

Abstract
A review of previous stabilization of α/β-hydrolase fold enzymes revealed many different strategies, but no comparison of strategies on the same enzyme. For this reason, we compared five strategies to identify stabilizing mutations in a model α/β-hydrolase-fold enzyme, salicylic acid binding protein 2, SABP2, to reversible denaturation by urea and to irreversible denaturation by heat. The five strategies included and one location-agnostic approach (random mutagenesis using error-prone PCR), two structure-based approaches (computational design (Rosetta, FoldX) and mutation of flexible regions) and two sequence-based approaches (addition of proline at locations where a more stable homolog has proline and mutation to consensus). All strategies identified stabilizing mutations, but the best balance of success rate, degree of stabilization, and ease of implementation was mutation to consensus. A web-based automated program that predicts substitutions needed to mutate to consensus is available at http://kazlab.umn.edu.
Graphical Abstract
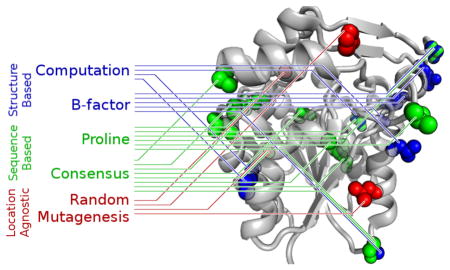
Introduction
Nature’s enzymes are often not stable enough for applications, so stabilization is a common goal of protein engineering. For example, the first engineering of an industrial enzyme stabilized subtilisin to oxidation by bleach for application as a laundry detergent additive.1 Besides stability to chemical modification, protein stability may refer to ability to maintain activity at ambient temperatures for a long time, maintain activity at high temperatures or in solutions containing organic solvents or other denaturants. Thus, there are different types of stability and a protein may be stable by one measure, but not by another.
Protein unfolding and folding are complex processes involving both random and cooperative steps, but simple models can approximate the process.2,3 For this paper, we treat protein un-folding as a single-step, cooperative process, as a simplification to ease comparison between different proteins even though proteins with several domains may unfold stepwise, Figure 1.
Figure 1.

Protein unfolding disrupts structure and function and may involve additional, irreversible steps. In normal conditions (top), a protein is primarily in its native folded form (left), and equilibrates with its unfolded form (right). Denaturants, e.g., heat or urea, shift the folding-unfolding equilibrium toward unfolding. Heat-induced protein unfolding is usually irreversible because it includes both unfolding and irreversible aggregation (middle). Unfolding by chemical denaturants like urea usually measures the reversible folding-unfolding transition (bottom).
The simplest way to measure enzyme stability is by heat denaturation. A sample is heated, then cooled and the remaining catalytic activity measured. A more stable enzyme will require a longer heating time or heating to a higher temperature to lose half of its activity. Heating usually favors unfolding, while upon cooling, some of the enzyme refolds, while some may not. Since loss of activity involves both unfolding and aggregation, connecting molecular changes to changes in this heat stability is difficult.
To compare stabilities of two proteins measured by heat denaturation, the ratio of the half-lives for the two proteins yields the difference in Gibbs free energy of activation for the activity loss according to:
| EQUATION 1 |
where R is the gas constant and T is the temperature where half-life was measured. In some cases, researchers reported only residual activity after incubation at elevated temperatures. For these cases, we assumed that inactivation followed first order kinetics and calculated the half-lives according to:
| EQUATION 2 |
| EQUATION 3 |
where A0 is initial activity, A is the residual activity after heating, t is the incubation time and k is the first order rate constant. The changes in half-lives were converted to free energies of activation using equation 1 above.
Another way to measure enzyme stability is measuring unfolding with increasing concentrations of a denaturant like urea. Urea changes hydrophobic interactions by changing the solvent properties of water and disrupts the protein’s native secondary structures by forming hydrogen bonds to the protein’s backbone amides. Many spectroscopic techniques, including circular dichroism and intrinsic fluorescence, can detect unfolding due to increasing concentrations of urea. The change in ratio of folded to unfolded protein can be used to calculate Gibbs free energy (ΔG) for the transition from folded to unfolded. Extrapolation of these values to a concentration of zero denaturant yields an estimate of the change in Gibbs free energy in the absence of denaturant, ΔGH2O.4
These two methods measure different aspects of protein stability. First, unfolding by heat and unfolding by urea are mechanistically different. Heat unfolds by partly by increasing the energetic contribution of entropy to both the folded and unfolded state, while urea unfolds by changing the solvent properties. Proteins differ in their resistance to the two mechanisms. Second, measuring heat-induced unfolding usually involves exposure to heat for a short time followed by cooling and assay for remaining activity. Loss of activity involves both unfolding at elevated temperature and an inability to refold upon cooling. This irreversible heat-induced unfolding measures kinetic stability. In contrast, urea-induced unfolding involves incubation to allow unfolding to come to equilibrium. This unfolding is reversible and therefore measures thermodynamic stability.
To narrow the scope of the problem, this manuscript focuses on the α/β-hydrolase fold superfamily.5–7 α/β-Hydrolase fold enzymes include lipases, esterases, and hydroxynitrile lyases. Since industry often uses these enzymes, numerous groups have engineered these enzymes for higher stability. A literature search revealed 56 reports of such protein engineering, Figure 2 & Table S1. The list only includes the amino acid substitutions that stabilized the enzyme and omits examples of immobilization or changes in solvent or reaction conditions. The use of different stability assays and different proteins makes an entirely fair comparison impossible. Nevertheless to identify broad trends, we converted stabilization data into ΔΔG values making assumptions and approximations noted in Table S1. The stabilization values are not normalized for differing numbers of substitutions or the size of the protein. We omitted examples that strongly decreased catalytic activity, but did not otherwise consider the effect on enzymatic activity. The most commonly stabilized enzymes are lipases (35 reports), but the others include esterases (15 reports), hydroxynitrile lyases, haloalkane dehalogenases, acetylcholinesterase, and epoxide hydrolase. When the reports include several stabilized variants, the comparison uses only the best one from each report.
Figure 2.

Summary of reported stabilization of α/β hydrolases. On average (black horizontal lines), location agnostic methods (typically error-prone PCR) yielded larger stabilizations than structure or sequence-based design. Circles with grey centers and numbers are the number of amino acid substitutions for that example, while circles without numbers correspond to 1–3 substitutions; Δ13 indicates deletion of 13 amino acids. Table S1 lists details for each example.
Protein engineering strategies vary in complexity and prerequisite knowledge, yet there are few examples of side-by-side comparisons of different strategies.8 This work seeks to begin to remedy this oversight in the literature by comparing various strategies used to stabilize proteins. First, requiring the most knowledge, is structure-based design. This approach uses the structure to model molecular interactions and predict specific substitutions. Alternatively, researchers may use the structure to identify flexible regions of the structure and then use trial and error to find suitable substitutions (a semi-rational approach). For example, using a combination of molecular modeling methods Floor and colleagues identified stabilizing substitutions in the haloalkane dehalogenase LinB. Combining 12 of these stabilizing substitutions increased the half-life at 60 °C over 200-fold (ΔΔG‡ = 3.5 kcal/mol).9 As an example of the semi-rational approach, the structure of Bacillus subtilis lipase A identified which regions were most flexible. Targeting random substitutions to these regions combined with screening identified a variant with a 490-fold (ΔΔG‡ = 4.0 kcal/mol) longer half-life at 55 °C.10 Similarly, Ahmad and coworkers targeted all loop regions and termini in this lipase reasoning that these regions are more flexible and increased the half-life 844-fold (ΔΔG‡ = 4.4 kcal/mol).11 Only approximately one third (11/30) of structure-based approach reports yielded large stabilizations (>2 kcal/mol), which corresponds to a >29-fold increase in stability at room temperature.
Second, requiring less knowledge, is sequence-based design. Sequence-based design uses sequence information to identify residues that differ from homologs. One way to use this sequence information is to replace amino acids in the target protein with those found in a more stable homolog. One of the five examples in Table S1 used this approach. Another way to use sequence information is to replace amino acids in the target protein with amino acids conserved in homologs, regardless of whether the homologs are more stable or not.12–14 This mutation-to-consensus approach hypothesizes that evolution conserves important residues. While some conserved residues contribute to catalysis, others contribute to stability15 so conserved residues are more likely to contribute to stability than non-conserved residues. Where the target protein differs from the consensus, mutation to the consensus residue is more likely to stabilize the protein than a random substitution. Only three reports used this consensus approach for α/β-hydrolase fold enzymes,16–18 but we find it especially reliable, see below. Using sequence-based approaches to pick substitutions at positions identified by structural approaches,19,20 were classified as structure-based since this ‘smart library’ approach requires structural information.
Third, location agnostic methods use random mutagenesis or large single-substitution libraries throughout the protein. Screening identifies the more stable mutations from these large libraries. These approaches are most useful when little is known about the protein. Location agnostic methods yielded the largest increases in stabilization. Two-thirds (14/21) of the random mutagenesis reports yielded >2 kcal/mol improvements. Two examples of location agnostic approaches are systematic site-saturation mutagenesis and error-prone PCR. Systematic site saturation mutagenesis creates and tests all possible single substitution variants. This approach identified eight stabilizing substitutions in a haloalkane dehalogenase. Combining these substitutions created a variant that was 30,000 times more stable than the parent (ΔΔG‡ = 7.2 kcal/mol).21 This is the largest stabilization reported for an α/β-hydrolase fold enzyme. Error-prone PCR also creates substitutions throughout a protein, but creates an average of only 5.7, not 19 amino acid substitutions because error-prone PCR is unlikely to change more than one nucleotide in a codon. Two or three nucleotide substitutions are needed to change codons to encode all possible amino acids so error-prone PCR creates an incomplete library.22 Error-prone PCR identified substitutions in Bacillus subtilis lipase A that increased stability by 22 °C (ΔΔG‡ = 6.2 kcal/mol).23
Location agnostic approaches appear to give the highest increases in stability followed by structure-based approaches and lastly sequence-based approaches. Variants created by location agnostic approaches increased the protein stability by an average of 3.1 ±1.9 kcal/mol, while structure or sequence based approaches increased stability by an average of 2.0 ±1.4 or 1.2 ±0.5 kcal/mol, respectively. The differences in these averages are at the edge of statistically significant. Comparison of the three by one-way ANOVA indicates that the differences are statistically significant for the 22 structure-based, 4 sequence-based, and 21 location agnostic reports (p = 0.02 where p = 0.05 is the cutoff for statistical significance) but pairwise comparison by a post-hoc Tukey’s range test indicates they are just outside the limit for statistical significance. The differences are not due to different numbers of substitutions since comparison of the improvement per substitution also yields the same ranking of the methods. The location agnostic approaches identified substitutions that increased stability by an average of 0.8 kcal/mol per substitution, while structure or sequence based approached identified substitutions with an average increase of 0.6 and 0.4 kcal/mol per substitution, respectively.
Over all, stabilizing mutations are common and occur anywhere in the protein. Stabilizing substitutions occurred in both lid and catalytic domains, surface exposed areas and buried regions. Stabilizing substitutions occurred in flexible regions like loops, C and N-termini, and also in secondary structures like helices and β-strands.
One generalization is that adding a disulfide link between the lid and catalytic domains often stabilized α/β-hydrolase fold enzymes. Researchers discovered these disulfide links either by structure-based design (eight examples) or by sequence-based design (one example). In the structure-based design, researchers first identified residue pairs that satisfy geometric requirements for a disulfide link and subsequently used different methods to narrow the choices. Pikkemaat and coworkers24 chose a pair where molecular dynamics modeling revealed a flexible region of the lid. Han and coworkers25 chose pairs where molecular modeling predicted the most stabilization, and Yu and coworkers26 chose the only two that involved lid residues because their previous work25 found stabilizing disulfide links there. Yamaguchi and coworkers27 chose the disulfide because homologs contained a similar cross-link, so we classified it as sequence-based design. One limitation of disulfide links to stabilize proteins is the added complexity of expression in E. coli. To form disulfide bonds with the normally reducing intracellular environment requires either specific E. coli strains, or the insertion of additional chaperone proteins.28 Because of this added complexity, we do not attempt to add disulfide bonds in this work.
While previous work provides examples of stabilization, it does not identify which strategies are best since they involve different approaches on different proteins. The best protein engineering approaches would reliably predict stabilizing substitutions, yield large increases in stability, and be easy to implement. In this paper, we compare five different methods (two structure-based, two sequence-based, and one location agnostic method) side by side on one protein - a plant esterase, salicylic acid binding protein 2 (SABP2) as a model α/β-hydrolase fold enzyme. For a fair comparison we limited each method to single substitution mutants. This comparison of methods on a single protein is an important next step in the critical comparison of engineering strategies and elimination of less effective strategies. Similar comparisons with other proteins are needed before making broad conclusions. SABP2 has typical stability for a protein from mesophiles (ΔΔGH2O = 6.9 kcal/mol).29
Methods
General
Reagents were from Sigma Aldrich unless otherwise noted. Distilled water was further purified by ion exchange (Milli-Q). DNA primers for the polymerase chain reaction were from Integrated DNA Technologies (Coralville, IA) and plasmid DNA was sequenced by ACGT (Wheeling, IL) using BigDye terminator version 3.1. Other molecular biology reagents were: Pfu DNA polymerase (Agilent Technologies, La Jolla, CA), restriction enzymes DpnI, BamHI, and XhoI (New England Biolabs, Beverly, MA), DNA marker and SDS-PAGE protein marker (Invitrogen Life Technologies, Grand Island, NY), isopropyl-β-D-thiogalactopyranoside (IPTG, Gold Biotechnology, St. Louis, MO), and ampicillin (Roche Diagnostics). Recombinant proteins with C-terminal hexahistidine tags were purified from the cell lysate by Ni-affinity chromatography (Ni-NTA, Qiagen) according to the manufacturer’s instructions. Protein concentrations were measured by the Bradford dye-binding assay at 595 nm using the Bio-Rad reagent and five dilutions of bovine serum albumin as a standard. Protein gels were run on sodium dodecyl sulfate polyacrylamide gradient gels (NuPage 4–12% gradient Bis-Tris gel from Invitrogen) using BenchMark protein ladder (5 μL/lane) as the standard. DNA gels were run using 0.8% ultrapure agarose with 1×TAE buffer and 1 kb DNA ladder as a standard. All modeling of salicylic acid binding protein 2 started with chain A from pdb entry: 1Y7I.30
Site-directed mutagenesis
Site-directed mutagenesis was done using whole plasmid mutagenesis similar to the QuikChange approach (Agilent). A high fidelity DNA polymerase extended a pair of overlapping mutagenic primers to yield linear strands with complementary overlapping ends that can cyclize to yield a nicked circular DNA. The template was a pET-21a(+) expression vector containing the wild-type SABP2 gene (pET-21a(+)-SABP2),31 which was amplified using the primers in Supplementary Table S2. The thermocycling reaction mixture contained 1–2 ng DNA template, 1 μL each of forward and reverse primer (10 μM), 1 μL of 10 mM dNTPs, 5 μL Pfu 10X buffer, 0.5μL Pfu DNA polymerase and sterile water to a final volume of 50 μL. Since two primers overlap, newly synthesized strands cannot serve as templates for subsequent cycles, thus, the thermocycling reaction amplifies the plasmid linearly instead of exponentially; it is not a chain reaction and should not be called a PCR. The thermocycling program for plasmid amplification was 95 °C for 1 min, 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 7 min, and a final extension at 72 °C for 10 min using an OpenPCR thermocycler (Chai Biotechnologies, Santa Clara, CA). After confirming with gel electrophoresis that the amplification succeeded, the methylated parental DNA strands were fragmented with a restriction enzyme specific for methylated DNA (DpnI, 5 U, 37 °C, 1 h). A portion of the reaction mixture (3 μL) was transformed into chemically competent E. coli DH5α by heat shock at 42 °C for 1 min. The transformed cells were spread on a lysogeny broth (LB) agar plate containing 100 μg/mL ampicillin and incubated at 37 °C overnight. Colonies that appeared were picked and grown in LB-ampicillin media (3 mL) overnight. Plasmid DNA was isolated from this culture using QIAprep MiniPrep kit and sequenced to confirm that the DNA contained the desired mutation.
Protein expression and purification
Plasmid pET-21a(+)-SABP2 containing wild type or mutant SABP2 gene was transformed into chemically competent E. coli BL21 (DE3) for protein expression. The pET-21a vector contains a T7 inducible promoter, an N-terminal tag (MetAlaGlyAspPro) and a C-terminal 6-His tag. Recombinant cells were grown at 37 °C until the OD600 reached 1.4–1.6 in modified Terrific Broth (TBg); 12 g/L Bacto™ tryptone, 24 g/L Bacto™ yeast extract, 9.86 g/L glucose (in place of the standard glycerol), with added 2.3 g/L KH2PO4 and 12.5 g/L K2HPO4, containing 100 μg/mL ampicillin. Cultures were cooled to 17 °C, then 1 mM IPTG was added to induce protein expression, the cells were incubated overnight and then harvested by centrifugation at 5000×g for 15 min at 4 °C. Supernatants were discarded and cells were resuspended in lysis buffer (50 mM sodium phosphate buffer, 500 mM NaCl, 20 mM imidazole, pH 8.0). Cells were lysed by sonication (Branson Digital Sonicator) on ice at 40% amplitude for 5 min carried out in 3 s pluses alternated with 6 s cooling. The cell extract was centrifuged at 12,000×g at 4 °C for 30 min to remove cell debris. Proteins were purified from the supernatant by Ni-affinity chromatography using a gravity column with 1 mL of regenerated Ni-NTA resin for up to 500 mL of culture, with washes of lysis buffer (50 mL), wash buffer (50 mL, lysis buffer containing 50 mM imidazole), and eluted with elution buffer (lysis buffer containing 125 mM imidazole). The fractions containing pure enzyme were combined and concentrated using a centrifuge tube containing an ultrafiltration membrane. Using the same centrifugal filtration, the buffer exchanged by enzyme storage buffer, 5 mM BES (N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid) pH 7.2. A 250-mL culture of E. coli typically yielded twenty mg of purified enzyme.
Structure-based Computational Design of More Stable Variants
Rosetta Design
Nine individual 7 to 21-residue secondary structure regions of SABP2 were redesigned using the RosettaDesign server.32,33 The regions were selected by first excluding residues near the active site, and then selecting contiguous regions. Regions were divided at turns between secondary structural elements. Regions were redesigned independently. Amino acids within a region were allowed to mutate to any amino acid if buried, or any hydrophilic amino acid if solvent exposed. Substitutions within each region that contributed most to the improved predicted energy were tested again with RosettaDesign in which only the single residue was redesigned. All residues within 4 Å of redesigned amino acid(s) were allowed to repack to better accommodate mutations.
RosettaDesign server was used with the following parameters specified in the “.res” files: Default natural amino acid, with natural conformation (NATRO), target residue(s) mutated to any amino acid, using rotamer database, plus 0.5 and 1.0 standard deviations (ALLAA EX 1 EX 2) except where residue was solvent exposed, in which case amino acid selection was limited to polar residues (PIKAA RNDEQHKSTY EX 1 EX 2). Residues within 4 Å of mutated amino acid(s) were designated to use native amino acid, and explore rotamer database and natural conformation to allow packing with mutated residues (USE_INPUT_SC EX 1 EX 2). To calculate change in energy scores caused by the substitution, the energies were compared to a control where the same rotamer freedom was given to all the same residues as the mutant calculations, but all residues were specified as natural amino acid.
FoldX
The FoldX algorithm (version 3.0 beta5.1, available from http://foldx.crg.es/) uses a full atomic description to estimate the folding free energy of proteins.34,35 The X-ray structure of SABP2 (PDB:1Y7I30) was preprocessed using “RepairPDB” command to minimize the energy of the structure, followed by the “BuildModel” command to evaluate the effect of various substitutions.
Sequence-based Design of More Stable Variants
Consensus Sequences
Consensus Finder is a web tool implemented in Python. Users input protein sequences in FASTA format. Suggested mutations are generated in the web browser, and a compressed file containing detailed results is made available for download. The detailed results contain a text file with the suggested mutations, aligned and trimmed sequences in FASTA format, the consensus sequence in FASTA format, and comma-separated values text files containing counts and frequencies of each amino acid at each position of the input sequence.
Consensus Finder automates a number of processes to generate the suggestions. Processing occurs via a Python script then invokes a BLASTp search of the NCBI “nr” database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with the input sequence, downloading the resulting sequences, removing over-represented sequences using CD-HIT Suite,36 aligning the sequences using Clustal Ω,37,38 and trimming the alignment to the size of the input sequence. The counts and frequencies of each amino acid at each position corresponding to the input sequence are calculated, and any positions where the input sequence differed from the consensus are suggested for mutation if the strength of the consensus is over a specified threshold. The specified threshold is given either as the minimum absolute amino acid frequency of the consensus or as the ratio of the consensus frequency and the frequency of the amino acid found at the same position in the query sequence. Use of the ratio may help to eliminate rare and destabilizing residues found in the query protein.
SABP2 was analyzed with Consensus Finder to identify places where its sequence could be mutated to consensus. We returned the top 200 results from the BLASTp search. Sequences that were >90% identical were grouped, and one representative from each group was selected, resulting in 116 sequences. Minimum consensus threshold was set at 70% for making mutations. Amino acid alignments were visualized with SeaView39 and the amino acid distribution was visualized as a “logo” using WebLogo 3.40
Random mutagenesis
Library construction using error-prone PCR (epPCR)
Random mutagenesis was carried out using Genemorph® II Random Mutagenesis Kit (Stratagene) including the polymerase Mutazyme II, which reduces the bias for mutations at AT versus GC sites. Plasmid pET-21a(+) containing SABP2 gene as used as a template in the ep-PCR. Forward primer 5′ AGG AGA TAT ACA TAT GGC GGG GGA and reverse primer 5′ GGC TTT GTT AGC AGC CGG ATC TC flank the SABP2 gene to confine the mutations to only this gene. Low mutation frequency (0–4.5 mutations/kbase) experiments used 500 ng of plasmid DNA. The PCR thermal cycle was 95 °C for 1 min, 54 °C for 1 minute and 72 °C for 1 min for a total of 30 cycles. The PCR-mutated genes and SABP2 plasmid were digested with restriction enzymes BamHI and XhoI and purified by gel electrophoresis on a 1% agarose gel. The desired bands were cut from the gel and isolated using the QIAquick Gel Extraction kit (Qiagen). The PCR mutated SABP2 was ligated into the pET-21a(+) vector backbone using T4 DNA ligase (New England Biolabs, Beverly, MA) and the resulting plasmid was transformed into E.coli BL21 electrocompetent cells by electroporation. The cells were grown overnight at 37 °C on LB plates containing ampicillin (100 μg/mL) and IPTG (150 μl of a 800 μM solution per plate, spread and dried prior to use). The ligation and transformation were repeated several times to obtain a total of ~2000 colonies.
Library Screening
A primary screen identified colonies producing active esterase.41 Colonies were covered with a layer of molten agarose (0.4%, w/v) containing 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES)-NaOH buffer (50 mM, pH 7.5), β-naphthyl acetate (0.28 mg/mL) and Fast Blue RR salt (10 mM). Approximately half of the colonies (1000 total) turned blue within minutes indicating esterase activity and were picked using pipet tips for secondary screening a 96-well master plate containing TBg (200 μL per well). Each secondary screening plate also included wild type SABP2 as a control. The 96-well master plates were shaken overnight at 800 rpm at 37 °C. Using a 96-metal-pin replicator, this master plates was used to inoculate protein expression plates: 96-deep-well plated containing IPTG (80 μM) and TBg (1 mL/well). The cultures were shaken at 800 rpm for 20 h at 37 °C to express the proteins, then the cells were harvested by centrifugation at 2,000 × g and the supernatant was discarded. The cell pellets were washed with phosphate saline buffer (500 μL per well, 50 mM phosphate, 200 mM NaCl, pH 7.4) and resuspended in 150 μL of BugBuster protein extraction reagent (Novagen, Merck Millipore), 0.5 mg/mL lysozyme (EMD Chemicals) and 1.3 U/mL DNase (Invitrogen). The cells were shaken (500 rpm) and incubated in room temperature for one hour for cell lysis and DNA digestion. Another 600 μL of 5 mM BES buffer (pH 7.2) was added to the suspension to increase the volume of the lysate. Lysate was centrifuged at 2,000 × g and the supernatant was used directly for screening. The initial esterase activity in the supernatant was determined by adding an aliquot (30 μL) of each supernatant to a reaction mixture (70 μL) consisting of BES buffer (5 mM, pH 7.2), 10 vol% acetonitrile and 0.3 mM pNPAc (p-nitrophenyl acetate) and monitoring the increase in absorbance at 404 nm at room temperature for 10 min. Another aliquot of each supernatant was heated to 50° C in a thermocycler for 15 min, cooled on ice, and the esterase activity measured as above. The ratio of activity remaining after heat treatment to the initial activity estimated the thermostability. Variants that retained more than three standard deviations more activity than wild type were further characterized.
Saturation mutagenesis
Whole-plasmid mutagenesis where a polymerase extends mutagenic primers to copy the entire pET-21a(+) plasmid was used for site-saturation mutagenesis of SABP2 at position 212 (method similar to QuikChange). Thermocycling using the high fidelity DNA polymerase Pfu linearly amplified the both strands of the plasmid to yield overlapping strands. The overlapping primers below include an NNK degenerate codon, which yields a mixture of 32 codons that encodes for all 20 amino acids at least once. The thermocycling reaction mixture contained 80 ng DNA template, 1.5 μL DMSO, 1 μL each of forward and reverse primer (100 μM), 5 μL 10X cloned Pfu reaction buffer, 2.5 μL 10 mM dNTPs, 1 μL cloned Pfu DNA polymerase (Stratagene) and autoclaved deionized water to a final volume of 50 μL. The thermocycling program was 25 cycles of denaturation at 95 °C for 1 min, annealing at 55 °C for 1 min and DNA synthesis at 72 °C for 10 min. To increase the efficiency of mutagenesis, the parental DNA, which was isolated from E. coli and was therefore methylated, was fragmented with DpnI, a restriction endonuclease that cleaves only methylated DNA, by adding 5 units of DpnI to the reaction mixture and incubating overnight at 37 °C. The DNA was isolated from the total 50 μL reaction using a PCR purification kit (Invitrogen) and 5 μL was transformed into E. coli BL21 (DE3) electrocompetent cells by electroporation. The bacteria were spread onto LB agar plates containing 100 μg/mL ampicillin and incubated at 37 °C overnight. About 350 colonies (corresponds to >99% probability that any of the 20 possible amino acid variants was tested at least once) were obtained, picked from the agar plates and screened for thermostability as described above for the error prone PCR variants.
Enzyme Characterization
Esterase activity
Hydrolysis of p-nitrophenyl acetate (pNPAc) was measured at pH 7.2 and room temperature and was corrected for spontaneous hydrolysis. The assay was performed in a 96-well microtiter plate with reaction volume of 100 μL (light path length = 0.29 cm) containing 0.3 mM pNPAc, 10 vol% acetonitrile, 4.2 mM BES (pH 7.2) and ~1 μg of enzyme. The release of p-nitrophenoxide (ε404 nm = 16,600 M−1cm−1; this value accounts for its incomplete ionization at this pH) was measured spectrophotometrically (SpectraMax plus-384 plate reader, Molecular Devices, Sunnyvale, CA). Reported values are the mean of three independent experiments. Steady state kinetic constants were determined with the same method with pNPAc concentrations varying from 0.3 up to 15 mM as needed and a nonlinear fit of the data to the Michaelis-Menten equation using the solver in Microsoft Excel or LibreOffice.42
Protein unfolding in urea
A solution of purified SABP2 (100–300 μg/mL) in BES buffer (5 mM, pH 7.2) with the concentrations of urea ranging from 0 to 6.2 M was incubated 24 hours at 4 °C. The intrinsic tryptophan fluorescence was measured at 329 nm with a SpectraMax GEMINI XS plus-384 plate reader (Molecular Devices, Sunnyvale, CA) using excitation light of 278 nm. These wavelengths maximized the observed decrease in fluorescence as the protein unfolded. Comparing the measured fluorescence to the fluorescence for the completely folded or unfolded forms indicated the equilibrium amount of each form.
SABP2 unfolds via at least one intermediate, resulting in overlapping unfolding curves, which leads to difficulties in calculating the Gibbs free energy of unfolding.29 To avoid these poor estimates, we report the concentration of urea required to cause 50% unfolding, [urea]½, which is less sensitive to these ambiguities.
The natural log of the ratio of unfolded to folded protein is a linear function of the concentration of denaturant (equation 4, derived from equations in reference 4), where Y is fluorescence, YF is fluorescence of fully folded protein, YU is fluorescence of fully unfolded protein, and m and b are the slope and intercept relating urea concentration. A custom Python script determined the values of YF, YU, b and m by a least squares fit to the measured fluorescence. When 50% unfolded, both sides of the equation equal zero. The [urea]½ value is the y intercept for the linear equation.
| EQUATION 4 |
Heat denaturation
Purified SABP2 solutions (0.2 mg/mL, 200 μL) were heated in a water bath at 60 °C for 15 min, then transferred to a water bath at 4 °C for 15 min. The esterase activity was measured at room temperature before and after heating and the half-life of esterase activity was calculated with the following equation:
| EQUATION 5 |
where T½ is the activity half-life at 60 °C, T is the incubation time (15 min), A0 is initial activity, and A15 is residual activity after 15 minute incubation.
Combinatorial gene library
The mutations initially identified as the most stabilizing were randomly combined to further increase stability using a gene shuffling approach. A set of oligonucleotides (Integrated DNA Technologies, Inc. Coralville, IA) were designed to randomly combine these 26 (24 locations) mutations (Table S5 lists mutations and degenerate codons; Table S2 lists oligonucleotide sequences). This arrangement of oligos used for creating the combinatorial gene library is based on the method described in Ness and colleagues43, with overlapping forward and reverse 60-mers containing degenerate nucleotides where possible to allow for any of the desired mutations. For example, “…ACA RCT CTT…” (R= A or G), which can code for …Thr-Ala-Leu… (WT) or …Thr-Thr-Leu… (stabilizing mutant). Where multiple degenerate nucleotides are required to reach all desired possibilities, additional, alternative oligomers were spiked in.
The library was generated with three separate PCR steps (two assembly, and one amplification), then ligated into pET-21a vector and transformed into E. coli BL21(DE3) using a method similar to that described by Ness and coworkers43 and Young and Dong.44 In the first assembly PCR, each possible set of four adjacent/overlapping oligos (2 forward, 2 reverse) were mixed in separate reactions to create nine separate 180 bp gene fragments, each overlapping 90 bp with the gene fragment on either side. The 20-μL reaction mixes contained oligo mix (2 μL, containing 2.0 μM of two “outside” oligos, 0.4 μM of two “inside” oligos), 2 μL Pfu buffer, dNTP mix (0.5 μL, 10 mM each), 0.5 μL Pfu polymerase, and 15 μL ddH2O. Reaction was carried out in OpenPCR thermocycler with 20 cycles of 94 degrees for 20 s, 45 degrees for 15 s, and 72 degrees for 30 s. A 5-μL aliquot from each reaction was mixed, and cleaned up with Pure-LINK PCR Purification kit (Invitrogen) and eluted with 45 μL elution buffer. The second assembly step combined 180 bp gene fragments, with 90 bp overlaps, into complete genes. A 22.5-μL aliquot of the cleaned up fragment mix was combined with 5 μL Pfu buffer, 1.25 μL dNTP Mix (10 mM each), 1.25 μL Pfu polymerase, and 20 μL ddH2O for a total reaction volume of 50 μL. These were reacted with 20 cycles of 94 degrees for 30 s, and 68 degrees for 120 s in OpenPCR thermocycler. After the second assembly step, the full length assemblies were amplified with both 5′ and 3′ oligos to increase amount, and to increase the proportion of complete gene assemblies relative to partial gene fragments. A 40-μL reaction containing F amplification primer (2 μL, 10 μM, “GGCGGGGGATCCGATG”), R amplification primer (2 μL, 10 μM “GTGGTGCTCGAGGTTGTATTTMTG”), 4 μL Pfu buffer, dNTP mix (2 μL, 10 mM each), 2 μL Assembly mix, 1 μL Pfu polymerase, and 27 μL ddH2O was amplified with the following program: 95 °C for 60 s, 23 cycles of 95 °C for 30 s, 65 °C for 30 s, and 72 °C for 120 s, with a final elongation step of 72 °C for 420 s. Product was cleaned up with PureLINK PCR Purification kit, digested with XhoI and BamHI (50 μL cleaned up PCR product, 7 μL NEB3 buffer, 3.5 μL XhoI, 3.5 μL BamHI, 0.7 μL 10 mg/mL BSA, 3.5 μL ddH2O) overnight at 37 °C. This was cleaned up again with PureLINK kit and 40–80 fmols were ligated into 20 fmols gel-extracted-digested pET-21a in a 20-μL reaction with 1 μL T4 ligase in 2 μL T4 ligase buffer overnight at 4 °C. Ligated product was cleaned up and eluted in water. A 2–5 μL aliquot of ligation product was transformed into E. coli BL21(DE3).
A two-step screen identified highly stable mutants. The cells were plated, and first screened for esterase activity, as above. Those showing activity were grown in a liquid culture, lysed and the supernatant was screened for thermal stability as described above, except that secondary thermal stability screening was done at 60 °C to increase stringency since variants were expected to be more stable that in the epPCR library. Those that had significantly higher thermal stability were expressed in a larger format and the purified proteins were more thoroughly characterized, as described above. In total, we screened ~500 combinations. This screen was far from complete, and many beneficial combinations were certainly missed.
Results
Wild-type SABP2
Wild-type SABP2 unfolds in urea stepwise with at least one intermediate. Fitting this unfolding to a three state model yielded a stabilization energy of 6.9 kcal/mol,29 which lies within the range of 5–15 kcal/mo1 for most globular proteins.45,46 The complexity of this three-state model makes it difficult to use the model to measure changes in stability. To simplify the approach, we compared the concentration of urea required to unfold half of the protein, “[urea]1/2”. This approach assumes that the unfolded protein structure is similar in each case.47 A concentration of 2.23 M urea unfolds half of wild-type SABP2 and a more stable variant would require a higher concentration. This urea-unfolding is mostly reversible so this metric measures the thermodynamic stability of SABP2.
To measure irreversible inactivation of SABP2, we heated the enzyme to a set temperature for 15 min, cooled it to room temperature and measured the amount of remaining esterase activity. Wild-type SABP2 had a half-life at 60 °C of 3.5 minutes,29 which is typical for globular proteins from mesophiles.48 This activity loss upon heating metric measures SABP2’s propensity to unfold and ability to refold versus aggregate and thus its kinetic stability.
Single amino acid substitutions that increase the stability of wild-type SABP2
Each method identified numerous single amino acid substitutions that stabilized SABP2 as measured by either urea-induced unfolding or activity loss after heating, Table 1. Mutations are sorted by amino acid number, and stabilizing effects are indicated by values and lengths of the blue/red bars in last two columns. The two structure-based methods, computational and flexibility based, predicted thirteen and fifteen stabilizing mutations respectively. Only four of the thirteen computationally predicted mutations stabilized SABP2 by at least one measure of stability, while nine mutations (at five positions) of the fifteen predicted by flexibility stabilized SABP2 by at least one measure. The two bioinformatics-based approaches, mutation to consensus and copying proline from a stable homologue, predicted twelve and seven stabilizing mutations, respectively. Nine mutations to consensus and six mutations to homologous prolines stabilized SABP2 to either urea or temperature. The location agnostic approach of random mutagenesis identified six stabilizing substitutions.
Table 1.
Single amino acid substitutions that stabilize SABP2 toward denaturation in urea solution or by heat.

|
Consensus = replacement by the most common residue found in homologs; Proline Theory = introduction of proline in locations found in a stable homolog; Random Mutagenesis = substitutions identified by error prone PCR followed by screening for increased stability; Computation = predicted stabilizing substitutions using both RosettaDesign and FoldX; B-factor = substitutions in highly flexible regions as identified by B-factor data in crystal structure; Literature = natural stabilizing variant in Manihot esculenta HNL.17
Enzymatic efficiency/kcat toward hydrolysis of p-nitrophenyl acetate as measured by release of p-nitrophenol at 404 nm, ε = 16,500 M−1 cm−1 in 10 vol% acetonitrile, pH 7.2 in 4.5 mM BES buffer.
irreversible unfolding calculated by comparison of half-life values at 60 °C, derived from measurement of residual esterase activity after 15 min incubation at 60 °C, assayed as above. Mutations that increased half-life at least 25% were counted as stabilizing.
unfolding in varied concentrations of urea measured after 24 h incubation. Incubated in 5mM BES at pH 7.2 with protein concentration of 0.1–0.3 mg/mL, urea concentrations from 0–6.2 M. Unfolding detected by loss of inherent tryptophan fluorescence at 329 nm measured with excitation at 278 nm. Wild type SABP2 showed a [urea]½ value of 2.2 M urea. Mutations that increased [urea]1/2 by at least 0.5 M were counted as stabilizing.
Mutations previously identified in reference 29.
Overall, stabilizing substitutions occurred throughout the protein as for the other α/β-hydrolase fold enzymes in the literature survey. Stabilizing substitutions in SABP2 occurred on the surface, at dimer interfaces, at buried sites and in both the lid (residues 111-184) and catalytic domains (residues 1-110 and 185-262). Substitutions that stabilized SABP2 to unfolding in urea correlated poorly with those that stabilized SABP2 to irreversible unfolding upon heating: R2 for 30 mutants was 0.08, see figure S2F. Some mutations even stabilized SABP2 by one metric, but destabilized it by the other. Three mutations predicted by computational approach and one predicted by flexibility resulted in insoluble protein, suggesting large destabilizing effects. All substitutions that yielded solubly expressed protein maintained esterase activity. The activity (kcat/Km) ranged between 39% (T228S) and 470% (K188R) of wild-type levels. The sections below provide further details on each of the three approaches.
Structure Based Design: Computationally identified substitutions
We used both RosettaDesign and FoldX to predict stabilizing substitutions. Both computational approaches model physical interactions of the atoms in the folded form, but take different approaches to include the effect of the unfolded protein. The FoldX algorithm includes energy terms that model physical interactions that contribute to folding such as bumping, hydrogen bonds, solvation and protein flexibility. Weighting these terms to fit the measured stabilities of >1000 proteins and variants ensures realistic predictions.34 The Rosetta Design algorithm instead uses a knowledge-based approach starting with known protein structures33 and weighting structures that occur more often as more favorable. Both algorithms previously predicted changes that increased protein stability.49
Predictions from these two algorithms often disagreed and correlated very poorly with each other (R2 = 0.03; Figure S2-E). One reason for the disagreement may be the high rate of false positive predictions. For example, of the sixteen substitutions predicted by RosettaDesign to stabilize cytosine deaminase, only three proved stabilizing and the degree of stabilization was small, an increase in apparent Tm of ~2 °C for each.50 To avoid these false positives, we searched for predictions where both algorithms agreed. First, we redesigned with RosettaDesign, then rechecked the 20 mutations predicted to be the most stabilizing with FoldX. FoldX agreed with RosettaDesign prediction in only eight cases. We tested these eight as well as five mutations that were predicted to be stabilizing only by RosettaDesign. Of these 13 mutations, the only four that increased the stability of SABP2 as measured by either heat-induced or urea-induced unfolding were L60R, S115A, C249A, and S251V. Both RosettaDesign and FoldX predicted that three of these four (not L60R) would be stabilizing. Of the four, the replacement of cysteine 249 with an alanine resulted in the greatest increase in thermal stability, increasing the half-life at 60 °C 3.6 fold from 3.6 minutes to 12.6 minutes. Of the four, the replacement of leucine 60 with an arginine resulted in the greatest increase in stability as measured by urea-induced unfolding, increasing the urea concentration needed to unfold half of the protein by 0.5 M, from 2.23 M to 2.74 M.
To further test the computational approach, we predicted the expected stabilization for all of the mutants, regardless of how they were identified. The predictions by RosettaDesign correlated poorly in the wrong direction with measured values for thermodynamic stability (R2 = 0.24, 43 mutants) and not at all with kinetic stability (R2 =0.02, 36 mutants), Figs, S2-B&D. Similarly, the predictions by FoldX did not correlate with measured values for thermodynamic or kinetic stability (R2 = 0.03 from 43 mutants and 0.06 from 36 mutants respectively, Figure S2-C&A). The poor match of computational approaches to measured thermodynamic stability may be due to the more complex unfolding of this protein, while the poor match to kinetic stability may be due to the role of aggregation in stability, see discussion section.
Structure Based Design: Substitutions in regions with high B-factor
Some protein regions are flexible because they lack strong interactions to nearby amino acids and may be the first regions to unfold. One approach to protein stabilization is to target flexible regions, as indicated by high temperature factors, or B-factors, in the x-ray crystal structures, for mutagenesis.10 The B-factors vary linearly with the mean square displacement of an atom around its equilibrium position as a result of thermal motion and positional disorder.10,51
Three loops (nine amino acids) in SABP2 had high B-factors, Figure 3. FoldX suggested fifteen possible substitutions at these nine positions. At five of these positions, at least one substitution stabilized SABP2. Mutating alanine 71 to proline, serine 141 to asparagine or proline, glutamate 144 to any of phenylalanine, tyrosine, or methionine, lysine 188 to arginine, and alanine 189 to either asparagine or serine stabilized SABP2 at least slightly as measured by one or both methods. Particularly stabilizing was A189S which increased half-life at 60° C by a factor of 3.4, while A71P, S141N, S141P, E144F, K188R, and A189N increased thermal stability by factors of 2.5–3. E144F, K188R, A89S, and A189S also increased tolerance to urea by 0.3–0.4 M.
Figure 3.

Location of stabilizing mutations in SABP2. Residues that increased stability when mutated are shown as spheres. The most flexible regions according to B-factors from x-ray crystal structure (yellow/orange/red regions; warmer colors indicate higher B-factor) are the three flexible loops that were targeted for mutagenesis and the C- and N-termini, which were not modified. The product (salicylic acid), as well as catalytic serine (81) and histidine (238), are shown as sticks. The entrance to the active site is from the right in this view. Chain A from pdb:1Y7I30
Bioinformatics Based Design: Mutation to consensus
We identified amino acids residues in SABP2 that differed from the most commonly occurring amino acid at the equivalent location in homologs (the consensus amino acid). A BLAST search for homologs of SABP2 identified 116 closely related sequences and identified ninety-one positions where there was at least a 70% consensus of a single amino acid, Figure S1. All but one of these highly conserved sites was in the catalytic domain. The catalytic domain is more highly conserved than the lid domain, so the bias of conserved sites for the catalytic domain is expected. The sequence of SABP2 differed from these ninety-one residues at 12 positions (all in the catalytic domain). These twelve residues were individually mutated to match the consensus sequence using site-directed mutagenesis.
Of the twelve tested, eight of these substitutions (G17A, S70P, S165P, F197Y, Q221M, A230V, E243S, and E244K) stabilized SABP2 as measured by either urea-induced unfolding or thermal unfolding. The most stabilizing to heat unfolding was the substitution of glutamine 221 with the consensus methionine, which increased the half-life at 60° C by a factor of 6.6 to 23 minutes. The most stabilizing substitution to unfolding in urea that was identified by consensus was E244K, which increased the tolerance to urea by 0.74 M urea.
Bioinformatics Based Design: Mutation to Homologous Proline
One rational approach to stabilize proteins is substitution of selected amino acid residues with proline. Substitutions with proline residues reduce the flexibility of the denatured form.52 This decrease in flexibility reduces the conformational entropy of the unfolded form, shifting the equilibrium toward the folded protein.
We reported details of these experiments elsewhere,29 but provide a brief summary here. To choose locations for the proline residues, we aligned the amino acid sequences of SABP2 and the orthologous hydroxynitrile lyase from Manihot esculenta (45% amino acid identity). This hydroxynitrile lyase is more than 10-fold more stable than SABP2 to heat denaturation: t½ = 2.7 h vs. 0.25 h at 50 °C.53 In six locations this stable ortholog contained a proline residue that was absent from SABP2. Each of these residues in SABP2 were mutated to proline and characterized. We also included one additional mutation to proline that was not previously reported. The serine at position 111 was mutated to proline to match that found in another homologue, PNAE from Rauvolfia serpentina (Accession: Q9SE93.1; 55% identity). Although the heat stability of this homolog has not been tested, this plant enzyme expressed efficiently in E. coli suggesting that it is a stable protein.54 We also copied the N114G substitution from a natural variant of M. esculenta hydroxynitrile lyase that is more stable.17
Four of the six proline substitutions from M. esculenta and the one from R. serpentine all increased stability. Substitution with proline at positions 46, 70, 111, 115, and 215 each stabilized SABP2 to heat-induced denaturation. The consensus approach described below also identified the S70P substitution. Conversions of residues 46, 70, 111, and 115 to proline increased the half-life of SABP2 when heated by a factor of 2–3. E215P showed the largest increase in stability as measured by urea-induced unfolding, requiring an extra 1.0 M urea to induce unfolding.
Location agnostic design: Random mutagenesis
Error prone PCR (epPCR) identified four stabilizing single amino acid substitutions. Error prone PCR of the SABP2 gene generated an average of 1.1 nucleotide changes as indicated by sequencing of plasmids from by 10 randomly chosen colonies. Colorimetric screening of the colonies with naphthyl acetate showed that about half of the colonies produced active esterases. Approximately 1000 of these active colonies were further screened for increased thermal stability. Active colonies were picked, grown in 96-deep-well plates and the crude cell lysates were assayed for esterase activity before and after heating to 50 °C for 15 min. Nineteen variants showed residual activity at least 3 standard deviations above that of wild type SABP2. The nine best mutants were Q162H/S173T, S19T, G4R/A230V (A230V was also identified by the consensus method), Y94F/N123K, H257Q, A100V, F150L/A189V/Q162H, A172V and L65F/G212C. Average mutation rate for the improved variants was 1.7 nucleotide changes per gene, slightly higher than the average mutation rate in the library (1.1). The three variants with half-lives at least double that of wild type enzyme, were selected for further characterization (F150L/A189V/Q162H, L65F/G212C, and A172V). These six substitutions were created individually. Substitutions F150L, G212C, and A230V were the substitutions that increased half-life by at least 50%. F150L and G212C also increased urea tolerance (0.8 M and 1.3 M more urea tolerated). Substitutions L65F stabilized SABP2 only to urea (1.5 M more urea tolerated).
Error prone PCR rarely creates multiple base changes in a codon, so only some amino acid substitutions are possible. Since G212C was one of the best hits found by epPCR, we tested additional substitutions at this location to see if better ones were missed. Screening 350 clones (>99% probability of testing each of the twenty possible amino acids at least once) identified five additional substitutions that increased the thermostability of SABP2, although none as much as G212C. The other stabilizing mutants were: G212A, G212I, G212L, G212M and G212V, Table S4. Four of these variants have higher esterase activity compared to wild type (9% to 216% higher kcat/KM), but G212M has 80% lower esterase activity.
Overall
All the methods found numerous substitutions that stabilized SABP2 to heat inactivation or urea unfolding or both. The best four substitutions to slow heat inactivation were S165P, Q221M, A230V, and C249A, which all increased the half-life at 60° at least 3.5-fold over wild type. A different set of four substitutions best prevented urea unfolding: F150L, G212C, E215P, and E244K, which increased the urea concentration needed to unfold half of the protein by at least 0.7 M over wild type. Most of these eight best substitutions were discovered by either the mutation to consensus or by random mutagenesis. The other strategies (computational design, mutation of flexible regions; addition of proline) each only identified 0 or 1 of these top hits.
Another group of eight substitutions stabilized SABP2 to both heat (at least 2.5 longer half-life) and urea (tolerate at least 0.2 M more urea). Three of these substitutions – G212C, Q221M, A230V – were also among the best above, but five other substitutions are new: L60R, E144F, K188R, A189N, A189S.
Some mutations were stabilizing by one stability measure, but destabilizing by another measure. Eight mutations that increased thermal stability decreased stability to urea-induced unfolding. These differences emphasize that these assays measure different aspects of enzyme stability.
Combining mutations
Among the limited screen of ~500 variants, we identified combinations of mutations that were more stable than any single mutant by screening random combinations of twenty-six stabilizing mutations at twenty-four locations. The best six combinations contained 7 to 19 substitutions and showed at least 40-fold longer half-lives than wild type at 60 °C, Table S6. By comparison, the best single substitution, Q221M, increased the half-life 6.6-fold. Each of the 26 single mutations in the screen was present in at least one of the six best combinations suggesting that they all contribute to stability. None of the 26 single substitutions were present in all of the best six combinations suggesting that no one substitution was especially stabilizing.
Discussion
All of the methods identified stabilizing mutations. This is consistent with the findings of others (Table S1). However, the degree of success, limitations, and amount of effort vary significantly between the methods.
The computational methods performed poorly. Neither computational approach correctly predicted stabilizing and destabilizing substitutions (R2 of 0.02–0.24, Figure S2). Potapov and colleagues55 also reported similar poor correlations (R2 of 0.25 for FoldX, and 0.07 for Rosetta) for a large set of >2000 substitutions. One possible reason is simplifications made by the computation methods. For example, the RosettaDesign web server does not include advanced options like backbone movement or an alternative energy function that is more tolerant of overlapping atoms, either of which increase the accuracy of Rosetta Design.56 However, initial computations with more advanced settings also gave poor correlations.
It is more likely that the complexities of this protein make the computations especially difficult. The computation methods do give accurate predictions for other proteins, so the problem may be α/β-hydrolase-fold enzymes. One challenge in all stability calculations is modeling the unfolded state. The computations assume that the protein unfolds completely,56 but the unfolding of SABP2 involves at least one intermediate. Since unfolding or aggregation of this intermediate destabilizes SABP2, omitting this intermediate from the computations likely leads to unreliable results. Multi-step unfolding is common for multi-domain proteins, so current computations methods are unlikely suitable for such cases.
Another reason for the poor performance of computational methods in predicting kinetic stability is that aggregation of the unfolded protein is not modeled by computation methods. For example, substitution A281E in CAL-B decreases its melting temperature from 58 to 51 °C suggesting a decrease in thermodynamic stability. However this substitution increases the half-life of this enzyme at 70 °C 22-fold demonstrating a reduced propensity to aggregate. This substitution makes a hydrophobic region of the enzyme less hydrophobic and less likely to aggregate even if the thermodynamic stability decreases.
Like previous efforts to stabilize α/β hydrolase fold proteins, we found that while all the implemented techniques found stabilizing mutations, there is a tradeoff between prerequisite knowledge (structure), amount of screening required, and whether special modeling methods are needed. The optimal approach would be a compromise between extensive screening required for random mutagenesis and the large amount of prerequisite structure and modeling expertise needed for computational approaches. Two such compromise methods are mutation to proline and mutation to consensus. Both use relatively simple criteria to maximize the positive results of a screen. The relatively simple rule of “if residue at position n differs from the strong consensus (e.g., 70%) at position n in a set of similar sequences, then mutate to consensus”, was simple and successful with 75% of mutations we tried. Creating the consensus sequence requires a number of tedious steps, so we created a simple program, Consensus Finder, to automate the process. Consensus Finder can be run on our server or the code can be downloaded at http://kazlab.um-n.edu. Several other web tools can also automatically predict stabilizing substitutions in proteins using the consensus sequence approach,57,58 but they require a three dimensional structure. Consensus finder is also easy to use by the non-expert, while allowing customization if desired.
Our results suggest that, with the exception of exhaustive site saturation mutagenesis, several methods are needed to identify most of the stabilizing mutations for a protein. Only seven locations were identified for mutation by more than one method (70, 94, 111, 115, 189, 230, 244), and only three of these were mutated to the same residue by both identifying methods (S70P, Y94F, A230V). The limited overlap can be partially attributed to different biases in the different methods. For example, the consensus approach favors substitutions in the interior of the protein since surface residues vary more during evolution. In contrast, we intentionally avoided residues near the active site when using computational methods to avoid enzyme inactivation. Thus, the computation approach favored residue near the surface. This limited overlap also suggests that this search for stabilizing substitutions was far from exhaustive and that there are likely many other stabilizing mutations possible. This work suggests that utilizing multiple strategies may be the best way to impart a larger degree of stabilization.
Combining stabilizing single substitutions yielded further increases in stability, but the combined effects were inconsistent and hard to predict. Most combinations showed at most, only minor gains and many were even less thermal stable than wild type. However, some combinations showed remarkable improvements not only over wild type, but also over any single substitution. There was no clear pattern of which combinations were highly stabilizing and which were not. All single mutations were able to combine with others to form highly stable combination mutants, yet none were found in all of the highly stable mutants. The effect of any particular mutation seems to be highly dependent on what other mutations are present. Since combining stabilizing mutations, identified in a wild type background one at a time can result in unpredictable results, a step-wise combination of identified stabilizing mutations inadvisable, as is making one combination mutant with all “good” mutations. A more successful strategy would be either a combinatorial approach like we have used or an iterative approach, where stabilized mutants are used as a starting point for repeated assessment of new mutations.
Supplementary Material
Acknowledgments
We thank the U.S. National Science Foundation (Grant CHE-1152804), U.S. National Institutes of Health (Grants 1R01GM102205-01 and 5T32 GM08347), and China National Natural Science Foundation (Grant 31470793; fellowship to J.H.) for funding, Jürgen Pleiss (Stuttgart U., Germany) and Brian Kuhlman (U. of North Carolina, Chapel Hill) for assistance with computations, and Daniel Klessig (Boyce Thompson Institute for Plant Research, Ithaca, NY) for the plasmid encoding SABP2.
Footnotes
Supporting Information Table of stabilizing mutations in α/β hydrolase fold enzymes reported in the literature. Primers used for mutagenesis and combinatorial library. Complete data for stabilizing point mutations in SABP2 from different methods in text and spreadsheet format. Mutations and characterization of combinatorial library hits. Logo of consensus sequence. Comparison of predicted versus measured effects of substitutions on stability of SABP2.
References
- 1.Estell DA, Graycar TP, Wells JA. Engineering an enzyme by site-directed mutagenesis to be resistant to chemical oxidation. J Biol Chem. 1985;260:6518–6521. [PubMed] [Google Scholar]
- 2.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Snow CD, Sorin EJ, Rhee YM, Pande VS. How well can simulation predict protein folding kinetics and thermodynamics? Annu Rev Biophys Biomol Struct. 2005;34:43–69. doi: 10.1146/annurev.biophys.34.040204.144447. [DOI] [PubMed] [Google Scholar]
- 4.Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–98. doi: 10.1016/0167-7799(90)90146-o. [DOI] [PubMed] [Google Scholar]
- 5.Holmquist M. Alpha beta-hydrolase fold enzymes structures, functions and mechanisms. Curr Protein Pept Sci. 2000;1:209–235. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 6.Jochens H, Hesseler M, Stiba K, Padhi SK, Kazlauskas RJ, Bornscheuer UT. Protein engineering of α/β-Hydrolase fold enzymes. ChemBioChem. 2011;12:1508–1517. doi: 10.1002/cbic.201000771. [DOI] [PubMed] [Google Scholar]
- 7.Kourist R, Jochens H, Bartsch S, Kuipers R, Padhi SK, Gall M, Böttcher D, Joosten HJ, Bornscheuer UT. The α/β–hydrolase fold 3DM database (ABHDB) as a tool for protein engineering. ChemBioChem. 2010;11:1635–1643. doi: 10.1002/cbic.201000213. [DOI] [PubMed] [Google Scholar]
- 8.Kazlauskas RJ, Bornscheuer UT. Finding better protein engineering strategies. Nat Chem Biol. 2009;5:526–529. doi: 10.1038/nchembio0809-526. [DOI] [PubMed] [Google Scholar]
- 9.Floor RJ, Wijma HJ, Colpa DI, Ramos-Silva A, Jekel PA, Szymański W, Feringa BL, Marrink SJ, Janssen DB. Computational library design for increasing haloalkane dehalogenase stability. ChemBioChem. 2014;15:1660–1672. doi: 10.1002/cbic.201402128. [DOI] [PubMed] [Google Scholar]
- 10.Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed. 2006;45:7745–7751. doi: 10.1002/anie.200602795. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad S, Kumar V, Ramanand KB, Rao NM. Probing protein stability and proteolytic resistance by loop scanning: A comprehensive mutational analysis. Protein Sci. 2012;21:433–446. doi: 10.1002/pro.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann M, Pasamontes L, Lassen SF, Wyss M. The consensus concept for thermostability engineering of proteins. Biochim Biophys Acta. 2000;1543:408–415. doi: 10.1016/s0167-4838(00)00238-7. [DOI] [PubMed] [Google Scholar]
- 13.Pantoliano MW, Whitlow M, Wood JF, Dodd SW, Hardman KD, Rollence ML, Bryan PN. Large increases in general stability for subtilisin BPN’ through incremental changes in the free energy of unfolding. Biochemistry. 1989;28:7205–7213. doi: 10.1021/bi00444a012. [DOI] [PubMed] [Google Scholar]
- 14.Khersonsky O, Rosenblat M, Toker L, Yacobson S, Hugenmatter A, Silman I, Sussman JL, Aviram M, Tawfik DS. Directed evolution of serum paraoxonase PON3 by family shuffling and ancestor/consensus mutagenesis, and its biochemical characterization. Biochemistry. 2009;48:6644–6654. doi: 10.1021/bi900583y. [DOI] [PubMed] [Google Scholar]
- 15.Echave J, Spielman SJ, Wilke CO. Causes of evolutionary rate variation among protein sites. Nat Rev Genet. 2016;17:109–121. doi: 10.1038/nrg.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Wu G, Feng S, Liu Z. Improved thermostability of esterase from Aspergillus fumigatus by site-directed mutagenesis. Enzyme Microb Technol. 2014;64–65:11–16. doi: 10.1016/j.enzmictec.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Yan G, Cheng S, Zhao G, Wu S, Liu Y, Sun W. A single residual replacement improves the folding and stability of recombinant cassava hydroxynitrile lyase in E. coli. Biotechnol Lett. 2003;25:1041–1047. doi: 10.1023/a:1024182228057. [DOI] [PubMed] [Google Scholar]
- 18.Patkar SA, Svendsen A, Kirk O, Clausen IG, Borch K. Effect of mutation in non-consensus sequence Thr-X-Ser-X-Gly of Candida antarctica lipase B on lipase specificity, specific activity and thermostability. J Mol Catal B Enzym. 1997;3:51–54. [Google Scholar]
- 19.Okrob D, Metzner J, Wiechert W, Gruber K, Pohl M. Tailoring a stabilized variant of hydroxynitrile lyase from Arabidopsis thaliana. ChemBioChem. 2012;13:797–802. doi: 10.1002/cbic.201100619. [DOI] [PubMed] [Google Scholar]
- 20.Jochens H, Aerts D, Bornscheuer UT. Thermostabilization of an esterase by alignment-guided focussed directed evolution. Protein Eng Des Sel. 2010;23:903–909. doi: 10.1093/protein/gzq071. [DOI] [PubMed] [Google Scholar]
- 21.Gray KA, Richardson TH, Kretz K, Short JM, Bartnek F, Knowles R, Kan L, Swanson PE, Robertson DE. Rapid evolution of reversible denaturation and elevated melting temperature in a microbial haloalkane dehalogenase. Adv Synth Catal. 2001;343:607–617. [Google Scholar]
- 22.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. Genome Res. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 23.Kamal MZ, Ahmad S, Molugu TR, Vijayalakshmi A, Deshmukh MV, Sankaranarayanan R, Rao NM. In vitro evolved non-aggregating and thermostable lipase: Structural and thermodynamic investigation. J Mol Biol. 2011;413:726–741. doi: 10.1016/j.jmb.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Pikkemaat MG, Linssen ABM, Berendsen HJC, Janssen DB. Molecular dynamics simulations as a tool for improving protein stability. Protein Eng. 2002;15:185–192. doi: 10.1093/protein/15.3.185. [DOI] [PubMed] [Google Scholar]
- 25.Han Z, Han S, Zheng S, Lin Y. Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface. Appl Microbiol Biotechnol. 2009;85:117–126. doi: 10.1007/s00253-009-2067-8. [DOI] [PubMed] [Google Scholar]
- 26.Yu XW, Tan NJ, Xiao R, Xu Y. Engineering a disulfide bond in the lid hinge region of Rhizopus chinensis lipase: Increased thermostability and altered acyl chain length specificity. PLoS ONE. 2012;7:e46388. doi: 10.1371/journal.pone.0046388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi S, Takeuchi K, Mase T, Oikawa K, McMullen T, Derewenda U, McElhaney RN, Kay CM, Derewenda ZS. The consequences of engineering an extra disulfide bond in the Penicillium camembertii mono-and diglyceride specific lipase. Protein Eng. 1996;9:789–795. doi: 10.1093/protein/9.9.789. [DOI] [PubMed] [Google Scholar]
- 28.De Marco A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb Cell Fact. 2009;8:26. doi: 10.1186/1475-2859-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Jones BJ, Kazlauskas RJ. Stabilization of an α/β-hydrolase by introducing proline residues: Salicylic acid binding protein 2 from tobacco. Biochemistry. 2015;54:4330–4341. doi: 10.1021/acs.biochem.5b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forouhar F, Yang Y, Kumar D, Chen Y, Fridman E, Park SW, Chiang Y, Acton TB, Montelione GT, Pichersky E, Klessig DF, Tong L. Structural and biochemical studies identify tobacco SABP2 as a methyl salicylate esterase and implicate it in plant innate immunity. Proc Natl Acad Sci USA. 2005;102:1773–1778. doi: 10.1073/pnas.0409227102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padhi SK, Fujii R, Legatt GA, Fossum SL, Berchtold R, Kazlauskas RJ. Switching from an esterase to a hydroxynitrile lyase mechanism requires only two amino acid substitutions. Chem Biol. 2010;17:863–871. doi: 10.1016/j.chembiol.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Kuhlman B. RosettaDesign server for protein design. Nucleic Acids Res. 2006;34:W235–W238. doi: 10.1093/nar/gkl163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaufmann KW, Lemmon GH, DeLuca SL, Sheehan JH, Meiler J. Practically useful: What the Rosetta protein modeling suite can do for you. Biochemistry. 2010;49:2987–2998. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 35.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: An online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 40.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reyes-Duarte D, Ferrer M, García-Arellano H. Functional-based screening methods for lipases, esterases, and phospholipases in metagenomic libraries. In: Sandoval G, editor. Lipases and Phospholipases. Humana Press; 2012. pp. 101–113. [DOI] [PubMed] [Google Scholar]
- 42.Kemmer G, Keller S. Nonlinear least-squares data fitting in Excel spreadsheets. Nat Protoc. 2010;5:267–281. doi: 10.1038/nprot.2009.182. [DOI] [PubMed] [Google Scholar]
- 43.Ness JE, Kim S, Gottman A, Pak R, Krebber A, Borchert TV, Govindarajan S, Mundorff EC, Minshull J. Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently. Nat Biotechnol. 2002;20:1251–1255. doi: 10.1038/nbt754. [DOI] [PubMed] [Google Scholar]
- 44.Young L, Dong Q. Two–step total gene synthesis method. Nucleic Acids Res. 2004;32:e59–e59. doi: 10.1093/nar/gnh058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pace CN. Conformational stability of globular proteins. Trends Biochem Sci. 1990;15:14–17. doi: 10.1016/0968-0004(90)90124-t. [DOI] [PubMed] [Google Scholar]
- 46.Pfeil W. Protein stability and folding: a collection of thermodynamic data. Berl Heidelb N Y 1998 [Google Scholar]
- 47.Scholtz JM, Grimsley GR, Pace CN. In: Chapter 23 Solvent denaturation of proteins and interpretations of the m value. Enzymology B-M, editor. Academic Press; 2009. pp. 549–565. [DOI] [PubMed] [Google Scholar]
- 48.Kumar S, Tsai CJ, Nussinov R. Thermodynamic differences among homologous thermophilic and mesophilic proteins. Biochemistry. 2001;40:14152–14165. doi: 10.1021/bi0106383. [DOI] [PubMed] [Google Scholar]
- 49.Dantas G, Corrent C, Reichow SL, Havranek JJ, Eletr ZM, Isern NG, Kuhlman B, Varani G, Merritt EA, Baker D. High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J Mol Biol. 2007;366:1209–1221. doi: 10.1016/j.jmb.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parthasarathy S, Murthy MRN. Protein thermal stability: Insights from atomic displacement parameters (B values) Protein Eng. 2000;13:9–13. doi: 10.1093/protein/13.1.9. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe K, Suzuki Y. Protein thermostabilization by proline substitutions. J Mol Catal B Enzym. 1998;4:167–180. [Google Scholar]
- 53.Guterl JK, Andexer JN, Sehl T, von Langermann J, Frindi-Wosch I, Rosenkranz T, Fitter J, Gruber K, Kragl U, Eggert T, Pohl M. Uneven twins: Comparison of two enantiocomplementary hydroxynitrile lyases with α/β-hydrolase fold. J Biotechnol. 2009;141:166–173. doi: 10.1016/j.jbiotec.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 54.Dogru E, Warzecha H, Seibel F, Haebel S, Lottspeich F, Stöckigt J. The gene encoding polyneuridine aldehyde esterase of monoterpenoid indole alkaloid biosynthesis in plants is an ortholog of theα/β hydrolase super family. Eur J Biochem. 2000;267:1397–1406. doi: 10.1046/j.1432-1327.2000.01136.x. [DOI] [PubMed] [Google Scholar]
- 55.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 56.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins Struct Funct Bioinforma. 2011;79:830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldenzweig A, Goldsmith M, Hill SE, Gertman O, Laurino P, Ashani Y, Dym O, Unger T, Albeck S, Prilusky J, Lieberman RL. Automated structucture-and sequence-based design of proteins for high bacterial expression and stability. Mol Cell. 2016;63:337–346. doi: 10.1016/j.molcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bendl J, Stourac J, Sebestova E, Vavra O, Musil M, Brezovsky J, Damborsky J. HotSpot Wizzard 2.0: Automated design of site-specific mutations and smart libraries in protein engineering. Nucleic Acids Res. 2016;44:W479–W487. doi: 10.1093/nar/gkw416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.