

Abstract
Significance: The molecular events that promote the development of pulmonary hypertension (PH) are complex and incompletely understood. The complex interplay between the pulmonary vasculature and its immediate microenvironment involving cells of immune system (i.e., macrophages) promotes a persistent inflammatory state, pathological angiogenesis, and fibrosis that are driven by metabolic reprogramming of mesenchymal and immune cells.
Recent Advancements: Consistent with previous findings in the field of cancer metabolism, increased glycolytic rates, incomplete glucose and glutamine oxidation to support anabolism and anaplerosis, altered lipid synthesis/oxidation ratios, increased one-carbon metabolism, and activation of the pentose phosphate pathway to support nucleoside synthesis are but some of the key metabolic signatures of vascular cells in PH. In addition, metabolic reprogramming of macrophages is observed in PH and is characterized by distinct features, such as the induction of specific activation or polarization states that enable their participation in the vascular remodeling process.
Critical Issues: Accumulation of reducing equivalents, such as NAD(P)H in PH cells, also contributes to their altered phenotype both directly and indirectly by regulating the activity of the transcriptional co-repressor C-terminal-binding protein 1 to control the proliferative/inflammatory gene expression in resident and immune cells. Further, similar to the role of anomalous metabolism in mitochondria in cancer, in PH short-term hypoxia-dependent and long-term hypoxia-independent alterations of mitochondrial activity, in the absence of genetic mutation of key mitochondrial enzymes, have been observed and explored as potential therapeutic targets.
Future Directions: For the foreseeable future, short- and long-term metabolic reprogramming will become a candidate druggable target in the treatment of PH. Antioxid. Redox Signal. 28, 230–250.
Keywords: : pulmonary hypertension, metabolism, aerobic glycolysis, mitochondria, hypoxia
Introduction
Pulmonary hypertension (PH) is a rapidly progressive, often fatal pulmonary vascular condition that affects the lungs and heart of >25 million individuals globally (57). Five distinct categories of PH have been identified on the basis of common clinical parameters, potential etiologic mechanisms, as well as pathological, pathophysiological, and therapeutic characteristics (166, 192). Despite PH class-distinct phenotypes, all categories are characterized by a complex panvasculopathy that recapitulates features similar to those observed in cancer (37, 78, 210, 211), including excessive proliferation, resistance to apoptosis, inflammation, and fibrosis mediated by a dramatic remodeling of the extracellular matrix. Borrowing from recent advancements in cancer (47, 122, 224) and immune research (119, 127, 128, 150, 151, 221), it has been suggested that PH phenotypes might be, in part, explained by a substantial metabolic/pro-inflammatory reprogramming of arterial cells and surrounding stromal cells, including fibroblasts and macrophages. Of note, in cancer research, it is now accepted that most oncogenes and mutated tumor suppressor genes play a primary role in metabolic regulation (106). Conversely, aberrant metabolism can promote epigenetic control of oncogenic reprogramming even in the absence of specific mutation, in response to hypoxic/cytokine-triggered stimuli, promoting a pro-inflammatory, hyperproliferative, and pro-fibrotic phenotype in cancer and immunity (80, 122, 150, 151). Studies on the metabolic reprogramming of activated immune cells taught us that such a complex phenotype can be observed in the absence of specific genetic mutations (150). This observation prompted cardiovascular investigators to wonder whether similar molecular underpinnings could contribute toward explaining PH etiology as well, fueling the debate around the metabolic theory of PH (56, 161, 201).
Even though Hanahan and Weinberg have included metabolic reprogramming as a hallmark of cancer only in recent years (78), seminal pioneering observations by Otto Warburg had pointed at the fact that hyperproliferating cells mostly rely on glycolysis to sustain their catabolic needs, even in the presence of oxygen (37, 62, 161, 210, 216, 233). In cancer cells, incomplete glucose oxidation through glycolysis is believed to provide the building blocks for anabolic reactions to sustain excess proliferation (216) by supplying the energy and substrates that are necessary to fuel DNA replication and gene expression, macromolecule biosynthesis (e.g., proteins and lipids for organelles and membrane lipid bilayers), ion gradients' homeostasis, and maintenance of cellular structure. Borrowing from cancer research, immunologists have noted similar metabolic phenotypes in highly proliferating immune cells [macrophages, lymphocytes (23, 149, 151, 204, 221)], observing that incomplete mitochondrial metabolism favored the accumulation of carboxylic acid intermediates—such as succinate—which were eventually found to participate in the stabilization of transcriptional factors at the crossroads between hypoxic and inflammatory responses such as hypoxia-inducible factor 1α (HIF1α) (85, 156). With the advent of omics technologies, in particular metabolomics, cardiovascular investigators had access to the tools that were necessary to confirm that similar changes occur in PH smooth muscle cells (SMCs), endothelial cells (ECs), and fibroblasts (Fibs) (60, 62, 68, 164, 233). Positron emission tomography (PET) imaging scans of 18F-fluorodeoxyglucose (18FDG) metabolism in vivo has further demonstrated increased glucose uptake and metabolism in PH patients as well as in the monocrotaline rat model of PH (121, 233). In line with 18FDG uptake experiments, gene-expression studies in pulmonary arterial fibroblasts, isolated from idiopathic pulmonary arterial hypertension (iPAH) patients, further provide supportive evidence about the role of glycolytic metabolic reprogramming in mediating the proliferative and pro-inflammatory phenotype observed in patients with pulmonary vascular pathologies (233). Overall, 18FDG uptake studies also showed that a similar metabolic phenotype is observed in perivascular mononuclear cells, which accumulate in the adventitial perivascular regions. In vivo studies in rats treated with monocrotaline revealed a correlation between remodeling of the pulmonary vasculature and lung 18FDG uptake. In this review, we will describe the main results from some of the pilot studies focusing on metabolic reprogramming in PH and lay the foundation for the concept of “hallmarks of PH” by focusing on the increasingly accepted “metabolic theory” of PH and its interplay with other hallmarks (Fig. 1) (161, 176, 236).
FIG. 1.

Borrowing from and noticing the similarities with the founding concepts of the hallmarks of cancer (78), here we propose and summarize the hallmarks of PH. ECM, extracellular matrix; HIF, hypoxia-inducible factor; PH, pulmonary hypertension.
Warburg-Like Glycolytic Reprogramming and Enzyme Isoform Switching in PH
In analogy to cancer, PH is characterized by a Warburg-like glycolytic reprogramming, excessive proliferation, and apoptotic resistance of ECs, SMCs, and Fibs, a phenotype that is also observed in the PH vessel wall (21, 37, 78). In PH, similar to cancer, glycolytic reprogramming provides the biological building blocks to support protein synthesis and the production of new cells. To provide for this increased biosynthetic demand, PH cells, similar to cancer cells, exhibit a higher uptake of nutrients such as glucose (201, 203). In addition, the metabolic pathways of PH cells are altered to allow production of macromolecules and withstand oxidative stress associated with inflammation and high rates of production (80, 201). Cytokines, hypoxia, and other mediators all contribute to the glycolytic reprogramming observed in cancer and PH, and they are mostly part of an interplay of signaling cascades with multiple feedback loops. For example, although hypoxic signaling is known to promote angiogenesis through HIF-mediated control of vascular endothelial growth factors (VEGF) (66), it has been recently observed that ischemic (32, 131, 196, 209) or hemorrhagic (39, 40, 193) hypoxia triggers succinate accumulation, which, in turn, can stabilize HIF and further promote VEGF signaling (134). Of note, stabilization of HIF signaling and regulation of the oxygen-sensitive subunits of HIF (191) may contribute to the observed increase in transcriptional expression of glycolytic enzymes that is observed in cancer cells and PH (120, 177, 188). In analogy to cancer, glycolytic reprogramming in fibroblasts from pulmonary hypertensive arteries (bovine and human), hereafter termed PH-Fibs, is, in part, explained by the expression of specific isoforms of glycolytic enzymes that promote faster fluxes through glycolysis, such as glucose transporter 1 (GLUT1), phosphofructokinase B3 (PFKB3), pyruvate dehydrogenase (PDH) kinase 1, and lactate dehydrogenase A (LDHA) (37, 108, 161, 164, 189).
Overexpression of glycolytic enzymes with moonlighting functions (58, 82) may also contribute to the increased resistance to apoptosis of cells, including PH-Fibs from the hypertensive vasculature. For example, it has been shown that the enzyme that catalyzes glucose phosphorylation at the initial step of glycolysis, hexokinase can bind to the voltage-dependent anion channel (VDAC) in the mitochondrial membrane, thus preventing cytochrome c release and apoptosis (162). Decreased expression of isoforms that favor further shuttling of triose phosphates toward the Krebs cycle (e.g., increased PKM2/PKM1 ratios) is an alternative mechanism explaining the glycolytic reprogramming of PH-Fibs (167). Isoforms 1 and 2 of pyruvate kinase M (PKM)—the enzyme responsible for the generation of pyruvate and ATP in late glycolysis—are characterized by differential activity, with PKM1 often being expressed in terminally differentiated tissues and showing constitutively high basal activity. Interest around the less active PKM2 isoform has grown on the appreciation of its preferential expression in proliferating cells or tissues with anabolic functions (cancer, embryonic cells) and cells with high intrinsic self-renewal properties. Notably, PKM2 is allosterically regulated between a high catalytically active tetramer and a low-activity dimer (48), with the latter being more common in highly proliferative cells such as PH-Fibs (Zhang et al., under review). Interconversion of the isoforms and allosteric regulation of PKM2 is favored by post-translational modifications [e.g., phosphorylation (33, 113) or oxidation at cysteine 358 (3)], or small-molecule metabolites [e.g., fructose 1,6-bisphosphate or serine (7)]. Activation of PKM2 can also be promoted through pharmacological intervention (e.g., through TEPP-46), a phenomenon that favors increased PK activity and, thus, increased pyruvate shuttling to the Krebs cycle. Slower phosphoenolpyruvate catabolism favors anabolic reactions and metabolic switches to alternative routes branching from glycolysis—such as serine synthesis (see Glucose Metabolism Beyond Glycolysis and the PPP)—instead of fueling further oxidation of carbon atoms through the Krebs cycle. PKM2 transcription is regulated, at least in part, by HIF1, with which PKM2 directly interacts to promote the transactivation of genes with HIF response elements in their promoters (113) and recruitment of p300 (114), a feed-forward mechanism to escalate the efficiency of glycolytic fluxes. Dimeric PKM2 can translocate into the nucleus, where it can phosphorylate nuclear proteins such as STAT3 and β-catenin (71, 93, 203). We recently found that PMK2/PKM1 ratio—mostly due to downregulation of PKM1 levels—is increased in PH-Fibs from calves and iPAH humans (232). In preliminary experiments, we found that the miR124/polypyrimidine-tract binding protein 1 (PTBP1) axis was responsible for the selective splicing of PKM2/PKM1 isoforms, and that genetic manipulations of miR124 (miR-124 mimic) or PTBP1 (siPTBP1) normalized the PKM splicing ratios in PH-Fibs, inhibiting cell proliferation and normalizing metabolic phenotypes (232).
Oxidation of pyruvate—a PKM byproduct—can be inhibited by phosphorylation of PDH in mitochondria by pyruvate dehydrogenase kinase (PDK), an enzyme that is directly regulated by HIF1α under hypoxic conditions (93). PDK can become independent from oxygen availability in cancers undergoing constitutive HIF activation or PDK mutations (81). PH-Fibs from calves exposed to chronic hypoxia and iPAH humans show similar metabolic adaptations (164). Pharmacological inhibition of PDK with dichloracetate increased glucose oxidation in the mitochondria and attenuated PH and vascular remodeling in the rat monocrotaline model (124). The same treatment also decreased the expression of the glucose transporter (Glut1), which is under HIF control (26) and usually upregulated in cells exhibiting high glycolysis. Dichloroacetate treatment also resulted in a decrease of 18FDG PET signals, reduced peripheral vascular muscularization, and prevented the accumulation of inflammatory cells (121, 124, 233). Overall, studies in the literature suggest that pharmacological correction or epigenetic regulation of the Warburg-like glycolytic phenotype is a viable strategy to ameliorate PH.
Glucose-6-Phosphate Dehydrogenase and the Pentose Phosphate Pathway in PH
In like fashion to cancer cells, increased glucose uptake can also fuel pathways branching from glycolysis such as the pentose phosphate pathway (PPP). Activation of the oxidative branch of the PPP in PH cells generates reducing equivalents (NADPH) to preserve the redox poise [NADPH is a necessary cofactor to restore oxidized glutathione back to its reduced form and is a key substrate for the catalysis of other antioxidant enzymes such as catalase (64, 96)]. Conversely, NADPH serves as a substrate for NADPH oxidase (NOX) to generate reactive oxygen species (ROS), a critical step in immune cell activation (see Reducing Equivalents in PH). Owing to the role that NADPH plays in anabolic reactions (e.g., de novo synthesis of fatty acids), a sustained activation of the oxidative branch of the PPP serves a key purpose in membrane homeostasis in highly proliferating cancer cells. In addition, as heoxoses are processed through the PPP (also known as the hexose monophosphate shunt), pentose phosphate compounds are generated, such as ribose phosphate, a sugar phosphate moiety for the de novo synthesis and salvage of nucleosides (the fundamental units of RNA and DNA). Of note, nucleosides and, in particular, purine homeostasis may play a key role in PH, not just from an energetic and replicative standpoint but also for extracellular signaling to promote angiogenesis and tissue oxygenation (102, 110, 133, 148, 195). NADPH plays a key role at the crossroads of survival and proliferation (88). In cancer cells, the interplay between AMP-activated protein kinase (AMPK) and NADPH levels is necessary in response to glucose starvation through a mechanism that involves increases in fatty acid oxidation through inhibitory phosphorylation of acetyl-CoA carboxylase 1 and 2 (88). In mitochondria, nicotinamide nucleotide transdehydrogenase (NNT) can transfer a proton from NADH to NADP+, therefore compensating for electron transport chain uncoupling resulting from the exposure to hypoxia (70). In doing so, NNT counteracts the accumulation of excess of NADH-reducing equivalents resulting from increased glycolysis and impaired electron transport activity at the complex I level of mitochondria observed in PH Fibs (see Mitochondrial Dysfunction and Superoxide Production).
Serving as a cofactor for nitric oxide synthase, NADPH participates in nitric oxide homeostasis and, thus, plays a role in the initial phase of PH development for vasoconstriction of pulmonary arteries (PAs) (105). Consistent with this consideration, in the African, Middle East, and Asian populations, a deficiency in the activity of the rate-limiting enzyme of the PPP, glucose 6-phosphate dehydrogenase (G6PD)—the most common enzymopathy in humans—is associated with increased susceptibility to red blood cell hemolysis (213), altered lipid metabolism and recycling of oxidized lipids, endothelial oxidative stress, and decreased NO bioavailability (65). G6PD is a major supplier of cellular NADPH (60% by G6PD +40% by isocitrate dehydrogenase [IDH]) for: anabolic reactions and NOXs-derived superoxide production (75). Alternatively, nicotinamide phosphoribosyltransferase (NAMPT) can transfer a phosphate group from phosphoribosylpyrophosphate to nicotinamide, participating in the synthesis of nicotiamide nucleosides and, thus, affecting the homeostasis of reducing cofactors. Recently, NAMPT has been shown as a candidate therapeutic target against pulmonary vascular remodeling in PH (27). Excess NADPH generation also contributes to pathogenic “reductive stress” in the cardiovascular system (90).
As previously discussed, numerous studies report increased glycolysis, glucose flux through the PPP, and the activity of NADPH-producing IDH-1 and -2 in PAs and lungs of idiopathic- and heritable-PAH patients (60, 230). Expression of enzymes in the glycolytic pathway (e.g., hexokinase-1, 6-phosphofructokinase, and pyruvate kinase M2) and the PPP (e.g., 6-phosphogluconolactonase, transaldolase-1, and transketolase) is upregulated in pulmonary artery smooth muscle cells (PASMCs) of PAH patients with BMPR2 exon1–8 deletion (230). Additional studies demonstrate that the expression and activity of G6PD is increased in: (i) endothelin-1-treated PASMCs from PAH patients with BMPR2 exon1–8 deletion (230); (ii) fibroblasts from idiopathic PAH patients and calves with severe hypoxic PH (108); (iii) hypoxic cultured rat PASMCs (28) and PASMCs of lambs exposed to increased pulmonary blood flow (13); (iv) ECs of monocrotaline-treated mice (199); and (v) lungs of hypoxia-induced and monocrotaline-induced PH rat models (28, 199). It has also been reported that G6PD and PPP activity is increased before PH develops (28, 171). This suggests that an increase of G6PD activity has a temporal relationship with PH.
G6PD deficiency is common in humans, and 400-point mutations have been found in this enzyme in different ethnic groups around the world. Epidemiological studies suggest that individuals who harbor a Mediterranean-type nonsynonymous mutation (single-nucleotide polymorphism in exon 6: dbSNP rs5030868) have 80% less G6PD activity as compared with normal individuals and are less likely to have vascular diseases (73), including sickle cell anemia-associated PH (38). Moreover, a Phase III clinical trial (NCT00581087) reports that dehydroepiandrosterone (DHEA), which potently inhibits G6PD activity among other actions in the cell (118, 185, 186), improves 6 min walk test and pulmonary hemodynamics in patients with COPD-associated PH (53). Conversely, low levels of DHEA-sulfate are associated with severity of PAH (218).
Gupte and Oka originally reported that G6PD and G6PD-derived NADPH mediate hypoxic pulmonary vasoconstriction (76) and hypoxia-induced contraction of bovine and rat PA (31, 73). More recently, it has been reported that: (i) hypoxia-induced contraction of isolated PAs from G6PD-deficient, as compared with wild-type, mice is blunted (74); and (ii) 6-aminonicotinamide, a competitive inhibitor of G6PD, prevents hypoxia-induced downregulation of contractile protein expression in isolated bovine PAs (31) and reduces expression of pro-inflammatory TNFα in hypoxic PAs and PASMCs (101). In addition, studies have shown that DHEA reduces pulmonary remodeling, pulmonary vascular resistance in hypoxia-induced PH and PAH rats, and heart failure in PAH rats (2, 15, 77, 144, 175). Collectively, these results suggest that G6PD and G6PD-dependent pathways potentially play a significant role in development of PH. However, whether upregulated G6PD contributes predominately toward increasing PA constriction or also to remodeling of PA in PH is unclear.
The PPP produces ribose sugar that is essential for de novo synthesis of RNA and DNA. In addition, G6PD-derived NADPH is essential for fatty acid metabolism and lipid synthesis that are required for the formation of membranes in proliferating cells, controlling the activities of cell cycle enzymes and of caspases that trigger apoptotic cell death. Therefore, NADPH has been considered to play a key role in stimulating proliferation and inhibiting apoptosis of cells (17). Ectopic expression of G6PD increases rat PASMC proliferation (28) and contributes to HIF1α-induced EC growth (105). Hyper-activation of G6PD in CD133+ progenitor cells promotes their growth (29). These cells play a critical role in tumorigenesis and potentially participate in the PA remodeling process in PAH (8). Consistently, it has been observed that G6PD-depleted embryonic stem cells proliferate at a reduced rate (61, 63). On the other hand, excess glucose-6-phosphate dehydrogenase activity and NADPH availability can promote, under certain conditions, apoptosis (143). Indeed, excess NADPH can promote the phosphorylation and thus the activation of the pro-aptoptotic caspase 2 by calcium/calmodulin-dependent kinase II (143).
Several studies also suggest that glucose flux is diverted into the PPP due to inhibition of phosphofructose kinase-1 in cancer cells (231) and increased expression of the pyruvate kinase isozyme M2 (165), and activation of the PPP supports K ras-induced, anchorage-independent cellular growth (223). In ongoing studies, Gupte et al. found that inhibition of G6PD with DHEA reduces hypoxia-induced phosphorylation of PDK in PASMCs (Fig. 2). Recently, they also found that genes critical for endothelial and SMC growth and function were differentially regulated in vascular tissues of a G6PD-deficient mouse as compared with wild-types [GEO Submission (GSE80972) NCBI tracking system #17863684 (30)]. Gene expression that is changed >1.5 Log2_fold in vascular tissue and that is required for the regulation of PA function and structure is demonstrated (Fig. 3). In PH, PASMC and EC proliferation is accompanied by decreased expression of pro-apoptotic genes (18). Therefore, collectively, these findings support the idea that increased G6PD expression and activity stimulated either by endothelin-1 and/or by hypoxia inhibits apoptosis and promotes proliferation of various cells (PASMC, ECs, and fibroblasts) and stem cells in lungs. These cells collectively mediate neomuscularization of arterioles and formation of complex cellular and fibrotic neointimal and plexiform lesions in distal PAs that, respectively, contribute to the progressive development of HPH and PAH.
FIG. 2.

Phosphorylation of PDH and gene expression in vascular tissue is regulated by G6PD. Phosphorylation of PDH subunits (E1–E3) was determined by In Cell Western blot assay kit (PhosphoPDH In-Cell ELISA Kit [IR]; Abcam, MA) in PASMCs cultured in normoxia (21% O2) and hypoxia (3% O2) treated with or without DHEA (100 μM). (A) As shown, phosphorylation of PDH, which inhibits acetyl CoA formation from pyruvate, was increased in hypoxic PASMCs and DHEA decreased hypoxia-induced phosphorylation of PDH. (B) Expression of genes critical in endothelial cells and PASMCs growth and function is modulated in vascular tissue of G6PD-deficient as compared with wild-type mice. *p < 0.05 T-test. DHEA, dehydroepiandrosterone; G6PD, glucose 6-phosphate dehydrogenase; PASMCs, pulmonary artery smooth muscle cells; PDH, pyruvate dehydrogenase.
FIG. 3.

G6PD inhibition increased phosphorylation of inositol triphosphate receptor and decreased angiotensin II-induced bovine PA contractions. (A) A representative Western blot of four different experiments demonstrates total and phosphorylated inositol triphosphate receptor (IP3R) at Ser1755, a PKG phosphorylation site in consensus GRRESLTS sequence (67), in angiotensin II (Ang II; 10 μM)-treated bovine PA in absence and presence of DHEA (100 μM) in hypoxia. (B) Summary data demonstrate that DHEA increased phospho-to-total IP3R ratio. (C) DHEA did not decrease Ang II-induced IP3 levels in bovine PAs. (D) Bovine PAs were precontracted with Ang II in hypoxia (0% O2). *p < 0.05 vs control T-test; #p < 0.05 vs Ang II T-test. DHEA treatment blocked Ang II-induced peak contraction. PA, pulmonary artery; PKG, protein kinase G.
G6PD role in redox homeostasis intercepts several signaling cascades, among which the cGMP-protein kinase G1 (PKG1)-signaling axis seems to provide a potential therapeutic window in PH (31, 101). In this regard, it was originally reported that G6PD inhibition with 6-aminonicotinamide or DHEA via TxR redox signaling oxidizes Cys42 residue and robustly increases PKG activity without significantly elevating intracellular total cGMP levels in isolated bovine PAs (31, 101). G6PD inhibition-elicited PKG1α-dependent signaling mediates: (i) expression of contractile proteins and (ii) reduction of pro-inflammatory TNFα in hypoxic PAs (31, 101). Further, it was recently proposed that G6PD inhibition-elicited PKG1α-dependent signaling blocks G-protein-coupled IP3 receptor signaling, supporting the idea that G6PD inhibition increases phosphorylation of IP3R, without decreasing Ang II-elicited IP3 levels, and simultaneously reduces Ang II-induced contraction of PAs in hypoxia (74) (Fig. 3). Therefore, it can be suggested that G6PD inhibition will increase PKG activity through a novel cGMP-independent mechanism that will potentially suppress nonresolving inflammation in lungs, induce apoptosis of PASMCs and other cell types, and reduce IP3-mediated Ca2+ release to prevent and reverse hypoxia-induced PH and PAH.
Glucose Metabolism Beyond Glycolysis and the PPP: From One-Carbon Metabolism to Epigenetic Regulation in PH
Other than the PPP, incompletely oxidized carbon backbones from glucose can be fueled into serine synthesis and one-carbon metabolism. These pathways are increasingly attracting a great deal of interest for their role in providing NADPH and carbon backbone substrates to fuel the biosynthesis of antioxidant compounds and nucleosides, which are necessary to sustain active proliferation. For example, glycine, cysteine, and deamination of glutamine to glutamate are all reactions occurring downstream to serine biosynthesis and provide the necessary building blocks for the tripeptide glutathione (25, 44, 100, 112, 115, 116, 159, 205, 229). Notably, fluxes through one-carbon metabolism and the serine synthesis pathway are regulated in cancer by isoform switching of late glycolytic enzymes (PKM2), for which serine is as an allosteric modulator (25). One-carbon metabolism (51) reactions also participate in S-adenosylmethionine/S-adenosylhomocysteine metabolism, which represents essential substrates for epigenetic histone modifications such as methylation events (125). Similarly, acetyl-CoA can be generated from carbon backbones derived from the incomplete oxidation of glucose, fatty acids, or pathways downstream to the reductive carboxylation of glutamine, which, in turn, promotes acetylation events (126). Epigenetic control in PH may depend on similar mechanisms (84, 92, 180). Notably, treatment with histone deacetylase inhibitors valproic acid and suberoylanilide hydroxamic acid seems to effectively rescue the metabolic reprogramming of PH-Fibs from chronically hypoxic calves or patients with iPAH (232, 234) (Zhang et al., under review). Epigenetic control might contribute toward explaining the long-term hypoxic-like phenotypes observed in PH-Fibs from chronically hypoxic calves or patients with iPAH cultured under normoxia, suggesting more effective potential therapeutic avenues to chronic PH (22, 234).
Reducing Equivalents in PH
Increased glycolytic fluxes in PH, similar to cancer or hypoxic cells, have been shown to promote excess lactate accumulation in the cellular and extracellular compartment. In cancer cells, excess lactate has been shown to stabilize NDRG3 protein, which binds to c-Raf and mediates hypoxic cell growth through the c-Raf-ERK pathway (104). In PH-Fibs, supraphysiological lactate accumulation triggers—in agreement with the law of mass action—the accumulation of excess reducing equivalents other than NADPH such as NADH, as gleaned through omics technologies and fluorescence lifetime imaging microscopy (108). Under hypoxic conditions, the lack of oxygen as a final acceptor of electrons in the electron transport chain may also contribute to the accumulation of reducing equivalents such as NADH and FADH2, which are otherwise regenerated to the relative oxidized forms under physiological conditions (Fig. 4). Under these circumstances, a reverse activity of succinate dehydrogenase (SDH) at complex II results in the supraphysiological accumulation succinate under ischemia; whereas at reperfusion, reverse electron flow may occur at the level of complex I, favoring the generation of high levels of ROS (163, 209). Independently from oxygen availability, supraphysiological levels of glycolytic intermediates such as fructose-1,6-bisphosphate can inhibit mitochondrial respiratory rate in highly glycolytic cells [Crabtree effect (45)].
FIG. 4.
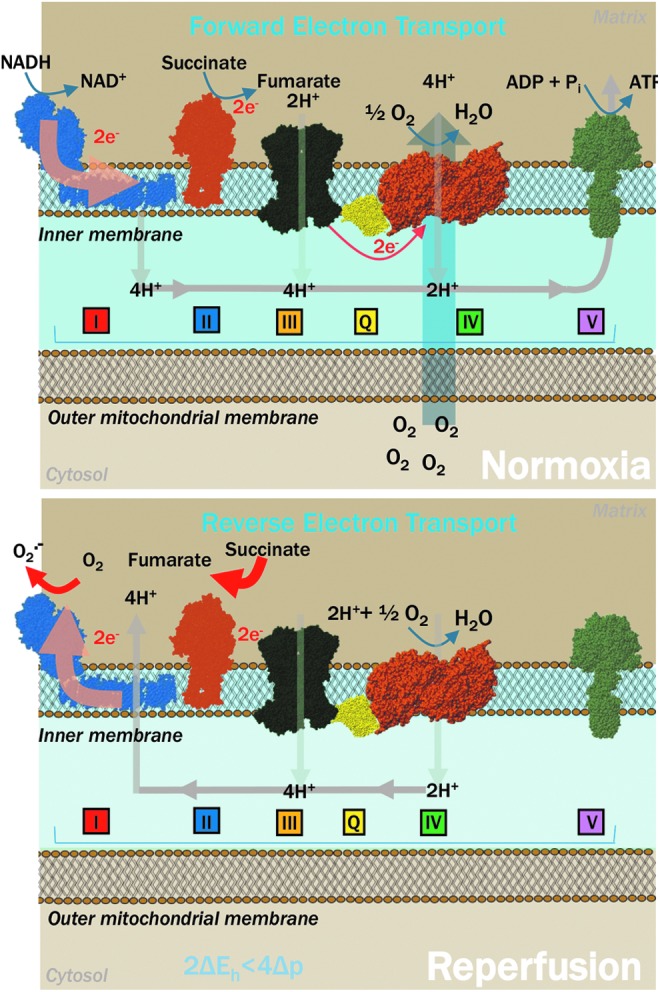
Decreased oxygen availability under hypoxic conditions promotes succinate accumulation owing to lack of oxygen as the final acceptor of electron at complex IV and reverse activity of succinate dehydrogenase at complex II. On reperfusion, rapid oxidation of succinate promotes a reverse-electron flow through complex I of the electron transport chain, a phenomenon that favors the accumulation of reactive oxygen species.
Mitochondrial or cytosolic isoforms of Krebs cycle enzymes can also contribute to NADH/NADPH homeostasis in cancer, ischemia, hemorrhage, and PH (32, 40, 108, 131, 193, 208, 209). Specific isoforms of malate or IDH and malic enzyme—either in cytosol or in mitochondria—can generate NADH or NADPH, depending on the isoform of the enzyme catalyzing the reaction, and NADP-dependent isoforms have been reported to be increased in PH patients (60). IDH1 and malic enzyme isoforms 2 and 3 (ME2, ME3) can generate NAD(P)H in mitochondria, whereas IDH2 and malic enzyme 1 (ME1) are producers in the cytoplasm. Cytoplasmic enzymes require exported citrate from mitochondria to generate oxaloacetate, malate, and isocitrate, respectively, for their activities, a phenomenon often observed in proliferating cells. Interestingly, IDH activity was found to be increased in PH patients, contributing to elevated NAD(P)H generation (60). Moreover, 70% of families with PAH (20) and 20% patients with idiopathic PAH bear mutations of bone morphogenic protein (BMPR2), a member of the TGF-β superfamily, that alter the activity of IDH (60).
Reducing equivalents: redox homeostasis
As anticipated from data presented in the previous paragraph, free NADPH plays a key role in anabolic reactions involving reductive synthesis of fatty acids, cholesterol, amino acids, and nucleotides, as well as in redox homeostasis by favoring reduction of oxidized proteins and antioxidant compounds such as glutathione. However, in many pathologies, PH included, NADPH can be used as a substrate to fuel the activity of NOXs, thus producing hydrogen peroxide. Free NADPH has mainly antioxidant properties, as it is used by glutathione (GSH) reductase or thioredoxin (TRX) reductases to regenerate GSH/TRXs, respectively. Because glutathione peroxidase have higher rate constants than peroxiredoxins (PRXs, its oxidized form is a substrate for TRX), but are less abundant, they were suggested to decompose mainly an enhanced pro-oxidative amount of hydrogen peroxide, whereas PRXs will rather terminate ROS signaling.
On the other hand, under pathological conditions when pro-oxidative cellular status is established, NADPH is widely consumed by NOXs to promote pro-oxidant reactions. These are multi-subunit complexes bound to plasma membranes associated with signaling domains that catalyze one-electron transfer from NADPH to molecular oxygen, thus generating ROS. Vascular NOXs produce superoxide, except for the NOX4 isoform, which primarily produces H2O2. Based on real-time PCR, NOX1, NOX2, and NOX4 isoforms were found to be expressed in ECs (predominantly NOX4), SMCs (predominantly NOX1,4), and Fibs (predominantly NOX4) in PAs (99). Expression of NOX5 was found to be present only in PA ECs and SMCs (158). All isoforms except NOX4 need to interact with additional proteins or undergo calcium-dependent activation. NOX4 is the only constitutively expressed NOX enzyme present in all layers of the PA. Its activity is regulated mainly by its expression (contains a HIF-binding site) and availability of reducing equivalents (46). Moreover, because of high Km for oxygen, it can respond directly to oxygen tension without any further intermediate signaling event (141). Because of increased glycolysis, PPP, and suppressed mitochondrial respiration all keeping NAD(P)H high, NOX4 activity is significantly elevated (109, 164). Indeed, NOX4 was suggested to participate in hypoxia-induced pulmonary vascular remodeling. It can signal through growth factors (TGF-β, PPAR-γ), cytokines, HIF, vasoactive agents (endothelin-1), or G-protein-coupled receptor agonists, allowing it to regulate enzymes and ion channels with proliferative, inflammatory, and apoptotic properties (9, 109). NOX4 expression was predominantly localized to the nucleus, where its cis-acting antioxidant response elements sequence can be regulated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2) under stress conditions (99). However, Veith et al. did not note any effect of NOX4 knockout on hypoxic pulmonary vasoconstriction or hypoxia-induced PH (217). Another NOX isoform, NOX2, found originally in phagocytes was suggested to contribute to PH, possibly though the regulation of inflammation (111). Although the role that NOXs play as sources of ROS production in PH pathology is well established, the expression and activity of individual isoforms, their regulation, and signaling properties in relation to PH development need to be further elucidated (164). Existing evidence strongly suggests that mitochondrial ROS interact with NOXs and other ROS sources to collectively induce a pro-oxidative redox state, which is further favored by the impairment of antioxidant capacity (superoxide dismutase, catalase, and glutathione peroxidase) of PH cells (6, 227).
Reducing equivalents: transcriptional control
More recently, a role has emerged for NAD(P)H in mediating transcriptional control in PH by controlling the activity of the NAD(P)H-dependent transcriptional repressor C-terminal-binding protein1 (CtBP1) (108) (Fig. 5). Dimerization, a phenomenon that activates CtBP1, is dependent on the relative levels of free NADH and NADH/NAD+ ratios (19). Activation of CtBP1 is relevant owing to its inhibitory interaction with gene-specific transcriptional factors, which is, in part, mediated by the recruitment of several epigenetic-modifying enzymes on the target genes (19). Recently, we found that increased expression of CtBP1 in fibroblasts generated from the PAs of chronically hypoxic calves or patients with iPAH mechanistically controlled the metabolic reprogramming of PH-Fibs toward aerobic glycolysis through a feed-forward mechanism. PH-Fibs exhibited increased free NADH, even under normoxic conditions, and were characterized by enhanced proliferation and inflammation. Genetic or pharmacological intervention to suppress CtBP1 expression or activity, respectively, exerted significant effects on cell phenotypes, as it (i) inhibited PH-Fibs proliferation (and promoted the upregulation of the cyclin-dependent kinase inhibitor genes, P21 and P15); (ii) promoted cell apoptosis through the upregulation of NOXA and PERP; and (iii) increased the expression of anti-inflammatory (HMOX1) markers (108). In addition, the transcriptional repressor CtBP1 exerts anti-apoptotic activity through inhibitory binding to the pro-apoptotic Bax or SIRT4, the repressor of glutaminolysis in mitochondria. In PH-Fibs, similar to cancer, the activation of glutaminolysis provides (i) alternative substrates for catabolism and anaplerosis; (ii) counteracts the acidification effect resulting from excess glycolysis with lactic fermentation (94, 220); and (iii) provides amine group moieties for the activation of nitrogen metabolism and downstream biosynthesis of polyamines, substrates for extracellular matrix remodeling [serum and urine desmosine and isodesmosine cross-linked amino acids are candidate biomarkers of PH (130)] and regulation of (pulmonary) fibrosis (10, 11, 228). The latter mechanism has been increasingly appreciated in cancer research in recent years (72, 142, 194). These observations are relevant in that they indicate the complex intertwinement at the transcriptional, metabolic, and epigenetic levels of proliferation, evasion of apoptosis, metabolic reprogramming, inflammation, as well as fibrosis—the various hallmarks of PH—in a similar fashion to the hallmarks of cancers.
FIG. 5.

Transcriptional regulation by CtBP1 in response to altered NADH/NAD+ homeostasis in PH (108). The crystal structure shows CtBP1 in complex with substrate MTOB (pdb: 4LCE). CtBP1, C-terminal-binding protein 1; MTOB, 4-methylthio 2-oxobutyric acid.
Metabolic Cross-Talk Between PH-Fibs and Macrophages
Recent studies in humans with PH and small and large animal models of PH have demonstrated a significant accumulation of macrophages in the vessel wall of the PAs, even down to small-size vessels (184, 198, 212). Experimental studies in rats by our group have demonstrated that macrophage recruitment and accumulation in the vessel wall is a prerequisite for vascular remodeling and development of PH, as clodronate-mediated ablation of circulating monocytes in hypoxia-exposed rats prevented macrophage accumulation and PH (69). A critical observation from humans with PH to various small and large animal models of PH was that macrophage accumulation remained largely restricted to the adventitial/perivascular compartment of the vessel wall, implying an important function for the adventitia and for adventitial macrophages in the vascular remodeling process and the pathogenesis of PH (184, 198). Subsequent studies by our group have identified that these adventitial macrophages express surface markers consistent with an alternative activation phenotype, which has been reported to be involved in tissue remodeling and fibrosis in other organs, such as the kidney (55, 219). Moreover, adventitial macrophages also express increased amounts of GLUT1 and CtBP1, therefore indicating their capacity for metabolic reprogramming and for cellular activation akin to the observations made in the adventitial fibroblasts described above (108). Macrophages are very important tissue cells and have been reported to contribute to tissue homeostasis across virtually all organs in the organism, but they are also known to be critical for both initiation of inflammation and resolution of inflammation (52, 98, 136, 137, 145, 146). They contribute to maintenance of tissue homeostasis through their ability to sense alterations in the microenvironment and to detect and respond to tissue-generated signals, such as cytokines, chemokines, oxygen tension, physical properties of the tissue composition, redox status, temperature, metabolites such as lactate, and even cathecholamines (34, 52, 54, 56, 98, 117, 136, 137, 140, 145, 146). Macrophages “read out” microenvironmental changes by undergoing metabolic reprogramming, which, in turn, is a prerequisite to enable a transcriptional response that is tailored to the inciting microenvironmental change (56, 129). As such, it has been established that pro-inflammatory activation of macrophages by lipopolysaccharide (LPS) and LPS in combination with interferon gamma (IFNγ) occurs as a function of metabolic rewiring to increased aerobic glycolysis and remodeling of the tricarboxylic acid (TCA) cycle, which is interrupted at the level of IDH and SDH with subsequent increases in citrate and succinate (89). It is believed that increased succinate levels can function within the mitochondria to increase ROS formation at complex I, which, in turn, promotes pro-inflammatory cytokine generation and that increased cytosolic succinate levels inhibit prolyl hydroxylase domains to stabilize HIF1, which, in turn, promotes expression of glycolytic enzymes and cooperates with PKM2 in regulating gene transcription, including that of pro-inflammatory IL1β (32, 35, 128). Intriguingly, the inflammasome, a signaling platform that regulates conversion of inactive pro-IL1 to active IL1b, localizes in close proximity to the mitochondria, as ROS derived from mitochondria activate inflammasome signaling (132). Moreover, hexokinase 1, a glycolytic enzyme increased through HIF1, has been shown to positively regulate inflammasome activity (132). Until recently, it was believed that pro-inflammatory macrophages activated by LPS could be reprogrammed or repolarized once the metabolic program was switched back to oxidative phosphorylation. However, recent studies have called this belief into question as the authors showed that pro-inflammatory macrophages activated by LPS/IFNg can be refractory to repolarization (215). On the other hand, recent studies have also shown that when metabolic reprogramming in LPS-activated macrophages is interrupted at the level of glycolysis or at the level of PKM2 and HIF1, these macrophages appear to repolarize toward increased expression of anti-inflammatory mediators and decreased expression of pro-inflammatory mediators (91, 157, 204). To what extent metabolic programs in tissue macrophages are actually regulating macrophage plasticity, that is, the ability to repolarize, needs to be investigated in further detail (56, 136). Regardless of this, the concept of repolarizing a pro-inflammatory macrophage into an anti-inflammatory macrophage through manipulation of a metabolic program is an attractive means to promote resolution of inflammation/fibrosis.
One important aspect of homeostatic tissue function of macrophages is their communication with other tissue cells, such as fibroblasts. We have recently documented that fibroblasts from animals and humans with PH have the ability to activate naive macrophages in vitro toward an alternative activation phenotype that is identical to the one observed within the adventitia of the PA [hereafter called PH-Fibs activated macrophages (55)]. Importantly, explanted remodeled adventitial tissue from animals with PH was also capable of activating macrophages to this phenotype (55). At the molecular basis, PH-Fibs activated macrophages display increased activation of STAT3 and HIF1 signaling and expression of downstream target genes that are important in promoting tissue remolding, such as Arginase1 (provide polyamines to sustain cellular proliferation), and VEGFa and glycolytic reprogramming such as GLUT1 and CtBP1 (55). Importantly, genetic inhibition of either STAT3 or HIF1 signaling in PH-Fibs activated macrophages attenuated their activation and decreased Arginase1 expression. Subsequent studies have revealed that this macrophage activation phenotype is, in part, mediated by PH-Fibs-derived paracrine IL-6, a canonical activator of STAT3 signaling in macrophages (55). Intriguingly, there is evidence of increased IL6, STAT3, and HIF1 signaling within the adventitia of both patients with PH and animals with PH, and IL6 has widely been reported to be an important cytokine in the pathogenesis of PH in patients and animal models (55, 197). Consistent with the above reference to STAT3 and HIF1's role in PKM2 signaling and metabolic reprogramming of cancer cells and fibroblasts, PH-Fibs activated macrophages also display increased glycolysis and remodeling of the TCA cycle with accumulation of succinate and lactate, and increased production of polyamines (metabolites of arginine metabolism by Arginase1). Recent studies have documented that pSTAT3 is sufficient to increase glycolysis in cells and that co-operation with HIF1 and PKM2 can result in a vicious feed-forward cycle that drives and maintains glycolytic reprogramming in cells (43). Therefore, within the adventitia, metabolic reprogramming of the PH-Fib occurs in synchrony with metabolic reprogramming of the macrophage in an IL6-STAT3-HIF1-dependent pathway (56). A recent study reported activation of a similar alternative activated macrophage phenotype (with increased expression of Arginase1 and HIF1 and VEGF) by paracrine signals derived from cancer cells. Intriguingly, activation of Arginase in these cancer cell activated macrophages was also dependent on HIF, similar to what we had observed in PH-Fibs activated macrophages, and expression of Arginase conferred a pro-tumorigenic phenotype on macrophages (34, 56). Ablation of Arginase1 in macrophages was associated with decreased tumor formation in vivo in mice. The mechanism was believed to be related to the ability of Arginase1 to generate polyamines as necessary factors to sustain tumor cell proliferation (34). Strikingly, expression of Arginase1 appeared to be mediated by cancer cell-derived paracrine lactate, the byproduct of highly proliferative cancer cells with metabolic reprogramming to glycolysis (34), thereby suggesting a similar metabolic synchrony between cancer cells and macrophages as we hypothesize exists between PH-Fibs and PH-fibroblast activated macrophages (56) (Fig. 6). Therefore, it appears that metabolically reprogrammed (cancer) cells can co-opt macrophage activation through paracrine signaling (such as lactate and/or cytokines) to increase tissue availability of metabolites, such as polyamines, to meet their demand for substrates that fuel proliferation and activation (Fig. 6). Thus, there appears to be a metabolic synergy between cancer cells and other associated tissue cells such as fibroblasts and macrophages, which form a basis for cellular proliferation and activation (Fig. 6).
FIG. 6.

Metabolic synergy between adventitial fibroblasts and macrophages in the vascular remodeling process in PH. In adventitial fibroblasts, epigenetic changes reflected in decreased miRNA124 expression enable increased PTBP1 signaling, resulting in increased expression of PKM2, which drives lactate formation and increases aerobic glycolysis while reducing TCA cycle. Increased glycolysis results in increased free NADH, which activates the transcriptional repressor CtBP1. Together, these alterations promote proliferation and apoptosis resistance concomitantly with an increased production of lactate, succinate, citrulline, IL-6, and other pro-inflammatory mediators, while suppressing anti-inflammatory mediator production. Adventitial macrophages respond to the increased concentrations of fibroblast-derived metabolites in the microenvironment whereby IL6, lactate, and succinate drive activation of STAT3 and HIF1, which, in turn, drive metabolic reprogramming similar to that in adventitial fibroblasts; utilization of citrulline feeds polyanine production by Arginase, which, in turn, is increased in response to IL6 and lactate. Thus, exchange of substrates and metabolites between macrophages and fibroblasts enables persistent metabolic reprogramming, cellular activation, and proliferation. PTBP1, polypyrimidine-tract binding protein 1; TCA, tricarboxylic acid.
We hypothesize that the vascular remolding process within the vessel wall of humans and animals with PH entails a similar metabolic synergy between fibroblasts and macrophages, whereby byproducts of metabolic programs in fibroblasts such as lactate and succinate can fuel metabolic programs in macrophages such that macrophages, in turn, engage a metabolic program that provides substrates that fuel the metabolism of the fibroblast (Fig. 6). Intriguingly, we have observed that fibroblast-derived lactate can synergize with fibroblast-derived IL-6 to greatly enhance macrophage activation and expression of Arginase1. Therefore, we suggest that within the PA adventitia there exists a metabolic synergy between the PH-Fibs and the adventitial macrophages, in which the fibroblasts co-opt the macrophage phenotype to sustain its increased proliferation and activation (Fig. 6). In addition, PH-Fibs produce not only increased amounts of lactate as a result of increased glycolysis but also increased amounts of citrulline (as a result of TCA and arginine-succinate shunt remodeling). When produced in excess, citrulline can be shunted out of cells and used by cells with a high citrulline demand (56, 168, 173). For example, macrophages expressing high amounts of Arginase1 can have a high demand for Arginine, which has been shown to become limiting in inflammatory conditions within the tissues (56, 168, 173). The high demand on Arginine can be met by importing citrulline, because it can serve as a precursor for arginine synthesis in macrophages (56, 168, 173). Therefore, polyamine synthesis through macrophage Arginase1 can be maintained despite low tissue arginine concentrations through utilization of fibroblast-derived citrulline. An important effect of this metabolic synergy is that the adventitia becomes autotrophic for arginine and to sustain cellular proliferation (56). Intriguingly, a similar role for citrulline has been shown to be a critical pathway to sustain the macrophage's ability to kill intracellular bacteria in conditions when arginine becomes limiting (168).
In addition to upregulation of Arginase 1, metabolic reprogramming in the PH-Fibs activated macrophages is also associated with increased succinate production, which has been reported to serve as a paracrine signal that can activate tissue cells; therefore, macrophage- and fibroblast-derived succinate can be potential paracrine signals through which macrophages sustain activation of PH-Fibs and vice versa (127). Moreover, signaling downstream of increased citrate as a result of TCA remodeling in the PH-Fibs activated macrophage can be utilized for epigenetic mechanisms that arrest macrophage activation in this phenotype (149). Future studies will have to be directed toward improving our understanding of how fibroblasts and macrophages engage in a metabolic symbiotic relationship to induce and maintain cellular activation and to define the signaling pathways and molecules that relay environmental change to metabolic programs and transcriptional responses. Further, it becomes more and more evident that the success of pharmacological interruptions to halt or prevent vascular remodeling will rely on the ability to target metabolic synergy and symbiosis within the cross-talk of macrophages and fibroblasts (Fig. 6).
The Other Facet of Metabolic Reprogramming in PH: Mitochondrial Dysfunction and Superoxide Production
As anticipated above, PH-Fibs as well as cells from all layers of the hypertensive PA wall are characterized by incomplete glucose oxidation through glycolysis and altered mitochondrial bioenergetics, in a similar fashion to cancer cells (68, 164). Mitochondria play a central role in energetics and ROS production, calcium homeostasis, cell survival, and apoptosis (Fig. 7). Accumulation of Krebs cycle intermediates provides critical substrates for anabolic reactions that are critical to highly proliferating cells such as PH-Fibs (164). In the absence of complete pyruvate oxidation, mitochondria rely on alternative carbon sources, such as fatty acids or amino acids, especially glutamine. In cancer cells, energy demands in terms of ATP production are mostly satisfied through glycolysis, which is estimated to contribute to 50%–70% of total ATP production (42, 123, 216). The so-called glucose-fatty acid cycle was first described by Randle more than 50 years ago (172) and recently expanded through molecular biology and omics investigations (83). Such mechanisms, first elucidated in cancer, have come in handy to test and confirm the upregulation of the expression of genes involved in fatty acid oxidation in lung tissue from PH patients (235). The relevance of such mechanisms in PH is clear when considering that mice lacking malonyl-coA decarboxylase (MDC) do not develop PH on chronic exposure to hypoxia (200). Notably, MDC can be inactivated through SIRT4-mediated deacetylation (103). Increased CtBP1 activity represses SIRT4 in PH-Fibs, allowing MDC to be active and drive fatty acid oxidation in mitochondria (108). This further strengthens the earlier considerations regarding the CtBP1/SIRT4 axis representing a candidate druggable target in PH. Alternatively, it has been noted that fatty acid uptake through CD36, but not oxidation, underlies intracellular fatty acid accumulation that fuels the biosynthesis of di- and tri-glycerides, key constituents of cellular membranes (202).
FIG. 7.

An overview of mitochondrial metabolism and its role in redox and energy homeostasis in PH. GDH, glutamate dehydrogenase; GSH, glutathione; PHD, prolyl hydroxylase domain; OAA, oxaloacetate; NOS, nitric oxide synthase; NOX, NADPH oxidase.
Recent findings suggest that PH-Fibs develop an addiction for glutamine, which, in turn, fuels polyamine synthesis to drive fibrosis (10, 11, 72, 194). Glutamine is the most abundant circulating amino acid as well as the main amine group donor in biochemical reactions, as it represents a key substrate for the synthesis of glutamate via glutaminolysis. Glutamate can, in turn, either be used as a substrate for the synthesis of glutathione or further processed to α-ketoglutarate (in part by transamination reactions that mitigate the excess of pyruvate through the synthesis of alanine); α-ketoglutarate can enter the Krebs cycle, serve an anaplerotic role for the synthesis of nonessential amino acids, or be used as a substrate for hydroxylase (see Fig. 7). Glutamine-derived ketolgutarate can also contribute to de novo lipogenesis by fueling reductive carboxylation reactions toward citrate (126). In cancer, it has been proposed that reductive glutamine metabolism is a function of ketoglutarate/citrate ratios, consistent with the law of mass action (59). Of note, even though tracing experiments with heavy labeled substrates will be necessary and are currently ongoing in our labs, our observations in PH-Fibs indicate increases in glutamine, ketoglutarate, and ketoglutarate/citrate ratios (107).
Under hypoxic conditions, α-ketoglutarate can give rise to the L enantiomer of 2-hydroxyglutarate (86, 147), a reaction that in cancer is catalyzed by IDH mutations (generating the D enantiomer), or a switch in the expression pattern of IDH isoforms in favor of NADPH-generating IDH1 (41). More recently, it has been noted that lactate dehydrogenase can catalyze ketoglutarate to L-2-hydroxyglutarate (87, 138). Of note, 2-hydroxyglutarate has been described as an oncometabolite and was found to elicit significant epigenetic changes that can contribute to cancer phenotype (190). TCA cycle intermediates, such as succinate, α-ketoglutarate, or fumarate, can regulate histone and DNA methylation (181). Therefore, carboxylic acids can contribute to the epigenetic regulation of DNA expression. Supraphysiological levels of succinate and fumarate can also replace α-ketoglutarate as a mock substrate for prolyl-hydroxylase, thereby preventing HIF1 hydroxylation and promoting its stabilization, thus enhancing the Warburg effect (97, 187).
In the previous sections, we described how the levels of reducing equivalents NADH/FADH2 in PH-Fibs were influenced by altered mitochondrial bioenergetics owing to uncoupling of the respiratory chain complexes I and II. Indeed, PH-Fibs mitochondria are hyperpolarized (164). There are several potential explanations for this observation. For example, mitochondrial respiratory chain complexes in PH-Fibs (complex I, III, and IV) may be pumping protons across the mitochondrial membrane. Alternatively, uncoupling proteins might be involved in the regulation of mitochondrial membrane potential. Studies using mitochondrial uncoupling protein 2-deficient (UCP2−/−) mice showed that increased ROS production, not by regulation of membrane potential itself, was the main factor influencing vascular remodeling with development of PH (155). On the other hand, spontaneous PH development in UCP2−/− mice could be, in part, explained by decreased mitochondrial calcium levels, which, in turn, would affect mitochondrial bioenergetics by targeting calcium-dependent enzymes (49). An alternative explanation involves the translocation of hexokinase II into mitochondria via a GCK3-β-dependent mechanism, an event that inhibits the VDAC to sustain mitochondrial hyperpolarization (50).
Electron flow through the respiratory chain with molecular oxygen as the final acceptor of electrons results in the generation of ROS, such as superoxide (36), that are mostly generated at the level of complex I and III (16, 135). Premature single-electron reduction of molecular oxygen mostly occurs when electron flow is either retarded (i.e., ATP synthase is inhibited, cytochrome c or coenzyme Q cycling is retarded) or there is substrate pressure (high load of NADH, FADH2) on respiratory complexes. Alternatively, stoichiometric mismatches can affect the correct assembly of electron transfer chain super complexes, which, in turn, can result in delay of electron flow on sites of the complexes mediating production of superoxide. As pointed out in the previous section, antioxidant redox couples (i.e., GSH/GSSG) are closely linked to the metabolic redox couples (NADH, FADH2), which also serve as the substrate of the respiratory chain (4). Superoxide can be formed within complex I in two distinct sites (207) (i) at the level of the flavin in the NADH-oxidizing site, producing superoxide in the mitochondrial matrix; or (ii) at the ubiquinone-reducing site, which pumps superoxide into mitochondrial intermembrane space. Production of superoxide into the mitochondrial intermembrane space by complex III is believed to arise from the quinol oxidizing site. complex III-driven superoxide may serve as a second messenger in cellular signaling; whereas complex I-generated superoxide is predominantly deleterious as it can react in high concentrations with mitochondrial DNA or other matrix components that are vulnerable to oxidative damage (12). Other sources of mitochondrial superoxide have been identified in other cell types such as complex II (169), glycerol phosphate dehydrogenase (152), PDH (196), or α-ketoglutarate dehydrogenase (208). The main sources of superoxide production during PH development remain to be elucidated.
Mutations of complex I and III may represent alternative sources of mitochondrial superoxide. Indeed, mutations in 9 out of 13 assembly factors of complex I are pathological in that they impair complex biogenesis (182). Murine embryonic fibroblasts are characterized by a direct downregulation of subunits of complex I (NDUFA4L2) (206), which may underlie increases in superoxide production. In PH-Fibs from chronically hypoxic calves, altered activity of complex I was explained by the downregulation of the assembly subunit NDUFS4, promoting complex I instability, an increased disconnection of electron influx of the NADH dehydrogenase module from the complex I holo-complex, mitochondrial hyperpolarization, and increased superoxide production (164). Consistently, NDUFS4 deficiency caused aberrant mitochondrial morphology and elevated ROS production in primary fibroblasts of NDUFS4 knockout mice, a model of Leigh syndrome (214). NDUFV1 gene knockdown partially inhibits complex I activity, partially reduces NAD+/NADH ratios, without drastically inhibiting oxidative phosphorylation, resulting in a significant enhancement of cell proliferation and metastasis (183). On the other hand, enhanced complex I activity can increase tumor cell autophagy and inhibit proliferation in vivo. In analogy to cancer investigations, complex I dysfunction seems to be involved in the switch of energy metabolism to glycolysis in SMC in PH, supporting the idea that maintaining complex I activity may be a potential therapeutic target for the treatment of PH (170).
Controversial evidence exists with respect to mitochondrial superoxide production in response to hypoxia in PA SMCs (5, 174, 222). In our study, we have confirmed accumulation of mitochondrial superoxide in PH-Fibs from chronically hypoxic calves grown and studied under normoxic conditions (164). Mitochondrial superoxide production from complex III plays a crucial role in the stabilization of HIF1α through the inhibition of HIF-prolyl-hydroxylases (24). Selective expression of the ROS scavenger PRX 5 in the mitochondrial intermembrane space of PH-Fibs suppressed ROS production and the acute activation of cytosolic calcium, which regulates vasoconstriction, the initial step in PH development (179).
Sirtuin 3 (SIRT3) protein, the predominant NAD+-dependent mitochondrial deacetylase, can regulate mitochondrial ROS production. Increased NADH/NAD+ ratios in PH-Fibs (108) together with increased glycolysis repress SIRT3 activity in human and rat PH cells. On the other hand, human iPAH has been associated with a loss-of-function SIRT3 polymorphism (160). In PH, SIRT3 targets, such as IDH2, SDH A, Lon protease, or NDUFA9 subunit of complex I, remain acetylated and inactivated (95). Inactive SIRT3 would thus result in the lack of deacetylation of its targets, resulting, for example, in the accumulation of succinate by less active SDH A or in the negative regulation of electron transport chain subunit assembly. In addition, since SIRT3 can regulate PDHA1, one of the components of PDH complex (153), decreased activity of SIRT3 in PH cells might underpin PDHA1 acetylation, a phenomenon that decreases PDHA1 activity and induces its phosphorylation, thereby preventing pyruvate metabolism into acetyl-CoA. Knockdown of SIRT3 in mice promotes the spontaneous development of PH (160). Notably, SMCs from PAs derived from SIRT3 knockout mice are characterized by enhanced stabilization of HIF1 and STAT3 phosphorylation, increased phosphorylation of PDH, and, thus, suppressed glucose oxidation in favor of glycolysis (160).
Finally, mitochondrial morphology is another critical factor influencing the efficiency of mitochondrial ATP synthesis. For example, in diabetes and selected neurological diseases (154), low respiratory mitochondrial bioenergetics can be also explained by a condensed state of cristae structure in situ (i.e., cristae expansion, matrix condensation) and mitochondrial fission. On the other hand, a higher respiratory capacity is observed in the presence of an orthodox cristae state in situ (i.e., matrix expansion, thin cristae) and fused mitochondrial morphology. For example, downregulated mitochondrial bioenergetics are observed in glycolytic hepatocellular cancer cells (HepG2), where mitochondria are characterized by a condensed cristae structure under chronic hypoxia (72 h) (164). Fusion and fission are metabolically controlled through GTPases (Opa1, Mfn1 and 2, Drp1) that can, in some cases, sense mitochondrial membrane potential (Opa1) or oxidative stress (226). Consistently, mitochondria from human PH SMCs or Fibs are fragmented and characterized by downregulated levels of pro-fusion proteins (Mfn2, Opa1) and upregulated levels of pro-fission proteins (Drp1, Fis1) (164, 178). Moreover, Cyclin B CDK1-dependent phosphorylation and activation of Drp1 was observed in PH mitochondria, whereas Mfn2 was deregulated by the transcriptional coactivator PGC1 or platelet-derived growth factor and endothelin-1 (178). Although further studies are mandatory, results to date seem to recapitulate observations in cancer (1, 14, 225) and point at a role of mitochondrial fission in promoting altered bioenergetics, increased proliferation, and resistance to apoptosis in PH-Fibs, an observation that may highlight potential therapeutic targets in the near future. Novel therapeutic avenues may involve the regulation of the mitochondrial chaperone family, as the MCJ/DnaJC15 DNAJ component MCJ has been shown to act as an endogenous mitochondrial repressor of the respiratory chain (79) by decreasing electron transfer efficiency at the level of supercomplex I of the electron transport chain (23). Silencing of MCJ expression, in turn, increases electron transport efficiency without increases in oxygen consumption rate, regulating CD8+ T-reg cell and macrophage proliferation and pro-inflammatory phenotype (23, 139).
Summary
In this review, we propose a role for metabolic reprogramming of resident and recruited cells within the microenvironment of the artery wall as a hallmark in PH (i.e., ECs, SMCs, fibroblasts, cells of the immune system). By reviewing the existing literature and comparing it with the vast body of knowledge on the so-called hallmarks of cancer, we identified the metabolic factors influencing pulmonary vascular remodeling in PH and suggested its synergistic role at the crossroads with other hallmarks, such as proliferation, resistance to apoptosis, inflammation, and fibrosis. By detailing molecular cascades involving the interconnectedness of transcriptional signaling, enzyme expression and activity, and metabolic phenotypes, we enlisted a series of promising therapeutic interventions that have been proposed to date (Fig. 8). For the foreseeable future, the identification of novel and more effective therapeutic strategies will be favored by an improved understanding of the metabolic basis of PH and the role that metabolic control plays in mediating PH development and progression, at the interface with the other hallmarks of PH.
FIG. 8.

A summary of metabolic pathways and potential therapeutic interventions in PH, as reviewed in this article. CHC, α-cyano-4-hydroxycinnamic acid; DCA, dichloroacetate; PDK, pyruvate dehydrogenase kinase.
Abbreviations Used
- 18FDG
18F-fluorodeoxyglucose
- AMPK
AMP-activated protein kinase
- CtBP1
C-terminal-binding protein 1
- DHEA
dehydroepiandrosterone
- EC
endothelial cell
- Fibs
fibroblasts
- G6PD
glucose 6-phosphate dehydrogenase
- GLUT1
glucose transporter 1
- GSH
glutathione
- HIF
hypoxia-inducible factor
- HMOX
heme oxygenase
- IDH
isocitrate dehydrogenase
- IFNγ
interferon gamma
- iPAH
idiopathic pulmonary arterial hypertension
- LDHA
lactate dehydrogenase A
- LPS
lipopolysaccharide
- MDC
malonyl-coA decarboxylase
- ME
malic enzyme
- NAD(P)H
nicotinamide adenine dinucleotide (phosphate), reduced form
- NAMPT
nicotinamide phosphoribosyltransferase
- NNT
nicotinamide nucleotide transdehydrogenase
- NOX
NADPH oxidase
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PA
pulmonary artery
- PASMCs
pulmonary artery smooth muscle cells
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PET
positron emission tomography
- PFKB3
phosphofructokinase B3
- PH
pulmonary hypertension
- PKG1
protein kinase G1
- PKM
pyruvate kinase M
- PPP
pentose phosphate pathway
- PRX
peroxiredoxin
- PTBP1
polypyrimidine-tract binding protein 1
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SIRT3
sirtuin 3
- SMC
smooth muscle cell
- TCA
tricarboxylic acid
- TRX
thioredoxin
- VDAC
voltage-dependent anion channel
- VEGF
vascular endothelial growth factor
Acknowledgments
This work has been supported by KONTAKT grant (LH 15071) from Czech Ministry of Education to LPH and by NIH Program Project Grant HL014985, NIH Axis Grant HL114887, NIH R01 HL125827, and Department of Defense Grant PR140977 to K.R.S., K.C.E.K., M.L., and H.Z. A.D. is a recipient of the Boettcher Webb-Waring Biomedical Research Award–Early Career grant.
References
- 1.Alirol E. and Martinou JC. Mitochondria and cancer: is there a morphological connection? Oncogene 25: 4706–4716, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, et al. . Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 304: H1708–H1718, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J, et al. . Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334: 1278–1283, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aon MA, Cortassa S, and O'Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta 1797: 865–877, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Archer SL, Huang J, Henry T, Peterson D, and Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 73: 1100–1112, 1993 [DOI] [PubMed] [Google Scholar]
- 6.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, et al. . Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–2671, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashizawa K, Willingham MC, Liang CM, and Cheng SY. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J Biol Chem 266: 16842–16846, 1991 [PubMed] [Google Scholar]
- 8.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, et al. . Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol 172: 615–627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, et al. . NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol 34: 1704–1715, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, et al. . Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 126: 3313–3335, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharya M. Stiff discipline for cells in pulmonary hypertension. Sci Transl Med 8: 358ec154, 2016 [Google Scholar]
- 12.Bleier L, Wittig I, Heide H, Steger M, Brandt U, and Dröse S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic Biol Med 78: 1–10, 2015 [DOI] [PubMed] [Google Scholar]
- 13.Boehme J, Sun X, Tormos KV, Gong W, Kellner M, et al. . Pulmonary artery smooth muscle cell hyperproliferation and metabolic shift triggered by pulmonary overcirculation. Am J Physiol Heart Circ Physiol 311: H944–H957, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boland ML, Chourasia AH, and Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol 3: 292, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnet S, Dumas-de-La-Roque E, Bégueret H, Marthan R, Fayon M, et al. . Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci U S A 100: 9488–9493, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466–472, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchakjian MR. and Kornbluth S. The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol 11: 715–727, 2010 [DOI] [PubMed] [Google Scholar]
- 18.Bull TM, Coldren CD, Geraci MW, and Voelkel NF. Gene expression profiling in pulmonary hypertension. Proc Am Thorac Soc 4: 117–120, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Byun JS. and Gardner K. C-terminal binding protein: a molecular link between metabolic imbalance and epigenetic regulation in breast cancer. Int J Cell Biol 2013: 647975, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai J, Pardali E, Sánchez-Duffhues G, and ten Dijke P. BMP signaling in vascular diseases. FEBS Lett 586: 1993–2002, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Carmeliet P. and Jain RK. Angiogenesis in cancer and other diseases. Nature 407: 249–257, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Cavasin MA, Stenmark KR, and McKinsey TA. Emerging roles for histone deacetylases in pulmonary hypertension and right ventricular remodeling (2013 Grover Conference series). Pulm Circ 5: 63–72, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Champagne DP, Hatle KM, Fortner KA, D'Alessandro A, Thornton TM, et al. . Fine-tuning of CD8(+) T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity 44: 1299–1311, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, and Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95: 11715–11720, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaneton B, Hillmann P, Zheng L, Martin ACL, Maddocks ODK, et al. . Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491: 458–462, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C, Pore N, Behrooz A, Ismail-Beigi F, and Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem 276: 9519–9525, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Sysol JR, Singla S, Zhao S, Yamamura A, et al. . Nicotinamide phosphoribosyltransferase promotes pulmonary vascular remodeling and is a therapeutic target in pulmonary arterial hypertension. Circulation 135: 1532–1546, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, and Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chettimada S, Joshi SR, Alzoubi A, Gebb SA, McMurtry IF, et al. . Glucose-6-phosphate dehydrogenase plays a critical role in hypoxia-induced CD133+ progenitor cells self-renewal and stimulates their accumulation in the lungs of pulmonary hypertensive rats. Am J Physiol Lung Cell Mol Physiol 307: L545–L556, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chettimada S, Joshi SR, Dhagia V, Aiezza A, Lincoln TM, et al. . Vascular smooth muscle cell contractile protein expression is increased through protein kinase G-dependent and -independent pathways by glucose-6-phosphate dehydrogenase inhibition and deficiency. Am J Physiol Heart Circ Physiol 311: H904–H912, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, et al. . Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, et al. . Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christofk HR, Vander Heiden MG, Wu N, Asara JM, and Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452: 181–186, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, et al. . Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, et al. . Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem 291: 14274–14284, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortassa S, O'Rourke B, and Aon MA. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta 1837: 287–295, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cottrill KA. and Chan SY. Metabolic dysfunction in pulmonary hypertension: the expanding relevance of the Warburg effect. Eur J Clin Invest 43: 855–865, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dahoui HA, Hayek MN, Nietert PJ, Arabi MT, Muwakkit SA, et al. . Pulmonary hypertension in children and young adults with sickle cell disease: evidence for familial clustering. Pediatr Blood Cancer 54: 398–402, 2010 [DOI] [PubMed] [Google Scholar]
- 39.D'Alessandro A, Moore HB, Moore EE, Wither M, Nemkov T, et al. . Early hemorrhage triggers metabolic responses that build up during prolonged shock. Am J Physiol Regul Integr Comp Physiol 308: R1034–R1044, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D'Alessandro A, Slaughter AL, Peltz ED, Moore EE, Silliman CC, et al. . Trauma/hemorrhagic shock instigates aberrant metabolic flux through glycolytic pathways, as revealed by preliminary 13C-glucose labeling metabolomics. J Transl Med 13: 253, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, et al. . Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 465: 966, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Demaria M. and Poli V. PKM2, STAT3 and HIF-1α: the Warburg's vicious circle. JAKSTAT 1: 194–196, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deschoemaeker S, Di Conza G, Lilla S, Martín-Pérez R, Mennerich D, et al. . PHD1 regulates p53-mediated colorectal cancer chemoresistance. EMBO Mol Med 7: 1350–1365, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Díaz-Ruiz R, Avéret N, Araiza D, Pinson B, Uribe-Carvajal S, et al. . Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in crabtree effect induction? J Biol Chem 283: 26948–26955, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Diebold I, Petry A, Hess J, and Görlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell 21: 2087–2096, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doherty JR. and Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest 123: 3685–3692, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong G, Mao Q, Xia W, Xu Y, Wang J, et al. . PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett 11: 1980–1986, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, and Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res 113: 126–136, 2013 [DOI] [PubMed] [Google Scholar]
- 50.Dromparis P, Sutendra G, and Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med Berl Ger 88: 1003–1010, 2010 [DOI] [PubMed] [Google Scholar]
- 51.Ducker GS. and Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab 25: 27–42, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, et al. . Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115: 56–65, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dumas de La Roque E, Savineau J-P, Metivier A-C, Billes M-A, Kraemer J-P, et al. . Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): a pilot study. Ann Endocrinol 73: 20–25, 2012 [DOI] [PubMed] [Google Scholar]
- 54.El Kasmi KC, Anderson AL, Devereaux MW, Vue PM, Zhang W, et al. . Phytosterols promote liver injury and Kupffer cell activation in parenteral nutrition-associated liver disease. Sci Transl Med 5: 206ra137, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, et al. . Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol 193: 597–609, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El Kasmi KC. and Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol 27: 267–275, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elliott CG, Barst RJ, Seeger W, Porres-Aguilar M, Brown LM, et al. . Worldwide physician education and training in pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest 137(6 Suppl): 85S–94S, 2010 [DOI] [PubMed] [Google Scholar]
- 58.Erez A. and DeBerardinis RJ. Metabolic dysregulation in monogenic disorders and cancer—finding method in madness. Nat Rev Cancer 15: 440–448, 2015 [DOI] [PubMed] [Google Scholar]
- 59.Fendt S-M, Bell EL, Keibler MA, Olenchock BA, Mayers JR, et al. . Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun 4: 2236, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, et al. . Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2: 201–213, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fico A, Paglialunga F, Cigliano L, Abrescia P, Verde P, et al. . Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis. Cell Death Differ 11: 823–831, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Fijalkowska I, Xu W, Comhair SAA, Janocha AJ, Mavrakis LA, et al. . Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–1138, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Filosa S, Fico A, Paglialunga F, Balestrieri M, Crooke A, et al. . Failure to increase glucose consumption through the pentose-phosphate pathway results in the death of glucose-6-phosphate dehydrogenase gene-deleted mouse embryonic stem cells subjected to oxidative stress. Biochem J 370(Pt 3): 935–943, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foley JF. The NADPH survival advantage. Sci Signal 5: ec156, 2012 [Google Scholar]
- 65.Forgione MA. and Loscalzo J. Oxidant stress as a critical determinant of endothelial function. Drug News Perspect 13: 523–529, 2000 [DOI] [PubMed] [Google Scholar]
- 66.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, et al. . Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16: 4604–4613, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Francis SH, Busch JL, Corbin JD, and Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62: 525–563, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freund-Michel V, Khoyrattee N, Savineau J-P, Muller B, and Guibert C. Mitochondria: roles in pulmonary hypertension. Int J Biochem Cell Biol 55: 93–97, 2014 [DOI] [PubMed] [Google Scholar]
- 69.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, et al. . Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol 168: 659–669, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gameiro PA, Laviolette LA, Kelleher JK, Iliopoulos O, and Stephanopoulos G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J Biol Chem 288: 12967–12977, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao X, Wang H, Yang JJ, Liu X, and Liu Z-R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 45: 598–609, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerner EW. and Meyskens FL. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer 4: 781–792, 2004 [DOI] [PubMed] [Google Scholar]
- 73.Gupte RS, Ata H, Rawat D, Abe M, Taylor MS, et al. . Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal 14: 543–558, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, et al. . Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, et al. . Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 288: H13–H21, 2005 [DOI] [PubMed] [Google Scholar]
- 76.Gupte SA, Li K-X, Okada T, Sato K, and Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther 301: 299–305, 2002 [DOI] [PubMed] [Google Scholar]
- 77.Hampl V, Bíbová J, Povýsilová V, and Herget J. Dehydroepiandrosterone sulphate reduces chronic hypoxic pulmonary hypertension in rats. Eur Respir J 21: 862–865, 2003 [DOI] [PubMed] [Google Scholar]
- 78.Hanahan D. and Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011 [DOI] [PubMed] [Google Scholar]
- 79.Hatle KM, Gummadidala P, Navasa N, Bernardo E, Dodge J, et al. . MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol 33: 2302–2314, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, et al. . Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol 35 Suppl: S129–S150, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hitosugi T, Fan J, Chung T-W, Lythgoe K, Wang X, et al. . Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell 44: 864–877, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huberts DHEW. and van der Klei IJ. Moonlighting proteins: an intriguing mode of multitasking. Biochim Biophys Acta 1803: 520–525, 2010 [DOI] [PubMed] [Google Scholar]
- 83.Hue L. and Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297: E578–E591, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huston JH. and Ryan JJ. The emerging role of epigenetics in pulmonary arterial hypertension: an important avenue for clinical trials (2015 Grover Conference series). Pulm Circ 6: 274–284, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imtiyaz HZ. and Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol 345: 105–120, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Intlekofer AM, Dematteo RG, Venneti S, Finley LWS, Lu C, et al. . Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab 22: 304–311, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, et al. . L-2-hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol 13: 494–500, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jeon S-M, Chandel NS, and Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485: 661–665, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, et al. . Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42: 419–430, 2015 [DOI] [PubMed] [Google Scholar]
- 90.Karimi Galougahi K, Ashley EA, and Ali ZA. Redox regulation of vascular remodeling. Cell Mol Life Sci 73: 349–363, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kelly B, Tannahill GM, Murphy MP, and O'Neill LAJ. Metformin inhibits the production of reactive oxygen species from NADH:ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem 290: 20348–20359, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim GH, Ryan JJ, Marsboom G, and Archer SL. Epigenetic mechanisms of pulmonary hypertension. Pulm Circ 1: 347–356, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim J, Tchernyshyov I, Semenza GL, and Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006 [DOI] [PubMed] [Google Scholar]
- 94.Kim JH. and Youn HD. C-terminal binding protein maintains mitochondrial activities. Cell Death Differ 16: 584–592, 2009 [DOI] [PubMed] [Google Scholar]
- 95.Kincaid B. and Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci 5: 48, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kirkman HN, Rolfo M, Ferraris AM, and Gaetani GF. Mechanisms of protection of catalase by NADPH kinetics and stoichiometry. J Biol Chem 274: 13908–13914, 1999 [DOI] [PubMed] [Google Scholar]
- 97.Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, and Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of hif. J Biol Chem 282: 4524–4532, 2007 [DOI] [PubMed] [Google Scholar]
- 98.Kotas ME. and Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell 160: 816–827, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, et al. . The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 10: 1139–1151, 2005 [DOI] [PubMed] [Google Scholar]
- 100.Labuschagne CF, van den Broek NJF, Mackay GM, Vousden KH, and Maddocks ODK. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 7: 1248–1258, 2014 [DOI] [PubMed] [Google Scholar]
- 101.Lakhkar A, Dhagia V, Joshi SR, Gotlinger K, Patel D, et al. . 20-HETE-induced mitochondrial superoxide production and inflammatory phenotype in vascular smooth muscle is prevented by glucose-6-phosphate dehydrogenase inhibition. Am J Physiol Heart Circ Physiol 310: H1107–H1117, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lapel M, Weston P, Strassheim D, Karoor V, Burns N, et al. . Glycolysis and oxidative phosphorylation are essential for purinergic receptor-mediated angiogenic responses in vasa vasorum endothelial cells. Am J Physiol Cell Physiol 312: C56–C70, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Laurent G, de Boer VCJ, Finley LWS, Sweeney M, Lu H, et al. . SIRT4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol Cell Biol 33: 4552–4561, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee DC, Sohn HA, Park Z-Y, Oh S, Kang YK, et al. . A lactate-induced response to hypoxia. Cell 161: 595–609, 2015 [DOI] [PubMed] [Google Scholar]
- 105.Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang Y-Y, et al. . Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem 278: 32100–32106, 2003 [DOI] [PubMed] [Google Scholar]
- 106.Levine AJ. and Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330: 1340–1344, 2010 [DOI] [PubMed] [Google Scholar]
- 107.Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, et al. . Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol 187: 2711–2722, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li M, Riddle S, Zhang H, D'Alessandro A, Flockton A, et al. . Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor C-terminal binding protein-1. Circulation 134: 1105–1121, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li S, Tabar SS, Malec V, Eul BG, Klepetko W, et al. . NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid Redox Signal 10: 1687–1698, 2008 [DOI] [PubMed] [Google Scholar]
- 110.Liu H, Zhang Y, Wu H, D'Alessandro A, Yegutkin GG, et al. . Beneficial role of erythrocyte adenosine A2B receptor–mediated AMP-activated protein kinase activation in high-altitude hypoxiaclinical perspective. Circulation 134: 405–421, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu JQ, Zelko IN, Erbynn EM, Sham JSK, and Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10, 2006 [DOI] [PubMed] [Google Scholar]
- 112.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 13: 572–583, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luo W, Hu H, Chang R, Zhong J, Knabel M, et al. . Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145: 732–744, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luo W. and Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2: 551–556, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, et al. . Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493: 542–546, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maddocks ODK, Labuschagne CF, Adams PD, and Vousden KH. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol Cell 61: 210–221, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Man K, Kutyavin VI, and Chawla A. Tissue immunometabolism: development, physiology, and pathobiology. Cell Metab 25: 11–26, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mannic T, Viguie J, and Rossier MF. In vivo and in vitro evidences of dehydroepiandrosterone protective role on the cardiovascular system. Int J Endocrinol Metab 13: e24660, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marelli-Berg FM, Fu H, and Mauro C. Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology 136: 363–369, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, and Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 9: 1084–1101, 2009 [DOI] [PubMed] [Google Scholar]
- 121.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, et al. . Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med 185: 670–679, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Martinez-Outschoorn UE, Lisanti MP, and Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol 25: 47–60, 2014 [DOI] [PubMed] [Google Scholar]
- 123.Mathupala SP, Ko YH, and Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta 1797: 1225–1230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McMurtry MS, Bonnet S, Wu X, Dyck JRB, Haromy A, et al. . Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95: 830–840, 2004 [DOI] [PubMed] [Google Scholar]
- 125.Mentch SJ. and Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci 1363: 91–98, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, et al. . Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481: 380–384, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mills E. and O'Neill LAJ. Succinate: a metabolic signal in inflammation. Trends Cell Biol 24: 313–320, 2014 [DOI] [PubMed] [Google Scholar]
- 128.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, et al. . Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470.e13, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mills EL. and O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol 46: 13–21, 2016 [DOI] [PubMed] [Google Scholar]
- 130.Minkin R, Sandhu G, Grosu H, Tartell L, Ma S, et al. . Desmosine and isodesmosine as a novel biomarker for pulmonary arterial hypertension: a pilot study. Am J Ther 2015. [Epub ahead of print]; DOI: 10.1097/MJT.0000000000000260 [DOI] [PubMed] [Google Scholar]
- 131.Miura G. Ischemia: succinate comes up roses. Nat Chem Biol 11: 3, 2015 [Google Scholar]
- 132.Moon J-S, Hisata S, Park M-A, DeNicola GM, Ryter SW, et al. . mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep 12: 102–115, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 133.Morgan JM, McCormack DG, Griffiths MJ, Morgan CJ, Barnes PJ, and Evans TW. Adenosine as a vasodilator in primary pulmonary hypertension. Circulation 84: 1145–1149, 1991 [DOI] [PubMed] [Google Scholar]
- 134.Mu X, Zhao T, Xu C, Shi W, Geng B, et al. . Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 8: 13174–13185, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Murray PJ. Macrophage polarization. Annu Rev Physiol 79: 541–566, 2017 [DOI] [PubMed] [Google Scholar]
- 137.Murray PJ. and Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, and Brookes PS. Acidic pH is a metabolic switch for 2-hydroxyglutarate generation and signaling. J Biol Chem 291: 20188–20197, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Navasa N, Martín I, Iglesias-Pedraz JM, Beraza N, Atondo E, et al. . Regulation of oxidative stress by methylation-controlled J protein controls macrophage responses to inflammatory insults. J Infect Dis 211: 135–145, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, et al. . Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480: 104–108, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nisimoto Y, Diebold BA, Cosentino-Gomes D, Constentino-Gomes D, and Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53: 5111–5120, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nowotarski SL, Woster PM, and Casero RA. Polyamines and cancer: implications for chemoprevention and chemotherapy. Expert Rev Mol Med 15: e3, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nutt LK. The Xenopus oocyte: a model for studying the metabolic regulation of cancer cell death. Semin Cell Dev Biol 23: 412–418, 2012 [DOI] [PubMed] [Google Scholar]
- 144.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, et al. . Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res 74: 377–387, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Okabe Y. and Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157: 832–844, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Okabe Y. and Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol 17: 9–17, 2016 [DOI] [PubMed] [Google Scholar]
- 147.Oldham WM, Clish CB, Yang Y, and Loscalzo J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab 22: 291–303, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Oliveira EC, Ribeiro ALP, and Amaral CFS. Adenosine for vasoreactivity testing in pulmonary hypertension: a head-to-head comparison with inhaled nitric oxide. Respir Med 104: 606–611, 2010 [DOI] [PubMed] [Google Scholar]
- 149.O'Neill LAJ. A critical role for citrate metabolism in LPS signalling. Biochem J 438: e5–e6, 2011 [DOI] [PubMed] [Google Scholar]
- 150.O'Neill LAJ, Kishton RJ, and Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 16: 553–565, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.O'Neill LAJ. and Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213: 15–23, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Orr AL, Quinlan CL, Perevoshchikova IV, and Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem 287: 42921–42935, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ozden O, Park S-H, Wagner BA, Yong Song H, Zhu Y, et al. . SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic Biol Med 76: 163–172, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pagano G, Talamanca AA, Castello G, Cordero MD, d'Ischia M, et al. . Oxidative stress and mitochondrial dysfunction across broad-ranging pathologies: toward mitochondria-targeted clinical strategies. Oxid Med Cell Longev 2014: 541230, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pak O, Sommer N, Hoeres T, Bakr A, Waisbrod S, et al. . Mitochondrial hyperpolarization in pulmonary vascular remodeling. mitochondrial uncoupling protein deficiency as disease model. Am J Respir Cell Mol Biol 49: 358–367, 2013 [DOI] [PubMed] [Google Scholar]
- 156.Palazon A, Goldrath A, Nizet V, and Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 41: 518–528, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, et al. . Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab 21: 65–80, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pandey D, Patel A, Patel V, Chen F, Qian J, et al. . Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol 302: H1919–H1928, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Parker SJ. and Metallo CM. Chasing one-carbon units to understand the role of serine in epigenetics. Mol Cell 61: 185–186, 2016 [DOI] [PubMed] [Google Scholar]
- 160.Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, et al. . Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab 20: 827–839, 2014 [DOI] [PubMed] [Google Scholar]
- 161.Paulin R. and Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res 115: 148–164, 2014 [DOI] [PubMed] [Google Scholar]
- 162.Pedersen PL. Voltage dependent anion channels (VDACS): a brief introduction with a focus on the outer mitochondrial compartment's roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr 40: 123–126, 2008 [DOI] [PubMed] [Google Scholar]
- 163.Pell VR, Chouchani ET, Frezza C, Murphy MP, and Krieg T. Succinate metabolism: a new therapeutic target for myocardial reperfusion injury. Cardiovasc Res 111: 134–141, 2016 [DOI] [PubMed] [Google Scholar]
- 164.Plecitá-Hlavatá L, Tauber J, Li M, Zhang H, Flockton AR, et al. . Constitutive reprogramming of fibroblast mitochondrial metabolism in pulmonary hypertension. Am J Respir Cell Mol Biol 55: 47–57, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, et al. . HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1–3 and PKM2. Stem Cells 32: 364–376, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, and Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol 308: L229–L252, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pullamsetti SS, Perros F, Chelladurai P, Yuan J, and Stenmark K. Transcription factors, transcriptional coregulators, and epigenetic modulation in the control of pulmonary vascular cell phenotype: therapeutic implications for pulmonary hypertension (2015 Grover Conference series). Pulm Circ 6: 448–464, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, et al. . Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe 12: 313–323, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, and Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287: 27255–27264, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rafikov R, Sun X, Rafikova O, Meadows ML, Desai AA, et al. . Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol 6: 278–286, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rafikova O, Meadows ML, Kinchen JM, Mohney RP, Maltepe E, et al. . Metabolic changes precede the development of pulmonary hypertension in the monocrotaline exposed rat lung. PLoS One 11: e0150480, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Randle PJ, Garland PB, Hales CN, and Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1: 785–789, 1963 [DOI] [PubMed] [Google Scholar]
- 173.Rapovy SM, Zhao J, Bricker RL, Schmidt SM, Setchell KDR, and Qualls JE. Differential requirements for L-citrulline and L-arginine during antimycobacterial macrophage activity. J Immunol 195: 3293–3300, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rathore R, Zheng Y-M, Li X-Q, Wang Q-S, Liu Q-H, et al. . Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem Biophys Res Commun 351: 784–790, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, et al. . Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64: 1266–1274, 2014 [DOI] [PubMed] [Google Scholar]
- 176.Rhodes CJ, Ghataorhe P, Wharton J, Rue-Albrecht KC, Hadinnapola C, et al. . Plasma metabolomics implicates modified transfer RNAs and altered bioenergetics in the outcomes of pulmonary arterial hypertension. Circulation 135: 460–475, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Robey IF, Lien AD, Welsh SJ, Baggett BK, and Gillies RJ. Hypoxia-inducible factor-1α and the glycolytic phenotype in tumors. Neoplasia 7: 324–330, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ryan J, Dasgupta A, Huston J, Chen K-H, and Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl) 93: 229–242, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sabharwal SS, Waypa GB, Marks JD, and Schumacker PT. Peroxiredoxin-5 targeted to the mitochondrial intermembrane space attenuates hypoxia-induced reactive oxygen species signalling. Biochem J 456: 337–346, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Saco TV, Parthasarathy PT, Cho Y, Lockey RF, and Kolliputi N. Role of epigenetics in pulmonary hypertension. Am J Physiol Cell Physiol 306: C1101–C1105, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Salminen A, Kaarniranta K, Hiltunen M, and Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal 26: 1598–1603, 2014 [DOI] [PubMed] [Google Scholar]
- 182.Sánchez-Caballero L, Guerrero-Castillo S, and Nijtmans L. Unraveling the complexity of mitochondrial complex I assembly: a dynamic process. Biochim Biophys Acta 1857: 980–990, 2016 [DOI] [PubMed] [Google Scholar]
- 183.Santidrian AF, LeBoeuf SE, Wold ED, Ritland M, Forsyth JS, and Felding BH. Nicotinamide phosphoribosyltransferase can affect metastatic activity and cell adhesive functions by regulating integrins in breast cancer. DNA Repair 23: 79–87, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, et al. . Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 897–908, 2012 [DOI] [PubMed] [Google Scholar]
- 185.Schwartz AG. and Pashko LL. Cancer prevention with dehydroepiandrosterone and non-androgenic structural analogs. J Cell Biochem Suppl 22: 210–217, 1995 [DOI] [PubMed] [Google Scholar]
- 186.Schwartz AG. and Pashko LL. Mechanism of cancer preventive action of DHEA. Role of glucose-6-phosphate dehydrogenase. Ann N Y Acad Sci 774: 180–186, 1995 [DOI] [PubMed] [Google Scholar]
- 187.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, et al. . Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7: 77–85, 2005 [DOI] [PubMed] [Google Scholar]
- 188.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol 76: 347–353, 2011 [DOI] [PubMed] [Google Scholar]
- 189.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol 76: 39–56, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shim E-H, Livi CB, Rakheja D, Tan J, Benson D, et al. . L-2-hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 4: 1290–1298, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Shimoda LA. and Laurie SS. HIF and pulmonary vascular responses to hypoxia. J Appl Physiol (1985) 116: 867–874, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, et al. . Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: D34–D41, 2013 [DOI] [PubMed] [Google Scholar]
- 193.Slaughter AL, D'Alessandro A, Moore EE, Banerjee A, Silliman CC, et al. . Glutamine metabolism drives succinate accumulation in plasma and the lung during hemorrhagic shock. J Trauma Acute Care Surg 81: 1012–1019, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Soda K. The mechanisms by which polyamines accelerate tumor spread. J Exp Clin Cancer Res 30: 95, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Song A, Zhang Y, Han L, Yegutkin GG, Liu H, et al. . Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat Commun 8: 14108, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, et al. . Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 24: 7779–7788, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, and Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104: 236–244, 28p following 244, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Stenmark KR, Tuder RM, and El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol (1985) 119: 1164–1172, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sun X, Kumar S, Sharma S, Aggarwal S, Lu Q, et al. . Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am J Respir Cell Mol Biol 50: 1084–1095, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, et al. . Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2: 44ra58, 2010 [DOI] [PubMed] [Google Scholar]
- 201.Sutendra G. and Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab 19: 558–573, 2014 [DOI] [PubMed] [Google Scholar]
- 202.Talati M. and Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ 5: 269–278, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tamada M, Suematsu M, and Saya H. Pyruvate kinase M2: multiple faces for conferring benefits on cancer cells. Clin Cancer Res 18: 5554–5561, 2012 [DOI] [PubMed] [Google Scholar]
- 204.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, et al. . Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496: 238–242, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tavana O. and Gu W. The hunger games: p53 regulates metabolism upon serine starvation. Cell Metab 17: 159–161, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, et al. . Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting complex I activity. Cell Metab 14: 768–779, 2011 [DOI] [PubMed] [Google Scholar]
- 207.Treberg JR, Quinlan CL, and Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 286: 27103–27110, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tretter L. and Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci 24: 7771–7778, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tretter L, Patocs A, and Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta 1857: 1086–1101, 2016 [DOI] [PubMed] [Google Scholar]
- 210.Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, et al. . Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl): D4–D12, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tuder RM, Davis LA, and Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 185: 260–266, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tuder RM. and Voelkel NF. Pulmonary hypertension and inflammation. J Lab Clin Med 132: 16–24, 1998 [DOI] [PubMed] [Google Scholar]
- 213.Tzounakas VL, Kriebardis AG, Georgatzakou HT, Foudoulaki-Paparizos LE, Dzieciatkowska M, et al. . Glucose 6-phosphate dehydrogenase deficient subjects may be better “storers” than donors of red blood cells. Free Radic Biol Med 96: 152–165, 2016 [DOI] [PubMed] [Google Scholar]
- 214.Valsecchi F, Grefte S, Roestenberg P, Joosten-Wagenaars J, Smeitink JAM, et al. . Primary fibroblasts of ndufs4(−/−) mice display increased ROS levels and aberrant mitochondrial morphology. Mitochondrion 13: 436–443, 2013 [DOI] [PubMed] [Google Scholar]
- 215.Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, et al. . Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep 17: 684–696, 2016 [DOI] [PubMed] [Google Scholar]
- 216.Vander Heiden MG, Cantley LC, and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Veith C, Kraut S, Wilhelm J, Sommer N, Quanz K, et al. . NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm Circ 6: 397–400, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ventetuolo CE, Baird GL, Barr RG, Bluemke DA, Fritz JS, et al. . Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med 193: 1168–1175, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, et al. . Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123: 1986–1995, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang L, Zhou H, Wang Y, Cui G, and Di L-J. CtBP maintains cancer cell growth and metabolic homeostasis via regulating SIRT4. Cell Death Dis 6: e1620, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, et al. . The transcription factor myc controls metabolic reprogramming upon t lymphocyte activation. Immunity 35: 871–882, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, et al. . Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res 99: 970–978, 2006 [DOI] [PubMed] [Google Scholar]
- 223.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, et al. . Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107: 8788–8793, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wellen KE. and Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol 13: 270–276, 2012 [DOI] [PubMed] [Google Scholar]
- 225.Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta 1817: 1833–1838, 2012 [DOI] [PubMed] [Google Scholar]
- 226.Willems PHGM, Rossignol R, Dieteren CEJ, Murphy MP, and Koopman WJH. Redox homeostasis and mitochondrial dynamics. Cell Metab 22: 207–218, 2015 [DOI] [PubMed] [Google Scholar]
- 227.Xu D, Guo H, Xu X, Lu Z, Fassett J, et al. . Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension 58: 303–309, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Xu H. and Pearl RG. Effect of l-glutamine on pulmonary hypertension in the perfused rabbit lung. Pharmacology 48: 260–264, 1994 [DOI] [PubMed] [Google Scholar]
- 229.Yang M. and Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer 16: 650–662, 2016 [DOI] [PubMed] [Google Scholar]
- 230.Yao C, Yu J, Taylor L, Polgar P, McComb ME, and Costello CE. Protein expression by human pulmonary artery smooth muscle cells containing a BMPR2 mutation and the action of ET-1 as determined by proteomic mass spectrometry. Int J Mass Spectrom 378: 347–359, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, et al. . Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337: 975–980, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhang H, Wang D, Li M, Plecita L, D'Alessandro A, et al. . Using genetics, epigenetics and small molecules to reverse metabolic reprogramming in adventitia fibroblasts for pulmonary hypertension therapy. In: B53. PHOUND IN TRANSLATION. New York, NY: American Thoracic Society, 2016, p. A3895 [Google Scholar]
- 233.Zhao L, Ashek A, Wang L, Fang W, Dabral S, et al. . Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 128: 1214–1224, 2013 [DOI] [PubMed] [Google Scholar]
- 234.Zhao L, Chen C-N, Hajji N, Oliver E, Cotroneo E, et al. . Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 126: 455–467, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhao Y, Peng J, Lu C, Hsin M, Mura M, et al. . Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS One 9: e88727, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhao YD, Chu L, Lin K, Granton E, Yin L, et al. . A biochemical approach to understand the pathogenesis of advanced pulmonary arterial hypertension: metabolomic profiles of arginine, sphingosine-1-phosphate, and heme of human lung. PLoS One 10: e0134958, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]