result example of PDBj‐BMRB integrated search. At first, each entry pane is displayed in collapsed form for summary, while detailed content can be displayed on demand. Here, content hits with the input keyword, “troponin”, are highlighted. Otherwise a “Hit context” menu appears in the pane and enables the user to identify where the keyword should hit in the source XML document that will serve as a clue to improve the original search query.
We have continuously added graphical contents in the pane. A notable improvement is the chart display for NMR experimental and derivative data, including validation of the assigned chemical shifts, various NMR relaxation analysis, and structural annotations, and so forth. Besides interactive HTML contents, tabular results, written in CSV, TSV, XML or JSON, are also available. They are suitable formats for saving the search results, importing into spreadsheet applications or data analysis tools for further data processing.
The bottom of page is allocated to the statistical panel of the search results, useful for grasping the tendencies of methodology, citation, and authors’ contribution.
Next, we have refreshed the BMRB entry page, each having a static URL such as http://bmrbdep.pdbj.org/bmr/bmr15400. The entry page basically follows the page design of the integrated search results page, except for that detailed contents are already expanded and having an integrated file export menu button, which allows users to retrieve various derivative data located on the BMRB mirror server (http://bmrb.pdbj.org) and PDBj‐BMRB data server (http://bmrbpub.protein.osaka-u.ac.jp).
PDBj‐BMRB data server has the role of releasing our derivative contents of the BMRB entries, originally via HTTP, rsync, or SPARQL endpoint. Recently, we have added several contents to enhance the integrated search service, as follows.
BMRB/JSON is another derivative representation of the NMR‐STAR data in JSON format that is smaller in size than the XML format for the same content. In fact, BMRB/JSON is on average about only 11% of the size of equivalent BMRB/XML. It is thus suitable for future development of web applications based on the NMR‐STAR data. The merits of JSON have been demonstrated by Molmil, for which PDBx/mmJSON has been developed.9 Furthermore, we prepared the schema of BMRB/JSON available at http://bmrbpub.protein.osaka-u.ac.jp/schema/mmcif_nmr-star.json.
The BMRB Relational DB snapshot (http://bmrbdep.pdbj.org/en/bmrb_rdb_snapshot.html) is an alternative RDB delivery service, which conforms to the original NMR‐STAR dictionary and contains remediation introduced while BMRB/XML was developed. It is provided as a compressed PostgreSQL dump image that can be transferred by the rsync protocol and restored using a single “pg_restore” command. Maintenance work can effectively be simplified, while unnecessary database replication can also be prevented. The schema of the RDB storing the original annotation of the dictionary is separately available at http://bmrbpub.protein.osaka-u.ac.jp/schema/mmcif_nmr-star.dic.schema.
Conclusion
The PDBj has developed several original services for the semantic web and for analyzing dynamic physico‐chemical properties and biological functions. In particular, as more large and complex structures are determined and registered in the PDB archive, much more complicated analyses will be required. The rapidly growing VR technology can also be applied for such analyses.
This work was supported by grants from the Database Integration Coordination Program from the National Bioscience Database Center (NBDC)‐JST (Japan Science and Technology Agency), the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from AMED, and JSPS KAKENHI [26440078] and [17K07364].
References
- 1. Kinjo AR, Suzuki H, Yamashita R, Ikegawa Y, Kudo T, Igarashi R, Kengaku Y, Cho H, Standley DM, Nakagawa A, Nakamura H (2012) Protein Data Bank Japan (PDBj): Maintaining a structural data archive and Resource Description Framework format. Nucleic Acids Res 40:D453–D460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kinjo AR, Bekker G‐J, Suzuki H, Tsuchiya Y, Kawabata T, Ikegawa Y, Nakamura H (2017) Protein Data Bank Japan (PDBj): updated user interfaces, resource description framework, analysis tools for large structures. Nucleic Acids Res 45:D282–D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berman HM, Henrick K, Nakamura H, Markley JL (2007) The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 35:D301–D303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berman HM, Burley SK, Kleywegt GJ, Markley JL, Nakamura H, Velankar S (2016) The archiving and dissemination of biological structure data. Curr Opin Struct Biol 40:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Velankar S, van Ginkel G, Alhroub Y, Battle GM, Berrisford JM, Conroy MJ, Dana JM, Gore SP, Gutmanas A, Haslam P, Hendrickx PMS, Lagerstedt I, Mir S, Montecelo MAF, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Sanz‐García E, Sen S, Slowley RA, Wainwright ME, Deshpande MS, Iudin A, Sahni G, Torres JS, Hirshberg M, Mak L, Nadzirin N, Armstrong DR, Clark AR, Smart OS, Korir PK, Kleywegt GJ (2016) PDBe: improved accessibility of macromolecular structure data from PDB and EMDB. Nucleic Acids Res 44:D385–D395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rose PW, Prlić A, Bi C, Bluhm WF, Christie CH, Dutta S, Green RK, Goodsell DS, Westbrook JD, Woo J, Young J, Zardecki C, Berman HM, Bourne PE, Burley SK (2015) The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res 43:D345–D356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Wenger RK, Yao H, Markley JL (2008) BioMagResBank. Nucleic Acids Res 36:D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Velankar S, Dana JM, Jacobsen J, van Ginkel G, Gane PJ, Luo J, Oldfield TJ, O'Donovan C, Martin M‐J, Kleywegt GJ (2013) SIFTS: structure integration with function, taxonomy and sequences resource. Nucleic Acids Res 41:D483–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bekker G‐J, Nakamura H, Kinjo AR (2016) Molmil: a molecular viewer for the PDB and beyond. J Cheminform 8:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wako H, Endo S (2013) Normal mode analysis based on an elastic network model for biomolecules in the Protein Data Bank, which uses dihedral angles as independent variables. Comput Biol Chem 44:22–30. [DOI] [PubMed] [Google Scholar]
- 11. Kinoshita K, Nakamura H (2004) eF‐site and PDBjViewer: database and viewer for protein functional sites. Bioinformatics 20:1329–1330. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki H, Kawabata T, Nakamura H (2016) Omokage search: shape similarity search service for biomolecular structures in both the PDB and EMDB. Bioinformatics 32:619–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valentini E, Kikhney AG, Previtali G, Jeffries CM, Svergun DI (2015) SASBDB, a repository for biological small‐angle scattering data. Nucleic Acids Res 43:D357–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawson CL, Baker ML, Best C, Bi C, Dougherty M, Feng P, van Ginkel G, Devkota B, Lagerstedt I, Ludtke SJ, Newman RH, Oldfield TJ, Rees I, Sahni G, Sala R, Velankar S, Warren J, Westbrook JD, Henrick K, Kleywegt GJ, Berman HM, Chiu W (2010) EMDataBank.org: unified data resource for CryoEM. Nucleic Acids Res 39:D456–D464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kinjo AR, Yamashita R, Nakamura H (2010) PDBj Mine: design and implementation of relational database interface for Protein Data Bank Japan. Database 2010:baq021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador‐Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Westbrook JD, Fitzgerald PMD (2009) The PDB format, mmCIF formats, and other data formats In: Bourne PE, Gu J, Eds. Structural Bioinformatics, 2nd edn Hoboken, New Jersey: John Wiley & Sons, pp. 271–291. [Google Scholar]
- 18. Westbrook J, Ito N, Nakamura H, Henrick K, Berman HM (2005) PDBML: The representation of archival macromolecular structure data in XML. Biooinformatics 21:988–992. [DOI] [PubMed] [Google Scholar]
- 19. Valasatava Y, Bradley AR, Rose AS, Duarte JM, Prlić A, Rose PW (2017) Towards an efficient compression of 3D coordinates of macromolecular structures. PLoS One 12:e0174846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bradley AR, Rose AS, Pavelka A, Valasatava Y, Duarte JM, Prlić A, Rose PW (2017) MMTF ‐ An efficient file format for the transmission, visualization, and analysis of macromolecular structures. PLoS Comput Biol 13:e1005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tama F, Sanejouand YH (2001) Conformational change of proteins arising from normal mode calculations. Protein Eng 14:1–6. [DOI] [PubMed] [Google Scholar]
- 22. Bahar I, Rader AJ (2005) Coarse‐grained normal mode analysis in structural biology. Curr Opin Struct Biol 15:586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobbins SE, Lesk VI, Sternberg MJ (2008) Insights into protein flexibility: The relationship between normal modes and conformational change upon protein‐protein docking. Proc Natl Acad Sci USA 105:10390–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wako H, Endo S (2011) Ligand‐induced conformational change of a protein reproduced by a linear combination of displacement vectors obtained from normal mode analysis. Biophys Chem 159:257–266. [DOI] [PubMed] [Google Scholar]
- 25. Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I (2007) Structural basis for transcription elongation by bacterial RNA polymerase. Nature 448:157–162. [DOI] [PubMed] [Google Scholar]
- 26. Wriggers W (2012) Conventions and workflows for using Situs. Acta Cryst D68:344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawabata T (2008) Multiple subunit fitting into a low‐resolution density map of a macromolecular complex using Gaussian mixture model. Biophys J 95:4643–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yokochi M, Kobayashi N, Ulrich EL, Kinjo AR, Iwata T, Ioannidis YE, Livny M, Markley JL, Nakamura H, Kojima C, Fujiwara T (2016) Publication of nuclear magnetic resonance experimental data with semantic web technology and the application thereof to biomedical research of proteins. J Biomed Semantics 7:16. [DOI] [PMC free article] [PubMed] [Google Scholar]