

Abstract
Background and Purpose
Endothelial cell‐mediated vasodilatation of cerebral arterioles is impaired in individuals with Type 1 diabetes (T1D). This defect compromises haemodynamics and can lead to hypoxia, microbleeds, inflammation and exaggerated ischaemia‐reperfusion injuries. The molecular causes for dysregulation of cerebral microvascular endothelial cells (cECs) in T1D remains poorly defined. This study tests the hypothesis that cECs dysregulation in T1D is triggered by increased generation of the mitochondrial toxin, methylglyoxal, by smooth muscle cells in cerebral arterioles (cSMCs).
Experimental Approach
Endothelial cell‐mediated vasodilatation, vascular transcytosis inflammation, hypoxia and ischaemia‐reperfusion injury were assessed in brains of male Sprague‐Dawley rats with streptozotocin‐induced diabetes and compared with those in diabetic rats with increased expression of methylglyoxal‐degrading enzyme glyoxalase‐I (Glo‐I) in cSMCs.
Key Results
After 7–8 weeks of T1D, endothelial cell‐mediated vasodilatation of cerebral arterioles was impaired. Microvascular leakage, gliosis, macrophage/neutrophil infiltration, NF‐κB activity and TNF‐α levels were increased, and density of perfused microvessels was reduced. Transient occlusion of a mid‐cerebral artery exacerbated ischaemia‐reperfusion injury. In cSMCs, Glo‐I protein was decreased, and the methylglyoxal‐synthesizing enzyme, vascular adhesion protein 1 (VAP‐1) and methylglyoxal were increased. Restoring Glo‐I protein in cSMCs of diabetic rats to control levels via gene transfer, blunted VAP‐1 and methylglyoxal increases, cECs dysfunction, microvascular leakage, inflammation, ischaemia‐reperfusion injury and increased microvessel perfusion.
Conclusions and Implications
Methylglyoxal generated by cSMCs induced cECs dysfunction, inflammation, hypoxia and exaggerated ischaemia‐reperfusion injury in diabetic rats. Lowering methylglyoxal produced by cSMCs may be a viable therapeutic strategy to preserve cECs function and blunt deleterious downstream consequences in T1D.
Abbreviations
- cECs
cerebral microvascular endothelial cells
- cSMCs
cerebral microvascular smooth muscle cells
- Glo‐I
glyoxalase‐I
- STZ
streptozotocin
- T1D
Type 1 diabetes
- VAP‐1
vascular adhesion protein‐1
Tables of Links
| TARGETS |
|---|
| Other protein targets a |
| TNF‐α |
| Enzymes b |
| VAP‐1, vascular adhesion protein‐1 |
| LIGANDS |
|---|
| ADP |
| Nitroglycerin |
| Insulin |
| Methylglyoxal |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Type 1 diabetes mellitus (T1D) is the syndrome that arises in children and adolescents when insulin‐producing beta cells of the pancreas are destroyed by their immune system (American Diabetes Association, 2010). Although a wide variety of forms of exogenous insulin is available to assist in the regulation of blood glucose in T1D to near physiological levels before and after meals (Pickup, 2015), tight glycaemic control for some is usually not achieved on a daily basis due to fears of hypoglycaemia. One of the consequences of this inadequate control is functional deterioration of the vasculature in end organs (Petitti et al., 2009).
The brain is one of the organs whose function is negatively affected by chronic or long‐term hyperglycaemia. Longitudinal, cross‐sectional studies and meta‐analyses over the past decade have confirmed declines in motor speed and psychomotor efficiency in individuals with chronic or long‐term T1D (Biessels and Reijmer, 2014; Moheet et al., 2015). These deficits are independent of hypoglycemia but correlate positively with microvascular complications, higher levels of haemoglobin A1C, oxidative stress and inflammation (Wessels et al., 2006; Kodl et al., 2008; Biessels and Reijmer, 2014; Moheet et al., 2015). Adults with chronic or long‐term T1D are also more likely to have microbleeds in brains and a stroke with long recovery times than non‐diabetic adults (Biessels and Reijmer, 2014; Woerdeman et al., 2014). The latter has been attributed in part to impaired microvascular perfusion and hypoxia, and/or disruption of microvessels and neurovascular units (Gilman 2006; Jackman and Iadecola, 2015).
Even before cognitive decline and/or a stroke occur, endothelial cell (EC)‐mediated dilator response of cerebral arterioles becomes compromised (Mayhan, 1989; Fujii et al., 1992; Brands et al., 2004). This early defect is thought to arise from increased generation of ROS that reduces the bioavailability and/or production of the EC‐derived relaxing factor NO (Shi and Vanhoutte, 2009). This surge in ROS in T1D has been attributed to increases in expression and/or activities of ROS‐producing enzymes (NADPH oxidase, cytochrome P‐450, xanthine oxidase, cyclo‐oxygenase and NO synthase) and/or a decrease in the activities of ROS‐degrading enzymes (Shi and Vanhoutte, 2009; Tomanek, 2015).
Recently, it was found that at supraphysiological concentrations, monocarbonyl and dicarbonyl species generated from glucose and fatty acid degradation can augment ROS production in cells by perturbing intracellular Ca2 + homeostasis and disrupting the electron transport chain of mitochondria (Pun and Murphy, 2012; Alomar, 2014). The most potent and best studied of these ROS‐stimulating or amplifying species is methylglyoxal (MG) (Baynes and Thorpe, 1999; Pun and Murphy, 2012; Shao et al., 2012). In all mammalian cells, methylglyoxal is constitutively generated, in high nanomolar or low micromolar amounts, from polyol pathway fluxes and oxidation of fatty acids, and it regulates a diverse array of cellular and physiological functions, including cell differentiation, proliferation, metabolism, apoptosis, anxiety, seizures and sleep (Hovatta et al., 2005; Distler et al., 2012; Distler et al., 2013; Jakubcakova et al., 2013). In T1D, methylglyoxal production increases, and it is assumed that the increase in blood methylglyoxal (Han et al., 2007) is responsible in part for the dysregulation of ECs in the vasculature of end organs, including the brain. However, to the best of our knowledge, the concentration of methylglyoxal in blood of patients and animals with T1D, typically ≤2 μM (Han et al., 2007), is significantly lower than the concentrations (well over 10μM) needed to potentiate mitochondrial ROS production, perturb intracellular Ca2 + homeostasis and impair endothelial function in vitro (Dhar et al., 2010, Shao et al., 2012). Moreover, erythrocytes contain high levels of the rate‐determining methylglyoxal‐degrading enzyme glyoxalase‐I (Glo‐I), and its cofactor reduced GSH (Sakhi et al., 2013; Mohanty et al., 2014) that rapidly degrade this diffusible electrophile and prevent its accumulation. This discrepancy led us to search for non‐blood sources of methylglyoxal that could be responsible for dysregulation of ECs in T1D.
Vascular adhesion protein‐1 (VAP‐1, AOC3, EC 1.4.3.21) is best known as an inflammation‐induced adhesin that is up‐regulated on high endothelial venules to aid in the extravasation of leukocytes from the blood into tissues (Salmi and Jalkanen, 2001). A less sialylated, but more glycosylated variant of VAP‐1, is also expressed on the membrane of smooth muscle cells (SMCs) where it functions as a Cu2 + amine oxidase enzyme that deaminates the glycine and threonine metabolite aminoacetone to produce methylglyoxal (Jaakkola et al., 1999). Earlier, Bono et al. (1999) reported increased VAP‐1 in SMCs of peripheral organs in non‐obese T1D mice. Wu and colleagues found that SMCs grown in high glucose can generate methylglyoxal (Liu et al., 2011). Nagaraj and colleagues also found that feeding‐control rats with aminoacetone increased methylglyoxal in aortic SMCs (Mathys et al., 2002). The present study was designed to determine if (i) VAP‐1 and methylglyoxal production are also up‐regulated in cerebral microvascular smooth muscle cells (cSMCs) of rats with diabetes induced by streptozotocin (STZ) that develop cerebral vascular complications comparable to those seen in T1D patients, and whether (ii) methylglyoxal produced by cSMCs would impair the functions of cerebral microvascular ECs (cECs) in cerebral arterioles in a rat model of T1D.
Methods
Synthesis of methylglyoxal
MG was synthesized using a modification of a procedure described earlier (Nemet et al., 2004). Briefly, Dowex 50 X 8 H+ ion exchange resin (5 g), methylglyoxal 1, 1‐dimethyl acetal (20 mL, 165) and water (300 mL) were refluxed for 1.5 h at 100°C. At the end of the reaction, unreacted methylglyoxal 1, 1‐dimethyl acetal and methanol were removed from the mixture using low pressure distillation (~20 mmHg with Vigreux column, 19 × 1.5 cm). methylglyoxal was removed from the ion exchange resin by filtration, and water was removed by freeze‐drying to afford a light pale yellow liquid (160 mL, yield of 60%). 1H NMR and 13C NMR spectroscopy in deuterated water (D2O) confirmed the structure of methylglyoxal (Nemet et al., 2004). The concentration of methylglyoxal was determined via titration with commercially available Glo‐I standard (1 unit of Glo‐I will react with 1.0 μmol of methylglyoxal to form 1.0 μmol of S‐lactoylglutathione).
Construction of adeno‐associated viruses
Rat Glo‐I (cat# RN208083, Origene, Rockville MD) was cloned into pZac 2.1 (a gift from Dr. Tsuneya Ikezu, Boston University MA) using Nhe1 and Xho1. The plasmid was transformed into the DH5α cells using heat shock and plated onto Luria‐Bertani (LB) agar plate containing ampicillin. The next day, single ampicillin‐resistant colonies were isolated and grown in LB broth for 24 h. Plasmid DNA was isolated from each colony using a Mini Prep kit according to the manufacturer's instructions (Qiagen, Valencia, CA, USA). Restriction enzyme mapping with Nhe1 and Xho1 and DNA sequencing confirmed the clones of interest. Clones of interest were amplified in LB broth (2 L) overnight and plasmids were isolated using Maxi Prep kits and sent to the University of Pennsylvania Vector Core facility for construction of viruses. The University of Pennsylvania Vector Core Facility created, harvested and purified rAAV2/9 containing Glo‐I driven with (AAV2/9 Endo‐Glo‐I) and GFP driven control of the endothelin‐1 promoter (AAV2/9‐GFP) under the Gene Therapy Resource Program, GTRP 1053.
Induction, verification and treatment of T1D
All animal care and experimental procedures for this study adhered to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee, University of Nebraska Medical Center. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
T1D was induced in anaesthetised male Sprague–Dawley rats using a single intravenous injection of STZ (vein under the tongue, 45 mg·kg−1 in 0.1 mL in citrate buffer pH 4.5). Aminals with blood glucose >15 mM were considered diabetic. Control animals were injected with same volume of citrate buffer only. Details of the procedures have been previously published (Shao et al., 2011; Shao et al., 2012).
Treatment protocol
One week after STZ injection, diabetic rats were randomly divided into four groups. The first group received a single intravenous injection of AAV2/9‐Endo‐Glo‐I (1.7 × 1012 viron particles·kg−1 in sterile physiological saline solution (PBS), Dia‐Endo‐Glo‐I), the second group was injected with AAV2/9‐Endo‐eGFP (Dia‐GFP), a third group was injected daily with 6–10 U·kg−1 of NPH insulin for 2 weeks, starting 5–6 weeks after STZ injection (Dia‐Ins), and a fourth group remained untreated for the duration of the 7–8 week study (Dia group). Rats injected with citrate buffer only (Con group) were divided into three groups 1 week after injection. The first and second groups were injected with AAV2/9‐Endo‐eGFP (Con‐GFP) and AAV2/9‐Endo‐Glo‐I (Con‐Endo‐Glo‐I), respectively, and the third group remained untreated (Con) for the duration of the study. Figure 1 details the experimental procedure along with the experiments.
Figure 1.
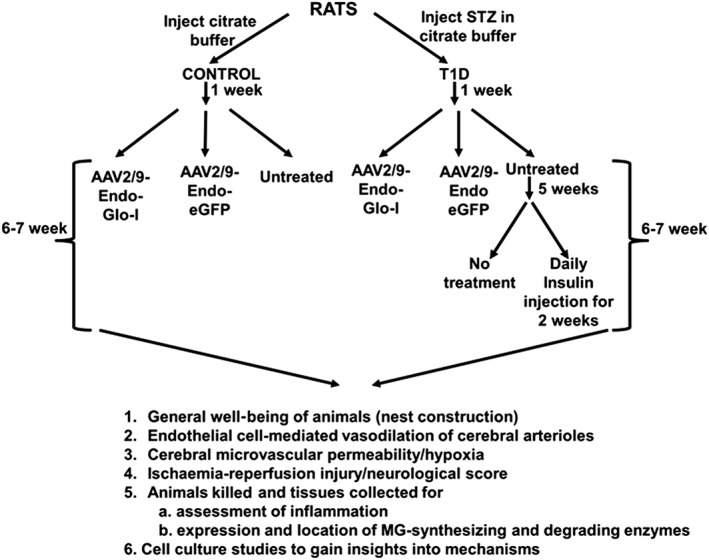
Overview of the experimental design for this study.
Overnight nest construction
The overall well‐being of Con, Dia, Con‐GFP, Con‐Endo‐Glo‐I, Dia‐GFP, Dia‐Endo‐Glo‐I and Dia‐Ins groups of rats were assessed at the end of the 7‐ 8‐week study protocol using overnight a nest construction assay (Deacon, 2012). For this, rats were transferred to clean, dry cages 2 h prior to start of the dark cycle. A piece of cotton (11 ± 0.5 g cotton, PetCo San Diego, CA) was placed in a corner of each cage. Next day, a 5‐point scale was used to assess overnight and score nest construction: shredding and movement of <10% of cotton to centre of cage = 1; shredding and movement of ~25% cotton to centre of cage = 2; shredding and movement of ~50% to the centre of cage = 3; shredding and movement of ~75% of cotton to the centre of cage = 4; shredding and movement of 100% to the middle of cage = 5.
Vasodilatation of cerebral arterioles
At the end of the 7‐ 8‐week protocol, craniectomies were performed on isoflurane‐anesthetized Con, Con‐GFP, Cont‐Endo‐Glo‐I, Dia, Dia‐GFP and Dia‐Endo‐Glo‐I rats over the left parietal cortex to visualize the cerebral microcirculation (Arrick et al., 2011). The cranial windows were suffused with artificial cerebrospinal fluid (ACF) (124 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 1 mM NaH2PO3, 26 mM NaCO3 and 5 mM glucose, pH of 7.3–7.5 at 37 degrees C) bubbled prior with 95% O2 and 5% CO2. Changes in diameter of a cerebral arteriole identified in the window (see insert between Figure 2C and D) in response to the endothelial NO synthase (eNOS)‐activating ligand, adenosine‐5‐diphosphate (ADP) (1 × 10−5 and 1 × 10−4 M) and the eNOS‐independent ligand, nitroglycerin (1 × 10−6 and 1 × 10−5 M) were determined using a video edge imaging system (Instrumentation for Physiology and Medicine, San Diego, CA).
Figure 2.
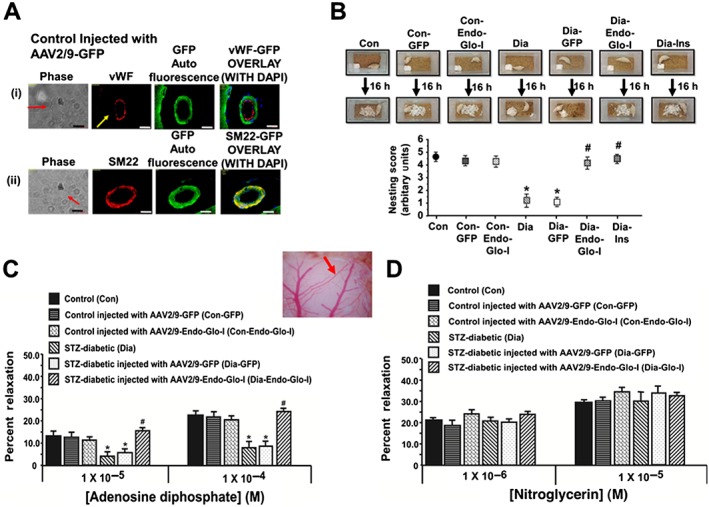
Panel A shows vWF, SM22‐α, DAPI and green auto fluorescence in cerebral microvessels from a control rat injected with AAV2–9‐GFP. Note the overlap between SM22‐α and GFP but not vWF and GFP, indicating that AAV2/9 targeted smooth muscle cells but not endothelial cells White bar at bottom of each image = 20 μm. Panels B shows overnight nest construction of animals used in the study. Graph below shows means ± SEM; n = 5–6 animals per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. Panels C and D show the responses of cerebral arterioles of animals to varying concentrations of the e‐NOS‐activating ligand ADP and the e‐NOS‐independent ligand nitroglycerin respectively. Insert between 2C and 2D shows the cerebral arteriole assayed in the study. Values shown are means ± SEM; n = 5–6 animals per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia.
Cranial windows were also prepared in a second group of Con rats and suffused with ACF containing 25 μM methylglyoxal for 30 min. After this time, the vasodilator response of the cerebral arteriole to ADP (1 × 10−5 and 1 × 10−4 M) or nitroglycerin (1 × 10−6 and 1 × 10−5 M) were measured using video imaging.
Cerebral ischaemia‐reperfusion injury
Transient middle cerebral artery (MCA) occlusion was utilized to assess ischaemia‐reperfusion insults and neurological scores using the procedure described earlier without modification (Zhao et al., 2008). For this, rats were anaesthetized with ketamine/xylazine. Anaesthesia was maintained with isoflurane (4%) in a gas mixture containing 30% oxygen/70% nitrogen via a nose cone. Rectal temperature was maintained at 37°C using a temperature controlled heating pad (TC‐1000 Temperature Controller, CWE). A Laser–Doppler flow probe (PeriFlux System 5000, Perimed) was attached to the right side of the dorsal surface of the skull (2 mm caudal and 5 mm lateral to the Bregma) to monitor regional cerebral blood flow (rCBF). A 10‐0 monofilament nylon suture was prepared by rounding its tip and coating with silicon. The right common and external carotid arteries were exposed and ligated. The MCA was occluded by inserting the filament from the basal part of the external carotid artery and advancing it cranially into the internal carotid artery to the point where the MCA branched off from the internal carotid artery. The onset of the MCA occlusion was determined by a rapid drop in rCBF. After this, the right MCA was occluded for 90 min, and reperfusion was initiated by removing the suture. The rats were allowed to recover for 24 h in the laboratory, and neurological deficit scores were assessed using the criteria detailed in Table 1 (Zhao et al., 2008). After this, the rats were anaesthetized with sodium thiobutabarbital (100 mg·kg−1) and decapitated. Brains were quickly removed and placed in ice‐cold sterile saline for 5 min and cut into six 2‐mm coronal sections. Sections were stained with 2% 2,3,5‐triphenyltetrazolium chloride. Images were digitalized, and the ischaemic lesions were quantified using Kodak Molecular Imaging Software. Infarct lesions corrected for cerebral oedema were expressed as percentage of the contra‐lateral hemisphere.
Table 1.
Neurological evaluation
| Parameters | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| Spontaneous activity (5 min) | No movement | Slight movement | Touches 1 or 2 sides of cage | Touches 3 or 4 sides of cage |
| Symmetry of movement | Left side: no movement | Left side: slight movement | Left side: moves slowly | Both side: move symmetrically |
| Response to vibrissae touch | – | No response on left side | Weak response on left side | Symmetrical response on left side |
| Floor walking | No walking | Walks in circles only | Curvilinear path | Straight path |
| Beam walking | Falls off of beam | Hugs beam | Stands on beam | Walks on beam |
| Symmetry of forelimbs (outstretching while held by tail) | Left side: no movement, no outreaching | Left side: slight movement to outreach | Left side: moves and outreaches less than right side | Symmetrical outreach |
| Climbing wall of wire cage | – | Fails to climb | Left side is weak | Normal climbing |
| Reaction to touch on either side of trunk | – | No response on left side | Weak response on left side | Symmetrical response |
Microvessel perfusion and permeability
At the end of the study protocol, Con, Dia, Dia‐Endo‐Glo‐I, Dia‐GFP and Dia‐Ins groups of rats were injected i.p. with heparin (100 unit·kg−1, i.p.); 5 min later, rats were anaesthetized (100 mg·kg−1 sodium thiobutabital i.p.), and BSA coupled to FITC (BSA‐FITC, 50 mg·kg−1 in sterile PBS buffer) was injected via a vein under the tongue (Carvey et al., 2005); 10 min after BSA‐FITC injection, animals were euthanized and decapitated, and brains were quickly removed and immersed in 4% paraformaldehyde for 16 hrs at 4°C. Brains were then transferred to 4% paraformaldehyde/30% sucrose solution for 24 h and then 30% sucrose solution for 24 h before storing at −80°C until analysed.
Cryoprotected brains were mounted using optimal temperature cutting medium and cut coronally into 30 μm slices from the olfactory bulb to the cerebellum on a cryostat and mounted onto pre‐cleaned glass slides. Slices were then washed three times with 1X PBS to remove mounting medium. Vectashield mounting medium with DAPI was then added, and slides were cover slipped and sealed. Microscopy (Zeiss ApoTome inverted fluorescence microscope or Zeiss 510 confocal microscope) was then conducted to determine the density of microvessels perfused with BSA‐FITC and impairment in vascular permeability. For measurement of the density of perfused microvessels, 200 X frames from three adjacent sections (defined regions) in Bregma approximately −3.3 were used. A perfused vessel must contain BSA‐FITC (green) in a length of ≥40 μm. Branched vessels were counted as one vessel. Impaired vascular permeability was detected when BSA‐FITC was present outside the confines of a microvessel.
Immunohistochemistry on paraformaldehyde‐fixed brain sections
Immunohistochemistry was conducted on paraformaldehyde‐fixed brains (see above) for the astrocyte marker protein, glial fibrillary acidic protein (GFAP). For this, 30 μm brain slices (Bregma −3.0 to −3.3 mm) were rehydrated with 1X PBS and permeabilized using 0.1% Triton‐X 100 for 10 min at room temperature and blocked with 10% normal goat serum for 1 h at room temperature. After this, slices were washed three times with 1X PBS (5 min) and incubated with anti‐GFAP antibodies (1:100) for 16 h at 4°C. At the end of the incubation, brain sections were washed three times with 1X PBS and incubated in the dark for 1 h with goat anti‐mouse‐Alexa Fluor 594 antibodies (1:200) at room temperature. Slices were then washed three times with 1X PBS. Vectashield mounting medium with DAPI was then added, and slides were cover slipped and sealed. Images were then taken using a Zeiss ApoTome inverted fluorescence microscope A or Zeiss 510 Confocal microscope. The number of activated astrocytes per 200 X frame was counted manually without knowledge of the treatments (blinded).
Immunohistochemistry on paraffin‐embedded brain slices
At the end of the study protocol, a second set of Con, Dia and Dia‐Endo‐Glo‐I animals were injected i.p. with heparin (100 unit·kg−1, i.p.); 3–5 min later, rats were anaesthetized (100 mg·kg−1 sodium thiobutabital, i.p.), the chest cavity was opened and animals were perfused intracardially with 100 mL of 1X PBS followed by 100 mL of 4% paraformaldehyde. After this, animals were decapitated, and brains were removed and immersed in 4% paraformaldehyde overnight at 4°C. Brains were then embedded in paraffin blocks, and serial coronal 5μm sections were cut using a sliding microtome. Brain sections were floated in a water bath at 38°C and placed onto glass slides. Slides were then drained, dried and stored at room temperature until use.
Immunohistochemical staining was conducted on paraffin‐embedded sections for the endothelial marker, vWF, the SMC marker, smooth muscle actin (SM22α), Glo‐I, VAP‐1, argpyrimidine (Arg, a surrogate marker for methylglyoxal) in brain microvessels, and for macrophage, neutrophils and microglia. For this, slides were heated in an oven at 65°C for 2 h. Paraffin was then removed with xylene (three changes, 10 min each) and rehydrated in decreasing concentrations of ethanol (100, 100, 95, 70 and 50%, and distilled water, 3 min each). Antigens were retrieved by incubating slides in 0.01 M citric acid (pH 6.39) for 40 min at 95°C. Sections were then cooled to room temperature for 20 min and then blocked in 10% normal horse serum for 1 h at room temperatureand incubated overnight at 4°C with the respective primary antibody combinations. Next day, sections were incubated with the corresponding secondary Alexa Fluor coupled antibodies or with HRP‐coupled antibodies, followed by 3,3‐diaminobenzidine (for 2–5 min). Sections were then washed with PBS and mounted with Prolong Gold Mountant Anti‐faid containing DAPI. Images were then taken with either a Zeiss ApoTome inverted fluorescence microscope A or Zeiss 510 Confocal microscope. A Nuance EX multispectral imaging system (Cambridge Research Instruments, Worburn, MA) fixed to a Nikon Eclipse E800 and image analysis software (Caliper Life sciences, Inc., a Perkin Elmer Company, Hopkinton, MA) was used for quantitate changes of Glo‐I, vWF, VAP‐1 expression and Arg level among different animal groups according to an earlier procedure (Dash et al., 2011). The number of activated microglia and macrophage and neutrophils was counted in three consecutive 200 X frames, without knowledge of the treatments (blinded).
Determination of methylglyoxal levels
Methylglyoxal concentrations in serum and brain homogenates from Con, Dia and Dia‐Endo‐Glo‐I groups of rats were determined using the procedure described earlier (Hasim et al., 2014). Briefly, brain (Bregma −2.7 to −3.5, ~100 mg), serum (500 μL) and cerebral microvessels (50 mg, freshly isolated) were placed in 3 X weight/volume of cold isolation buffer containing 0.3 M sucrose, 10 mM histidine and 230 μM phenylethylsufonyl fluoride (dissolved in 100% ethanol), pH 7.4, and placed on ice for 10–15 min to chill. At the end of this time, samples were sonicated for 3 × 5 s (15 s interval between sonication). The sonicated samples were mixed with equal volumes of perchloric acid (5.0 M) and placed on ice for 10 min. The samples were then centrifuged in an Eppendorf 5417R centrifuge for 10 min at 1300 × g. Supernatants (50 μL) were then added to 150 μL of 0.1 M O‐phenyldiamine in 1.6 M perchloric acid and allowed to react overnight at room temperature in the dark. The reaction product, 2‐methylquinoxaline, was extracted with three times with 2 mL each of chloroform, and water was removed from the chloroform with anhydrous sodium sulfate (0.5 g). Chloroform was then removed from each sample using a gentle stream of nitrogen. The dried samples were then resuspended in 500 μL methanol and analysed by HPLC (SCL‐Shimadzu 10 A) employing a Kinetex 5 μ C18 100R column with a mobile phase consisting of methanol : water:trifluoroacetic acid (52:48:0.1) at flow rate of 1 mL·min−1, and wavelength of 312 nm to determine the amount of 2‐methylquinoxaline in each sample. A calibration curve using commercially available 2‐methylquinoxaline from 0.5 to 100 μM was used for the quantification. Reaction efficiency and 2‐methylquinoxaline recovery was 90%.
Methylglyoxal levels in the culture media of human brain microvascular smooth muscle cells (ScienCell, Carlsbad, CA, USA) grown in 5.5 mM and 25.0 mM glucose for 0, 6, 12 and 24 h were also assayed, using the procedure described above.
Glo‐I activity
Glo‐I activities in brain tissues and cerebral microvessels from Con, Dia, Dia‐Endo‐Glo‐I and Dia‐GFP animals were determined spectrophotometrically (McLellan and Thornalley, 1989). Briefly, brain (Bregma −2.7 to −3.5, ~100 mg) and cerebral microvessels (~50 mg) were placed in 300 μL of cold isolation buffer (0.3 M sucrose, 10 mM histidine and 230 μM phenylethylsufonyl fluoride, pH 7.4) and placed on ice for 10–15 min to chill. Samples were sonicated for 3 × 5 s (15 s intervals wait in between sonications) and centrifuged for 10 min at 1300 × g, and protein concentrations in supernatants were determined. In a 1 mL quartz cuvette, 250 μL of 0.1 M NaPO4 buffer, pH 6.6, 233 μL of distilled water, 25 μL of 4 mM methylglyoxal and 50 μL of 2 mM reduced GSH were mixed, and the absorbance at 240 nm was measured. Thereafter, 500 μg of brain homogenate or 100 μg cerebral vessel homogenate was added, solutions were mixed and absorbance was recorded after 60 and 120 s at room temperature using a spectrometer (Smart Spec 3000, BioRad Inc, Burlingame CA).
A standard curve was generated using commercially available Glo‐I standard (1.63 units·ug−1) and used for quantification. For this, 250 μL of 0.1 M NaPO4 buffer, pH 6.6, 233 μL of distilled water, 25 μL of 4 mM methylglyoxal and 50 μL of 2 mM reduced GSH were mixed in a 1 mL quartz cuvette, and absorbance was measured at 240 nm. Thereafter, Glo‐I (0, 0.5, 1.0, 1.5, 2 and 5 units) was added, and absorbance changes were recorded after 60 and 120 s. Glo‐I activity in brain lysates was interpolated from Glo‐I calibration curve. One unit of enzyme corresponded to 1 μmol of S‐D‐lactoylglutathione formed min−1·mg−1 protein.
GSH and enzymes that regulate GSH production
GSH was measured in brain lysates using a commercial kit according to the manufacturer's protocol (Oxis Research, Portland, OR). Briefly, cut brain pieces (Bregma −2.7 to −3.5) from Con, Dia and Dia‐Endo‐Glo‐I animals were sonicated in 5% metaphosphoric acid and centrifuged at 4°C for 20 min (3000 × g) to precipitate proteins. Supernatants from each group of samples were collected, and total GSH [GSH + GSSG] was determined by measuring the formation of 2‐nitro‐5‐thiobenzoic acid at 412 nm (25°C) in the presence of 5,5′‐dithio‐bis‐(2‐nitrobenzoic acid), NADPH, GSH reductase and EDTA. GSSG was also determined by derivatizing supernatants with 1‐methyl‐2‐vinylpyridium trifluoromethane sulfonate and assaying the derivatized samples as above for total GSH. Standard curves for GSH and GSSG were constructed, and the concentration of GSH was calculated by subtracting the GSSG concentration from total GSH (GSH + GSSG). Measured concentrations of GSH and GSSG were expressed in μmol·L−1·mg−1 protein and as a ratio (GSH/GSSG).
GSH reductase and γ‐glutamylcysteine ligase were also measured in brain tissues from Con, Dia and Dia‐Endo‐Glo‐I animals. For GSH reductase activity, brain tissues were homogenized in ice‐cold Tris buffer (0.1 mol·L−1, pH 8.0, with 2 mM EDTA) and centrifuged at 4°C (6000 × g) for 30 min as described earlier (Carlberg and Mannervik, 1995). A 200 μL aliquot of the supernatant was added to a cuvette containing KH2PO4 buffer (0.2 M, pH 7.0) plus 2 mM EDTA, 20 mM GSSG and 2 mmol·L−1 NADPH. The change in absorbance at 340 nm was monitored for 5 min at 25°C using a spectrophotometer. A milliunit (mU) of GSH reductase activity was defined as the amount of enzyme catalysing the reduction of 1 nmol NADPH min−1.
γ‐Glutamylcysteine ligase activity was determined using the method of Seelig and Meister (1995). Brain tissue homogenates were prepared as above, and a 50‐μL aliquot of the supernatant from each of Con, Dia and Dia‐Endo‐Glo‐I animals was added to a reaction mixture containing 0.1 M Tris buffer and (in mM) 150 KCl, 5 Na2ATP, 2 phosphoenolpyruvate, 10 L‐glutamate, 10 L‐α‐aminobutyrate, 20 MgCl2, 2 Na2‐EDTA, 0.2 NADH, 17 μg pyruvate kinase and 17 μg lactate dehydrogenase. The change in absorbance at 340 nm was monitored for 5 min at 25°C, and γ‐glutamylcysteine ligase activity was expressed in milliunits, defined as the activity converting 1 nmol of NADH to NAD min−1.
Measurement of TNF‐α
Total RNA was extracted from brain tissues (Bregma −2.7 to −3.5, ~100 mg) using Trizol and purified using the spin column purification process, RNeasy Mini Kit according to the manufacturer's protocol (Qiagen; Germantown, MD). RNA samples were treated with DNase I to remove any contamination DNA. RNA concentrations and purity were measured at OD 260 and OD 280. Total RNA with OD260/OD280 greater than 1.8 (1 μg) were reverse‐transcribed into cDNA using MLV reverse transcriptase (Invitrogen Corporation, Carlsbad, CA, USA) in 20 μL volume. cDNA of each sample was used in Taqman real‐time PCR to obtain the CT value for TNF‐α in an ABI sequence detector (Applied Biosystems, Foster City, CA, USA). The real‐time PCR for β‐actin was run in parallel and served as the PCR control. Each sample was run in triplicate in PCR to determine gene expression of TNF‐α. Primers used for TNF‐α were 5′ ATG‐AGC‐ACT‐GAA‐AGC‐ATG‐AT and 3′ CTC‐FIG‐ATG‐GCA‐GAG‐AGG‐AG (Integrated DNA Technologies, Coralville, IA). Relative gene expression was calculated from the CT values using the Δ CT method.
Western blot analyses
Cerebral microvessels were isolated from rat brains using the method described by Kowaloff et al. (1980) with the exception that 50 μm nylon mesh filters were used to isolate microvessels. Isolated microvessels were lysed, and Western blot assays were conducted to determine steady state levels of Glo‐I, VAP‐I and actin. Brain (Bregma −2.7 to −3.5) lysates, prepared as described above, were also used to determine p‐65 (NF‐κB) and phospho‐p65 (Ser536, NF‐κB) levels using Western blot assays.
Nitric oxide generation in ECs
ECs were isolated from the abdominal aorta and used for assessment of ability to release the vasorelaxing factor NO. For this, animals were anaesthetised, abdominal cavities were opened and abdominal aortas were collected. A longitudinal incision was made on each aorta and a cotton swab was used to gently scrape the ECs and place then into polylysine‐coated dishes containing EC growth medium (Lonza, USA) for 2 h. After this time, cells were incubated with 10 μM 4,5‐diaminofluorescein diacetate (DAF‐2 diacetate; Sigma‐Aldrich) for 30 min and then washed three times with EC growth medium to remove extracellular DAF‐2‐diacetate. Cells were then placed on the stage of a Zeiss LSM 710 Meta laser scanning microscope, with excitation at 488 nm, and emissions were monitored at 516 nm, and basal DAF‐2 fluorescence was measured. Cells were then stimulated with acetylcholine (10 μM), and images were taken every 1 s for 5 min for changes in DAF fluorescence. For some experiments, ECs isolated from aortas were pretreated with 5 μM of the eNOS inhibitor L‐N G‐nitroarginine methyl ester before addition of acetylcholine. DAF‐2 fluorescence was quantified with Image J analysis software (http://rsbweb.nih.gov/ij/).
Measurement of ROS by electron paramagnetic resonance (EPR) spectroscopy
Freshly isolated cerebral microvascular vessels (≤50 μm in diameter, 50 μg protein) were resuspended in Krebs‐HEPES buffer (in mM: 99.0 NaCl, 4.69 KCl, 2.5 CaCl2, 1.2 MgSO4, 25 NaHCO3, 1.03 KH2PO4, 5.6 D‐glucose, 20 Na‐HEPES) supplemented with metal chelators DETC (2 μM) and deferoxamine (DF, 50 μM). Immediately after adding the spin probe 1‐hydroxy‐3‐methoxycarbonyl‐2, 2, 5, 5‐tetramethylpyrrolidine (CMH; 200 μM), microvessels were placed into 50 μL glass capillary tubes. Using a Bruker E‐scan EPR spectrophotometer (Bruker BioSpin GmbH, Rheinstetten/Karlsruhe, Germany), total ROS levels at 37°C were determined with the following EPR settings: field sweep 60.0 G; microwave frequency 9.746 GHz; microwave power 21.90 mW; modulation amplitude 2.37 G; conversion time 10.24 ms; time constant 40.96 ms.
Effects of methylglyoxal on viability of ECs and SMCs
Human cECs (hcECs) and human cerebral microvascular SMCs (hcSMCs; 1 × 106, ScienCell, Carlsbad, CA) were incubated with varying concentrations of methylglyoxal (1–1000 μM) for 24 h at 37°C. At the end of this time, cell viability was determined using MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay kit according to the manufacturer's instructions (EMD Millipore, Billerica, MA). Western blots were also conducted for expression of the claudin‐5 and occludin in hcECs exposed to 25, 50 and 100 μM methylglyoxal.
Relative levels of Glo‐I were also determined in hcECs and hcSMCs. For this, cells were harvested in isolation buffer (0.3 M sucrose, 10 mM histidine and 230 μM phenylethylsufonyl fluoride, pH 7.4) and sonicated for 3 × 5 s (15 s intervals wait in between sonication). Samples were then centrifuged for 10 min at 1300 × g. The supernatants were removed, and protein concentrations were determined. Western blots were then conducted to determine steady state levels of Glo‐I protein.
Effects of methylglyoxal on mitochondrial ROS production
The procedure described by Shao et al. (2012) was used without modification. Briefly, hcECs were loaded with the mitochondria‐localizing probe MitoTracker Green (100 nM, Life Technologies, Grand Island NY), followed by the fluorogenic, mitochondria‐targeted, ROS probe MitoSOX™ Red (2 μM), for 15 min each. After this, cells were washed three times and placed on the stage of a Zeiss LSM 510 Meta laser scanning microscope, with excitation at 488 nm to capture fluorescence images of MitoSOX Red. MitoTracker Green was excited at 488 nm, and emissions were monitored at 516 nm. Methylglyoxal (30 μM) was then added, and images were taken every 5 s for 10 min. For some experiments, bECs were pretreated with manganese (III) tetrakis (4‐benzoic acid) porphyrin chloride (MnTBAP), a cell‐permeable SOD mimetic for 30 min, and then, methylglyoxal was added. MitoSOX Red fluorescence was quantified with ImageJ analysis software (http://rsbweb.nih.gov/ij/).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data shown are means ± SEM. Differences among each of the seven groups of rats (Con, Con‐GFP, Con‐Endo‐Glo‐I, Dia, Dia‐GFP, Dia‐Endo‐Glo‐I and Dia‐Ins) were evaluated using Prism GraphPad 6 (La Jolla, CA), using one‐way ANOVA followed by Bonferroni's post hoc test to determine if there was significant difference between control and diabetic rats and whether these changes were attenuated with treatment. Results were considered significantly different if P < 0.05 (95% confidence interval).
Materials
Primary antibodies were obtained from AbCam Inc, Cambridge MA (von Willerbrand factor (vWF) sheep polyclonal, cat# ab11713; and smooth muscle actin α, SM22, rabbit polyclonal, cat# ab14106, goat polyclonal, cat# ab10135 and F4/80 (SP115), cat# MA5–16 363, IBA1 antibody, cat# ab5076), DAKO, Carpinteria, CA (vWF, rabbit polyclonal, cat# A0082), JaiCA, Shizuoka, Japan (argpyrimidine (MG surrogate), cat# JAI‐MMG‐030 N), Santa Cruz Biotechnology Inc., Santa Cruz, CA (Glo‐I antibodies FL‐184, rabbit polyclonal, cat# SC‐67 351; VAP‐1 antibodies, E‐19, goat polyclonal, cat # sc‐13 741; and actin antibodies 1–19, goat polyclonal cat# SC‐1616), CalBiochem/EMD Bioscience, Billerica, MA (VAP‐1 antibodies TK8–14, mouse monoclonal, cat# OP190), Millipore, Temecula, CA (anti‐glial fibrillary acidic protein, GFAP, rabbit monoclonal‐GA5, cat# MAB3402), Invitrogen Life Technologies, Grand Island NY (occludin, rabbit polyclonal, cat# 71–1500), Accurate Chemical and Scientific Cooperation, Westbury, NY (polymorphonuclear neutrophil (PMN, cat# AIL395) and Cell Signaling Technologies, Danves MA (NF‐κB p65 cat #8242 and phospho‐p65 (Ser536, NF‐κB), cat#3031). Secondary antibodies were obtained from Invitrogen Life Technologies (goat anti‐mouse IgG coupled to Alexa Fluor 594, cat# A‐11 032; chicken‐anti‐rabbit IgG coupled to Alexa Fluor 488, cat# A‐21 441; donkey anti‐goat IgG coupled to Alexa Fluor 488 cat# A‐11 055, donkey anti‐goat IgG coupled to Alexa 594 cat# A11058; donkey‐anti‐sheep IgG coupled to Alexa Fluor 594, cat# A‐11 016), Santa Cruz Biotechnology, Inc. Santa Cruz, CA (Donkey anti rabbit IgG‐HRP, cat# sc‐2305 Donkey anti goat IgG‐HRP cat# sc‐2304) and Vector Laboratories, Burlingame, CA (Fluorescein rabbit anti‐rat IgG cat# FI‐4000, and peroxidase‐conjugated anti‐mouse IgG).
BSA labelled with FITC (BSA‐FITC, cat# A9771), 2,3,5‐triphenyltetrazolium chloride (cat# T8877), Dowex 50 X 8 H+ ion exchange resin (cat # 217 492), methylglyoxal 1,1‐dimethyl acetal (cat# 170 216), sodium thiobutabarbital (Inactin® cat# T133), 3,4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT, cat# M2128) and Glo‐I (cat# G4252–500, nitroglycerin (cat # 146605) and adenosine‐5‐diphosphate (cat # 10905)) were obtained from Sigma‐Aldrich (St Louis, MO, USA). 3,3 diaminobenzidine Substrate Kit was from (Vector Laboratories, Burlingame, CA, cat# SK‐4100). Neutral protamine hagedorn (NPH) insulin was from Eli Lilly, Indianapolis, IN. All other reagents used were of the highest grade commercially available.
Results
Establishing that AAV2/9 targets cSMCs
Immunohistochemical and immunofluorescence assays were first conducted to confirm that our custom‐designed AAV2/9 virus exhibited selective tropism for SMCs in the cerebral microvasculature of rats following a single intravenous injection. Figure 2A shows that injecting Con rats with AAV2/9‐Glo‐I leads to increased GFP fluorescence in cSMCs. This green fluorescence did not overlap with the red immunofluorescence of vWF, a marker for ECs, but it did with SM22‐α, a marker for SMCs. Similar results were seen in more than 30 brain arterioles investigated from 5 rats. Having established our AAV2/9 exhibited preferential tropism for SMCs in the cerebral microvasculature (cSMCs) of rats, we then used this custom‐designed AAV2/9 to test our hypothesis that methylglyoxal generated by cSMCs of cerebral arterioles was dysregulating juxtaposed cECs.
General characteristic of animals used in the study
The general characteristics of animals used for this study are listed in Table 2. The results show that diabetic rats had significantly elevated blood glucose and HbA1c levels, serum thiobarbituric acids reactive substances, semicarbazide‐sensitive amine oxidase activity and serum and brain methylglyoxal concentration. They also had significantly lower body mass and blood insulin level. In Dia‐Endo‐Glo‐I and insulin‐treated animals, these measured parameters were partially restored.
Table 2.
General Characteristics of animals
| Parameter | Con | Con‐GFP | Con‐Endo‐Glo‐I | Dia | Dia‐GFP | Dia‐Endo‐Glo‐I | Dia‐Ins |
|---|---|---|---|---|---|---|---|
| n = 18 | n = 18 | n = 18 | n = 20 | n = 20 | n = 20 | n = 20 | |
| Body mass (g) | 415 ± 10 | 401 ± 11 | 388 ± 15 | 275 ± 15a | 290 ± 12a | 320 ± 10b | 390 ± 10b |
| Blood glucose (mmol·L−1) | 5.5 ± 1.6 | 6.3 ± 1.7 | 6.6 ± 2.4 | 25.6 ± 4.5a | 24.1 ± 3.5 | 22.9 ± 3.2 | 11.1 ± 3.3b |
| % Glycated haemoglobin (HbA1c) | 4.2 ± 0.3 | 4.3 ± 0.4 | 4.4 ± 0.5 | 7.6 ± 0.2a | 7.5 ± 0.3a | 7.2 ± 0.5 | 5.2 ± 0.2b |
| Serum insulin (ng·mL−1) | 0.9 ± 0.2 | 1.0 ± 0.1 | 0.9 ± 0.2 | 0.3 ± 0.1a | 0.3 ± 0.1a | 0.6 ± 0.1b | 1.2 ± 0.2b |
| Serum TBARS (nmol·mL−1) | 2.4 ± 0.3 | 2.5 ± 0.6 | 2.7 ± 1.0 | 12.0 ± 2.1a | 11.5 ± 1.9a | 7.5 ± 1.0b | 4.0 ± 1.1b |
| SSAO activity (units·ml−1·min−1) | 0.28 ± 0.02 | 0.26 ± 0.03 | 0.3 ± 0.02 | 0.71 ± 0.05a | 0.65 ± 0.06a | 0.60 ± 0.03b | 0.41 ± 0.05b |
| Serum methylglyoxal (μM) | 0.3 ± 0. | 0.3 ± 0.1 | 0.2 ± 0.1 | 1.2 ± 0.2a | 1.4 ± 0.2a | 1.2 ± 0.3 | 0.7 ± 0.2b |
| Brain methylglyoxal (μmol·(100 mg)−1) | 2.5 ± 0.4 | 2.4 ± 0.6 | 2.2 ± 0.5 | 30.2 ± 4.0a | 28.6 ± 3.8a | 1.2 ± 0.5b | 3.5 ± 0.6b |
P < 0.05, significantly different from control.
P < 0.05, significantly different from diabetic.
TBARS, thiobarbituric acid‐reactive substances; SSAO, semicarbazide‐sensitive amine oxidase.
Overnight nest construction
Nest building, which requires the complex interplay of attention, decision‐making, motor function, and orofacial and forelimb movement were used to assess overall well‐being of animals at the end of the study. After 7–8 weeks of diabetes, the ability of rats to shred the cotton and create a nest in the centre of the cage overnight was significantly compromised (Figure 2B). Administering insulin for 2 weeks after 6 weeks of T1D to achieve a euglycaemic state, restored nest construction capability (Figure 2B), indicating that impairment in nest construction resulted from the hyperglycaemia (T1D) and not from STZ toxicity. Administering a single dose of AAV2/9‐Endo‐Glo‐I 1 week after the onset of T1D (Dia‐Endo‐Glo‐I) preserved their ability to construct a nest overnight. Diabetic animals injected with a single dose of AAV2/9‐GFP 1 week after the onset of T1D continued to show impaired nest construction ability. Injection of AAV2/9‐GFP or AAV2/9‐Endo‐Glo‐I in control rats had no effects on their ability to construct nests.
EC‐mediated vasodilatation of cerebral arterioles
The responses of a cerebral arteriole to acute challenges with ADP or nitroglycerin were used to discern between impairment in EC‐mediated vasodilatation and SMC‐mediated vasodilatation respectively. The specific arteriole assayed in this study is shown in the insert between Figure 2C and D. Figure 2C shows an attenuated response of this arteriole from the Dia group of rats, to ADP but not to nitroglycerin (Figure 2D); establishing EC‐mediated but not SMC‐mediated vasodilatation was impaired in T1D. Administering a single injection of AAV2/9‐Endo‐Glo‐I 1 week after the onset of T1D blunted impairment in EC‐mediated vasodilatation of cerebral arterioles (Figure 2C). AAV2/9‐Endo‐Glo‐I administration did not alter nitroglycerin‐induced vasodilatation in the Dia group of rats (Figure 2D). Administering AAV2/9‐GFP to diabetic rats did not blunt endothelial dysfunction (Figure 2C). Administering AAV2/9‐GFP and AAV2/9‐Endo‐Glo‐I to the Con group of rats did not affect the vasodilatation of cerebral arterioles, mediated by cECs or by cSMCs (Figure 2C and D).
Suffusing the cranial window of Con rats with ACF containing 25 μM methylglyoxal for 30 min impaired EC‐mediated vasodilatation to an extent similar to that seen in the Dia group of rats. Interestingly, this amount of methylglyoxal did not attenuate SMC‐mediated vasodilatation of cerebral arteriole (Figures 3A), as seen in the Dia group of rats (Figure 2C and D).
Figure 3.
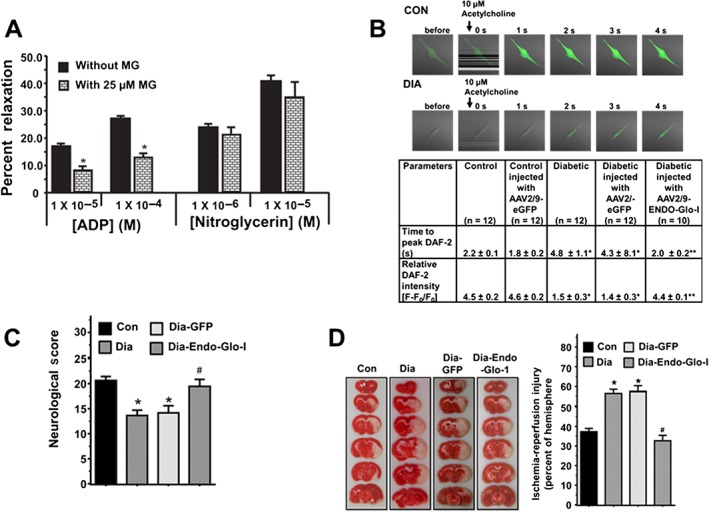
Panel A shows the responses of cerebral arterioles from Con animals pretreated with 25 μM methylglyoxal for 30 min to varying concentrations of the ADP and nitroglycerin. Values shown are means ± SEM; n = 6 separate animals. * P < 0.05, significantly different from no methylglyoxal pretreatment. Panel B upper panels show representative images for NO generation in live cells from Con and Dia aortic endothelial cells stimulated with 10 μM acetylcholine. Lower table shows time to peak NO production and the amount of NO generated (fluorescence amplitude, [(F‐F0)/F0]) following acetylcholine stimulation in aortic endothelial cells from Con, Con‐GFP, Dia, Dia‐GFP and Dia‐Endo‐Glo‐I animals. Values shown are means ± SEM; n = 12 cells from three separate animals. * P < 0.05, significantly different from control. ** P < 0.05, significantly different from Dia. Panel B shows neurological score of animals 24 h after a 1.5‐h MCA occlusion. Values shown are means ± SEM; n = 5–6 rats per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. Panel C shows quantification of infarct size in brains of animals 24 h after a 1.5‐h mid‐cerebral artery occlusion. Values shown are means ± SEM; n = 5–6 rats per group. * P < 0.05, significantly different from Con.
Nitric oxide production
The increased responsiveness of cerebral arterioles from AAV2/9‐Endo‐Glo‐I‐treated animals to the eNOS‐activating ligand ADP prompted us to investigate whether AAV2/9‐Endo‐Glo‐I‐treatment attenuated loss of production or bioavailability of NO in ECs. In Figure 3B, the upper panels show representative images for NO generation in live cell recordings from aortic ECs, taken from the Con or Dia groups of rats and stimulated with 10 μM acetylcholine. Time to peak NO production and the amount of NO generated (fluorescence amplitude, [(F‐F0)/F0]) following acetylcholine stimulation were significantly reduced in aortic ECs from the Dia animals compared with those from Con animals. Treating Dia animals with AAV2/9‐eGFP did not restore acetylcholine‐stimulated NO production in aortic ECs (Figure 3B, table below figure). However, treating Dia animals with AAV2/9‐Endo‐Glo‐I restored time to peak NO generation and the amount of NO to near that in control animals. Treating Con animals with AAV2/9‐eGFP did not alter time to peak acetylcholine‐stimulated NO or the amount of NO generated (Figure 3B, table below figure).
Ischaemia‐reperfusion injury following transient MCA occlusion
Mean neurological score averaged over the eight criteria listed in Table 1, 24 h after a transient MCA occlusion was significantly reduced in Dia rats compared with the the Con group of rats, with similar cerebral blood flow (rCBF) (Figure 3C). Ischaemia‐reperfusion injury in the occluded hemisphere of the Dia rats was also significantly larger than that in the Con group of rats (Figure 3D; P < 0.05). Injecting rats with AAV2/9‐Endo‐Glo‐I 1 week after onset of T1D blunted the exacerbated ischaemia‐reperfusion damage and significantly improved mean neurological score (Figure 3C and D). Injection of AAV2/9‐GFP did not improve neurological scores or reduce ischaemia‐reperfusion injury in Dia rats (Figure 3C and D). Injecting Con rats with AAV2/9‐Endo‐Glo‐I did not increase ischaemia‐reperfusion injury (data not shown).
Microvessel permeability and perfusion
Blunting of ischaemia‐reperfusion damage by AAV2/9‐Endo‐Glo‐I prompted us to investigate further, whether the density of microvessels perfused was altered in the Dia group of rats. When injected into a vein, the fluorescent‐labelled protein BSA‐FITC should distribute to all microvessels that are actively perfused, and its fluorescence within the confines of vessels was used as an index of the density (number) of perfused microvessels. In this study, the number of microvessels (≤10 μm) with BSA‐FITC per 200 X frame within Bregma −3.0 to −3.3 was significantly lower in Dia, compared with Con rats (Figure 4A and B). The number of microvessels (≤10 μm) perfused with BSA‐FITC per 200 X frame in Dia‐Endo‐Glo‐I rats was similar to those of Con animals. Injecting Dia rats with insulin also increased the density of microvessels, with BSA‐FITC per frame, in cortex and hippocampus but not thalamus (Figure 4A and B). Injecting Dia rats with AAV2/9‐GFP did not increase the density of microvessels perfused with BSA‐FITC (Figure 4A and B).
Figure 4.
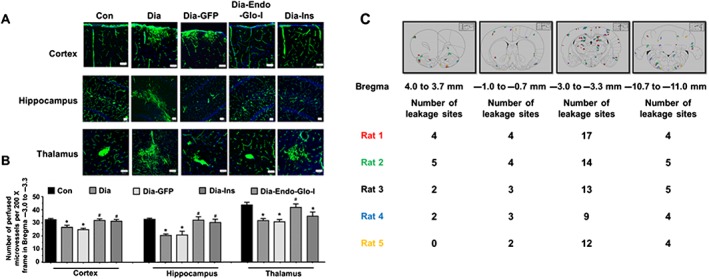
Panel A shows representative images of BSA‐FITC within the confines of microvessels in cortex, hippocampus and thalamus (Bregma −3.0 to −3.3) used as an index of the density of microvessel perfusion in Con, Dia, Dia GFP, Dia‐Endo‐Glo‐I and Dia‐Ins rats. White bar at bottom of each image = 50 μm. Panel B shows summary data from 5 animals per group as means ± SEM. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. Panel C shows sites of BSA‐FITC leakage in the various brain regions (upper panel) obtained from 5 separate animals. Data from each rat is colour‐coded for easy visualization. Table below shows the number of leakage sites within each region of the brain.
During analysis of microvessel perfusion within Bregma −3.0 to −3.3 of Dia rats, we also noticed BSA‐FITC was leaking from the microvessels, and this prompted us to scan the whole brain for BSA‐FITC transcytosis. In the five Dia rats investigated, the mean number of leakage sites within Bregma 4.0–3.7 mm was 2.6 ± 0.8; within Bregma 0.7 to −1.0 mm, 3.2 ± 0.4 leakage sites were detected; within Bregma −3.0 to −3.3 there were 13.0 ± 1.6 leakage sites; and within Bregma −10.7 to −11.0 mm there were 4.7 ± 0.1 leakage sites (Figure 4C). Data from each rat are colour‐coded for clarity, in Figure 4C.
More detailed analyses within Bregma −3.0 to −3.3 of Dia rats indicated that BSA‐FITC was not only leaking from microvessels with diameters ≤10 μm but was also leaking from a limited number of arterioles, ranging in size from ~20 μm to ~50 μm, resembling microbleeds (Figure 5B, lower panels). In brains of Con rats, little or no BSA‐FITC transcytosis was observed within Bregma −3.0 to −3.3 (Figure 5A). Injecting rats with AAV2/9‐Endo‐Glo‐I 1 week after the onset of T1D blunted BSA‐FITC transcytosis 7–8 weeks later (Figures 5C and 5D). Injecting Dia rats with AAV2/9‐GFP did not prevent BSA‐FITC transcytosis (Figure 5D). Injecting Dia rats with insulin for 2 weeks to achieve a euglycaemic state, blunted BSA‐FITC transcytosis in all brain regions except the thalamus (Figures 4A and B).
Figure 5.
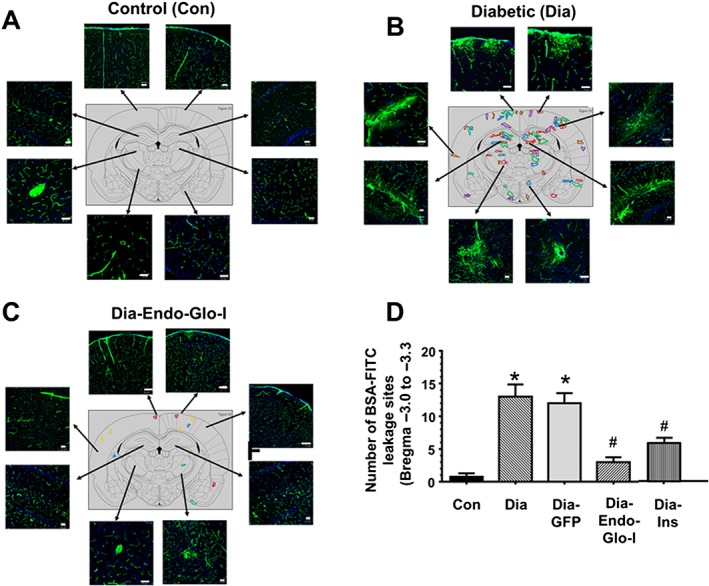
Panels A–C show representative BSA‐FITC images from different regions within Bregma −3.0 to 3.3 from Con, Dia and Dia‐Endo‐Glo‐I rats. Panel D shows the mean ± SEM of BSA‐FITC transcytosis sites within Bregma −3.0 to −3.3 from 6 rats per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. White bar at bottom of each image = 50 μm. Each colour in brain map represents a different animal.
Glial activation
Astrogliosis and microgliosis are markers of inflammatory responses in the brain (Zhang et al., 2010; Ma et al., 2016). To assess this, immunohistochemistry was conducted for GFAP and IBA1, and qPCR and Western blot analyses were used to determine the amount of the inflammatory marker, TNF‐α mRNA, and the phosphorylated form of oxidant transcription factor, NF‐κB (p‐p65) respectively. In brains from control animals, GFAP staining was punctate and confined to the cytoplasm of the astrocyte (Figure 6A, left). However, in brains of diabetic animals, GFAP staining transformed to ‘star‐shaped’, characteristic of activation (Figure 6A, middle). Note in the example shown, the star‐shaped GFAP staining in brain of Dia rats was around an arteriole (40 μm) from which BSA‐FITC was permeating (green, Figure 6A, middle panel). Figure 6B also shows a significant increase in the number of IBA1 positive cells in parenchyma of Dia rats. Data shown are from a thalamic region within Bregma −3.0 to −3.3, but similar data were seen in other regions of the brain with increased BSA‐FITC transcytosis. Injecting Dia rats with AAV2/9‐Endo‐Glo‐I significantly blunted the number of activated astrocytes and microglia (Figure 6A and B). Injecting Dia rats with AAV2/9‐GFP did not attenuate astrocyte and microglia activation (data not shown).
Figure 6.
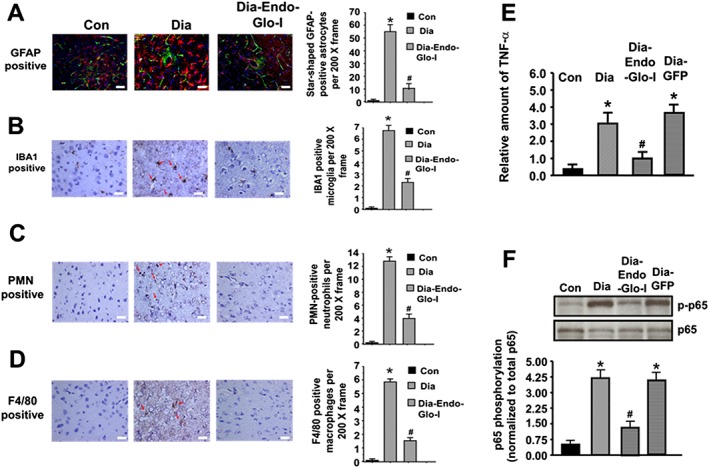
Panels A‐D show representative images for GFAP‐staining (activated astrocytes, red staining), activated microglia (IBA1 positive, brown staining, red arrows), neutrophils (PMN positive, brown staining, red arrows) and monocytes/macrophages (F4/80 positive, brown staining, red arrows) in the mid thalamus of Bregma −3.0 to −3.3 from Con, Dia and Dia‐Endo‐Glo‐I animals. Graphs on the right show means ± SEM; n = 5–6 rats per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. White bar at bottom of each image = 50 μm. Panel E shows relative amounts of TNF‐α mRNA in brain tissues (Bregma −2.7 to −3.5 mm) of Con, Dia, Dia GFP, Dia‐Endo‐Glo‐I and Dia‐Ins rats. Values shown in the graph are mean ± SEM; n = 5–6 rats per group, normalized to actin. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. Panel F shows representative autoradiograms for phosphorylated NF‐κB (p65) in brain homogenates (Bregma −2.7 to −3.5 mm) in Con, Dia, Dia GFP and Dia‐Endo‐Glo‐I rats. Values shown in the graph below are means ± SEM; n = 5–6 rats per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia.
Macrophage and neutrophil infiltration and inflammation
Microglia activation is the first step of inflammatory response in the brain, and this is usually followed by infiltration of immune cells such as neutrophils, macrophages/monocytes (Ma et al., 2016). To assess the latter, immunohistochemistry was conducted for monocytes/macrophages and neutrophils, infiltrating into the parenchyma. Figure 6C and D shows significant increases in the number of monocytes/macrophages and polymorphonuclear neutrophils (PMNs) in the parenchyma of Dia rats compared with control values. Images shown are from the thalamus within Bregma −3.0 to −3.3 of Dia rats, but similar infiltration was also seen at other BSA‐FITC transcytosis sites. Injecting rats with AAV2/9‐Endo‐Glo‐I 1 week after the onset of T1D blunted infiltration of monocytes/macrophages and PMNs in the parenchyma (Figure 6C and D). Injecting Dia rats with AAV2/9‐GFP did not attenuate infiltration of monocytes/macrophages and PMNs into the parenchyma (data not shown).
mRNA level for the inflammatory marker TNF‐α was also 3.0‐fold higher in brain (Bregma −2.7 to −3.5) of Dia rats compared with control (Figure 6E). Figure 6F also shows a fourfold increase in the amount of p‐p65 (NF‐κB) in Bregma −2.7 to −3.5 from 7–8 week Dia rats, compared with control. Injecting Dia rats with AAV2/9‐Endo‐Glo‐I significantly blunted p‐p65 and decreased the amount of TNF‐α mRNA. Injecting Dia rats with AAV2/9‐GFP only minimally attenuated the increase in p‐p65 and TNF‐α mRNA (Figure 6E and F).
Steady state levels of VAP‐1 and Glo‐I in brain microvessels
Western blot assays were then conducted to determine expression of VAP‐1 and Glo‐I proteins in isolated cerebral microvessels from Con, Dia and Dia‐treated rats. There was 30% reduction in Glo‐I protein and an 85% increase in VAP‐1 in cerebral microvessels from the Dia group of rats (Figure 7A). Administering AAV2/9‐Endo‐Glo‐I to Dia rats increased steady state level of Glo‐I protein threefold over Dia and 75% more than in the Con group of rats. Increasing Glo‐I protein in cSMCs also lowered VAP‐I protein. Administering AAV2/9‐GFP to Dia rats did not alter expression of Glo‐I and VAP‐1 levels in cSMCs. Treating Dia rats with insulin increased Glo‐I expression and decreased VAP‐1 expression in cerebral microvessels (Figure 7A).
Figure 7.
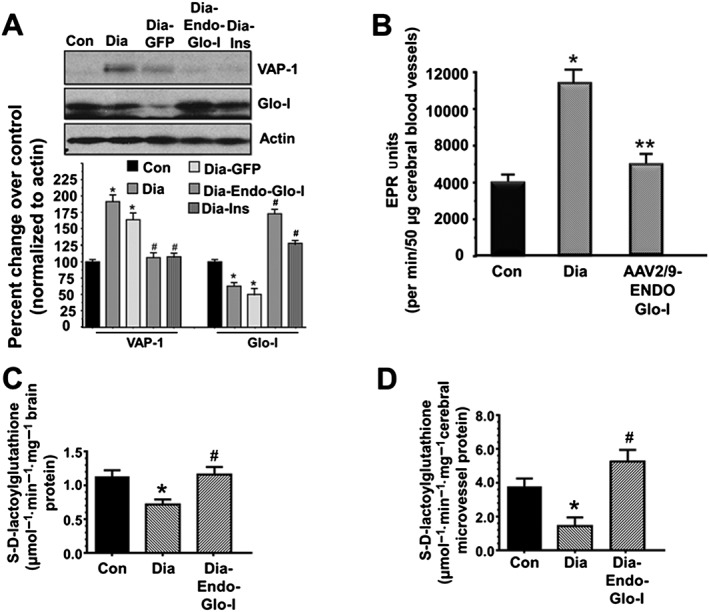
Panel A shows representative autoradiograms for VAP‐1, Glo‐I and actin in microvessels isolated from brains of Con, Dia, Dia GFP, Dia‐Endo‐Glo‐I and Dia‐Ins rats. Values shown in the graph are means ± SEM; n = 5–6 rats per group, normalized to actin. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. Panel B shows basal ROS levels in cerebral microvessels isolated from Con, Dia and Dia‐Endo‐Glo‐I rats. Values shown in the graph are means ± SEM; n = 5 rats per group. * P < 0.05, significantly different from Con. ** P < 0.05, significantly different from Dia. Panel C shows the ability of Glo‐I in brain homogenates (Bregma −2.7 to −3.5 mm) from Con, Dia and Dia‐Endo‐Glo‐I rats to metabolize methylglyoxal to produce S‐D‐lactoylglutahtione. Values shown in the graph are means ± SEM; n = 6 rats per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia. Panel D shows the ability of homogenates from cerebral microvessels (Bregma −2.7 to −3.5 mm) from Con, Dia and Dia‐Endo‐Glo‐I rats to metabolize methylglyoxal to produce S‐D‐lactoylglutahtione. Values shown in the graph are means ± SEM; n = 6 rats per group. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia.
Basal level of ROS in brain microvessels
After 7–8 weeks of T1D, total ROS in cerebral miscrovessels from Dia animals measured using EPR spectroscopy was 2.5‐fold higher than that in cerebral microvessels from Con animals (Figure 7B). Not surprising, increasing expression of Glo‐I in cSMCs to reduce ROS‐generating methylglyoxal attenuated total ROS in cerebral miscrovessels of Dia rats to near that in control animals (Figure 7B).
Levels of methylglyoxal and Glo‐I activity in brain and cerebral microvessels
Levels of methylglyoxal in the serum and brain of Dia rats were 4× and 15× higher, respectively, than that in Con animals (Table 2, lower rows). Glo‐I activity, determined by the formation of S‐D‐lactoylglutathione formation in Dia brain (Bregma −2.7 to −3.5), was 30% lower than that Con (Figure 7C). Injecting rats with AAV2/9‐Endo‐Glo‐I 1 week after the onset of T1D did not lower serum methylglyoxal. However, injecting Dia rats with AAV2/9‐Endo‐Glo‐I significantly reduced brain methylglyoxal level (Table 2). Mean Glo‐I activity in brain homogenates from AAV2/9‐Endo‐Glo‐I was similar to that in Con animals (Figure 7C). Injecting Dia rats with insulin for 2 weeks blunted serum and brain methylglyoxal levels and also restored brain Glo‐I protein and activity to near control levels (Table 2; Figure 7B). Injecting Dia rats with AAV2/9‐GFP did not increase Glo‐I activity and had only minimal effects on lowering serum and brain methylglyoxal levels (Table 2).
Glo‐I activity was also assessed in cerebral microvessels isolated from brains of Con, Dia and Endo‐Glo‐I‐Dia animals. Glo‐I activity in cerebral microvessels was also 30% lower than that Con (Figure 7D). Injecting Dia rats with AAV2/9‐Endo‐Glo‐I increased Glo‐I in cerebral microvessel homogenates and increased Glo‐I activity in cerebral microvessels to 32.5% higher than that in Con animals (Figure 7D).
Cell types in microvessels with altered VAP‐1, methylglyoxal and Glo‐I level
Immunohistochemistry was then used to delineate the cell type(s) in microvessels with altered Glo‐I, VAP‐1 and methylglyoxal levels. Immunoreactivity for vWF, a marker for ECs, was 62% lower in Dia rats compared with Con (Figure 8A, left). We did not detect any change in immunoreactivity for smooth muscle actin (SM22‐α, right). In cSMCs of cerebral microvessels with reduced vWF immunoreactivity, steady state level of the VAP‐1 was increased fivefold (Figure 8B, left). In cSMCS with elevated VAP‐1, the methylglyoxal surrogate argpyrimidine (Arg) was also increased nearly sixfold (Figures 8C and 8D (lower graph)). cSMCs of Dia rats also had 40% less Glo‐I immunoreactivity. Administering AAV2/9‐Endo‐Glo‐I to rats 1 week after the onset of T1D increased the steady state level of Glo‐I protein in cSMCs (Figure 8A, lower panels) but it did not increase Glo‐I protein in ECs (Figure 8A, left bottom right). Administering AAV2/9‐Endo‐Glo‐I to Dia rats also decreased Arg in cSMCs (Figures 8C, lower left). VAP‐1 immunoreactivity was also significantly blunted in cSMCs of Dia rats overexpressing Glo‐I protein (Figure 8C, panel C right side). Microvessels from Dia‐Endo‐Glo‐I rats with increased Glo‐I and reduced Arg in SMCs also contained higher levels of vWF immuno‐staining (Figure 8C, bottom left). These vessels also exhibited normal ADP‐mediated vasodilatation (Figure 2C).
Figure 8.
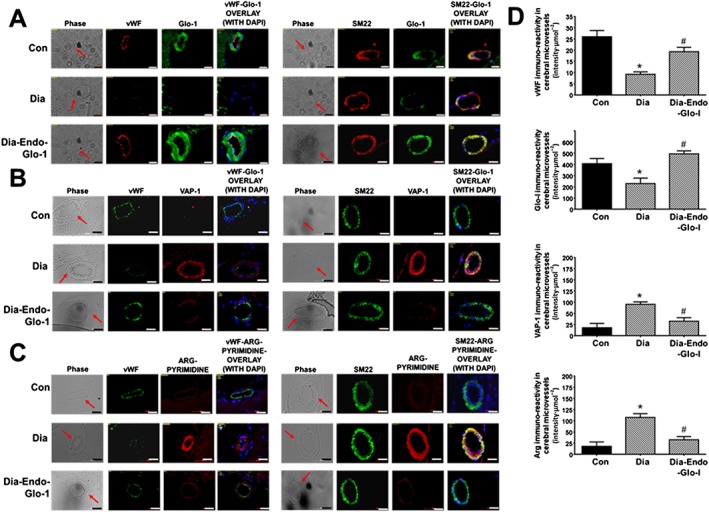
Panels A, B and C show representative immunohistochemical data for vWF, SM22‐α, DAPI, Glo‐I, VAP‐I and Arg in cortical microvessels in 20‐μm thick brain slices (Bregma −3.0 to −3.3) from Con, Dia and Dia‐Endo‐Glo‐I animals. White bar at bottom of each image = 20 μm. Graphs on the right (Panels D), starting from the top, show quantification of the amount of vWF, Glo‐I, VAP‐I and Arg in cortical arterioles in brains from Con, Dia and Dia‐Endo‐Glo‐I animals. Values shown are means ± SEM for 40 vessels from n = 5–6 rats. * P < 0.05, significantly different from Con; # P < 0.05, significantly different from Dia.
cSMCs produce methylglyoxal in high glucose
Having found increased VAP‐1 and the methylglyoxal surrogate in cSMCs of Dia rats, another series of experiments were then conducted to verify that cSMCs can indeed produce methylglyoxal when incubated in high glucose concentrations. Figure 9A shows no significant difference in the media from hcSMCs grown in 5.5 mM glucose over 24 h at 37°C in ambient air. However, when cSMCs were grown in 25 mM glucose over for 24 h, methylglyoxal level increased near sevenfold after 6 h but subsided after 12 and 24 h (Figure 9A). Adding 25 mM glucose to culture media and leaving in the incubator at 37°C in ambient air did not produce any significant increase in methylglyoxal (Figure 9A). Interestingly, we did not detect any significant change in VAP‐1 level in cSMCs grown in high glucose medium.
Figure 9.
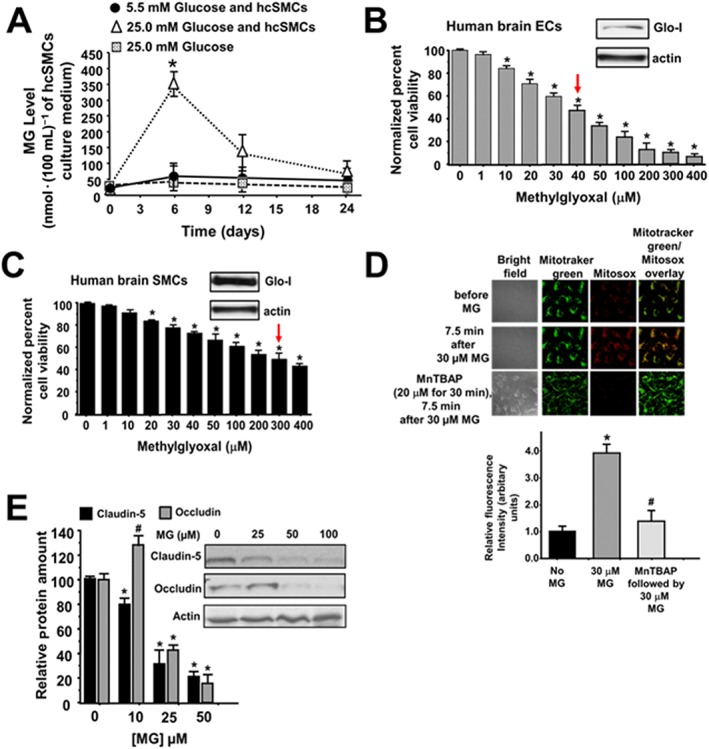
Panel A shows methylglyoxal levels in culture media from hcSMCs grown in 5.5 mM glucose and 25 mM glucose at 0.6, 12 and 24 h. Values shown in graph are for five independent experiments carried out using three separate preparations of hcSMCs. * P < 0.05, significantly different from 5.5 mM glucose. Panels B and C show viability of hcECs and hcSMCs 24 h after addition of a single addition of varying concentrations of methylglyoxal to the culture chambers. Values shown in graph are for eight independent experiments carried out using four separate preparations of hcECs and hcSMCs. * P < 0.05, significantly different from 0 μM methylglyoxal. Inset shows Western blots for Glo‐I and actin in hbECs and hbSMCs. Panels D shows representative images of changes in mitochondrial ROS in hbECs before and after exposure to 30 μM methylglyoxal. The third row also shows mitochondria ROS levels in hbECs that were pretreated for 30 min with 20 μM of MnTBAP, a cell permeable synthetic metalloporphyrin that exhibits SOD activity. For these studies, cells were loaded with the mitochondria‐localizing probe MitoTracker Green (100 nM), followed by the fluorogenic, mitochondria‐targeted, superoxide/ROS probe MitoSOX Red (2 μM), for 15 min each. Values shown in the graph below are for n = 5 independent experiments carried out using three separate preparations of hbECs. * P < 0.05, significantly different from 0 μM methylglyoxal. # P < 0.05, significantly different from 30 μM methylglyoxal. Panels E shows representative autoradiograms for claudin‐5 and occludin in hbECs, 24 h after adding a single addition of varying concentrations of methylglyoxal to the culture chamber. Values shown in the graph below are for n = 5 independent experiments carried out using three separate preparations of hcECs and hcSMCs.
GSH and enzymes that regulate GSH level
Because reduced GSH is a cofactor for Glo‐I, GSH and GSSH levels as well as the enzymes that synthesize GSH were also measured in brain tissues. In this study, GSH : GSSH ratio was significantly lower in the Dia rats compared with Con (1.5 ± 0.05, n = 5 brains, compared with 2.3 ± 0.3 for control, n = 6). Administering AAV2/9‐Endo‐Glo‐I to Dia rats increased GSH/GSSH to near Con levels (2.1 ± 0.4, n = 6). Administering AAV2/9‐GFP to Dia rats did not significantly alter GSH/GSSH ratio (1.6 ± 0.3, n = 5). Treating Dia rats with insulin for 2 weeks, after 6 weeks of diabetes also restored GSH/GSSH ratios to near Con levels (2.2 ± 0.1, n = 5).
In the brain of Dia rats (Bregma −2.7 to −3.5 mm) γ‐glutamylcysteine ligase (the rate‐limiting enzyme in the synthesis of GSH) and GSH reductase (the enzyme that reduces oxidized GSH) were decreased by 41 and 28%, respectively, compared with Con (289 ± 12 mU·mg−1 protein compared with 490 ± 18 mU·mg−1 protein and 37 ± 8 mU·mg−1 protein compared with 52 ± 7 mU·mg−1 protein). Administering AAV2/9‐Endo‐Glo‐I to Dia rats restored the activities of γ‐glutamylcysteine ligase and GSH reductase to near Con levels (503 ± 20 mU·mg−1 protein and 46 ± 10 mU·mg−1 protein respectively). Treating Dia rats with insulin for 2 weeks also restored the activities of γ‐glutamylcysteine ligase and GSH reductase to near control levels (515 ± 18 mU·mg−1 protein and 53 ± 9 mU·mg−1 protein respectively). Administering AAV2/9‐GFP to Dia rats did not significantly increase the activities of γ‐glutamylcysteine ligase and GSH reductase (290 ± 21 mU·mg−1 protein and 38 ± 11 mU·mg−1).
Viability of hcSMCs and hcECs exposed to methylglyoxal for 24 h
The present data, thus far, suggested that methylglyoxal produced by cSMCs was impairing the function of juxtaposed cECs in vivo. This prompted us to investigate why healthy cSMCs were more resistant to methylglyoxal than healthy cECs. Using Western blots, we found that hcSMCs contain 4.5‐fold more Glo‐I protein than hcECs, and that 7.5‐fold more methylglyoxal was needed to decrease the number of hcSMCs in culture by 50%, compared with hcECs (Figure 9B and C). Exposing hcECs to methylglyoxal also rapidly increased production of ROS in mitochondria (Figure 9D), and this effect was blocked by pretreating hcECs with the SOD mimetic MnTBAP for 20 min. Methylglyoxal also increased mitochondrial ROS production in cSMCs, albeit at higher concentrations. Exposing hcECs to methylglyoxal also decreased expression of the tight junction proteins claudin‐5 and occludin.
Discussion
There are several new findings in the present study. First, using immunohistochemistry, we show for the first time that methylglyoxal production (detected by its surrogate argpyrimidine) is up‐regulated ninefold in cSMCs of cerebral arterioles (≤50 μm) of STZ‐induced Dia rats. This increase in methylglyoxal arose from an increase in expression of VAP‐1 (fivefold) and a reduced expression of Glo‐I (50%). Second, using video imaging, we showed that cerebral arterioles with elevated VAP‐1 and methylglyoxal in their cSMCs were less responsive to EC‐mediated vasodilatation with ADP. Preventing methylglyoxal increase in cSMCs by restoring steady state levels of Glo‐I prevented impairment in EC‐mediated vasodilatation, suggesting for the first time that methylglyoxal produced by cSMCs is responsible for dysregulation of juxtaposed cECs. Because the catalytic domain of VAP‐1 is on the outside of SMCs, methylglyoxal is likely to be generated extracellularly, that is, in the microenvironment (Salmi and Jalkanen, 2001). This uncharged methylglyoxal can readily diffuse away from cSMCs and alter the function of neighboring cECs that contain low levels of the methylglyoxal degrading enzyme Glo‐I. Third, using the fluorescent labelled‐BSA and microscopy, we found a limited number of arterioles ranging in diameter from 20 μm to 50 μm that were leaky in the brain of Dia rats, resembling the microbleeds reported in patients with T1D (Biessels and Reijmer, 2014; Woerdeman et al., 2014). These vessels were detected primarily in the cortex and hypothalamus (Bregma −3.0 to −3.3 mm). Increasing expression of Glo‐I in cSMCs to lower methylglyoxal blunted BSA‐FITC transcytosis from these arterioles, suggesting for the first time that methylglyoxal produced in cSMCs may be causative for cerebral microbleeds at least in STZ‐induced Dia rats. Fourth, in the parenchyma surrounding BSA‐FITC transcytosis sites, gliosis and infiltration of immune cells (macrophage and neutrophils) were prominent. NF‐κB activation (p‐p65) and TNF‐α production were also increased in brain (Bregma −3.0 to −3.3), indicative of increased inflammation. Increasing Glo‐I protein in cSMCs of Dia rats blunted macrophage and neutrophil infiltration, astrogliosis and microgliosis, NF‐κB activation and TNF‐α production, identifying for first time a direct link between increased methylglyoxal production in cSMCs and brain inflammation. Fifth, using BSA‐FITC fluorescence, we also found that the density of perfused microvessels was reduced in the brains of Dia rats and that Dia rats were also more susceptible to ischaemia‐perfusion damage, employing the widely utilized transient MCA occlusion model of experimental stroke (Arrick et al., 2012). Blunting methylglyoxal up‐regulation in cSMCs of diabetic rats by restoring expression of Glo‐I not only increased the density of microvessels perfused (with BSA‐FITC), but it also minimized ischaemia‐reperfusion damage following transient MCA occlusion. These data indicate for the first time that methylglyoxal produced by cSMCs may be an initiating cause for hypoxia and increased susceptibility of the brain to ischaemia‐reperfusion damage following transient MCA occlusion.
Mechanisms responsible for increased VAP‐1 and reduced Glo‐I in cSMCs of diabetic rats
The specific mechanisms responsible for up‐regulation of VAP‐1 and for down‐regulating Glo‐I in cSMCs of Dia rats remains uncharacterized. What we know from the published literature is that VAP‐1 expression is induced in ECs in vivo by reactive carbonyl and oxygen species (RCS/ROS), IL‐1 and IL‐4, TNF‐α, IFN‐γ and lipopolysaccharide (Arvilommi et al., 1997). It is also known that the VAP‐1 gene (AOC3) is transcriptionally regulated by NF‐κB, AP‐1 and Jun (Bono et al., 1998). Whether the same molecules and transcription factors responsible for inducing expression of VAP‐1 in ECs also regulate expression of VAP‐1 on cSMCs remain to be determined. In this study, measurable changes in RCS/ROS and TNF‐α were detected subsequent to dysregulation of cECs. Therefore, ROS/RCS and TNF‐α are likely to be sustaining VAP‐1 expression rather that inducing its expression. Whether it is the increase in methylglyoxal produced via degradation of glucose/and or lipids within SMCs of Dia rats that is responsible for the initial induction of VAP‐1 expression remains to be determined.
In this study, we also found reduced Glo‐I protein in cSMCs of Dia rats. Glo‐I expression is transcriptionally regulated by the transcription factor Nrf2 (Xue et al., 2012). In the cytoplasm, Nrf2 forms a complex with Keap1 (Kelch‐like ECH‐associated protein 1). Under oxidative stress conditions, Keap1 dissociates from Nrf2 allow the latter to translocate from the cytoplasm to the nucleus where it induces expression of anti‐oxidant genes, including Glo‐I (Uruno et al., 2015). When oxidative stress or inflammation persists, as is the case with T1D, the actions of Nrf2 could be attenuated, preventing up‐regulation of Glo‐I expression in cSMCs. It should be mentioned that in addition to Glo‐I, three other enzymes are known to degrade methylglyoxal, albeit with higher K M values. They include aldehyde dehydrogenase, 2‐oxoaldehyde dehydrogenase and aldose reductase (Vander Jagt and Hunsaker, 2003). Additional work is needed to determine if expression of these enzymes is altered in the cSMCs of diabetic rats.
We also found that perfusing the cranial windows of controls rats with 25 μM methylglyoxal for 30 min attenuated ADP‐stimulated vasodilatation of arterioles to levels similar to that seen in T1D rats. These data led us to conclude that, at a minimum 25 μmol·L−1 methylglyoxal would be needed to be produced by the cSMCs into the microenvironment of cECs. How much VAP‐1 in cSMCs is required to achieve this concentration of methylglyoxal in vivo remains to be determined. It should be mentioned that in addition to ECs, the methylglyoxal generated vy the cSMCs could also affect the function of other neighbouring cells, including astrocytes and neurons.
Transcytosis, inflammation and microvessel perfusion in parenchyma
Under healthy conditions, substances from the blood enter into the brain parenchyma via fenestrated post capillary venules. If cECs in arterioles lose their ability to vasodilate, then it is possible that pressure can build up and transcytosis could increase. In this study, we found that BSA‐FITC transcytosis also occurred from select arterioles (20−50 μm) in the brain of diabetic rats. Leakage from these vessels resemble the microbleeds reported in some but not all regions of the brain of T1D patients (Biessels and Reijmer, 2014; Woerdeman et al., 2014). Why some vessels become ‘leakier’ than others in T1D is not well defined. One explanation could be heterogeneity in VAP‐1 expression and therefore the amount of methylglyoxal produced by cSMCs. Because methylglyoxal impairs the function of cECs in a concentration‐dependent manner, it seems logical the more VAP‐1 and methylglyoxal produced, the greater the damage to cECs. We also posit that the methylglyoxal produced in SMCs arterioles, and met‐arterioles could be diffusing and affecting the function of ECs in neighbouring capillaries.
In the present study, astrogliosis and microgliosis and macrophage/monocyte and neutrophil infiltration were prominent in the parenchyma around sites with increased BSA‐FITC transcytosis. In the brain region with the highest number of transcytosis sites (Bregma −3.0 to −3.3 mm), NF‐κB activation (p‐p65) and TNF‐α were increased threefold to fourfold, consistent with increased inflammation. Dominguez et al. (2015) also reported increased levels of other pro‐inflammatory cytokines and proteases, including IL‐1β, IL‐3 macrophage colony‐stimulating factor, IGF‐I and IGF‐binding protein‐3 in T1D rats.
To date, the cell types in the brain of T1D rats responsible for increasing TNF‐α remain undefined. Astrocytes, the major cell type by number in the brain, produce an array of chemokines and contain receptors for several pro‐inflammatory cytokines. However, recent in vitro studies indicate that while they respond to, they do not produce TNF‐α (Rama‐Rao and Kielian, 2015). It is tempting to speculate that activated microglia and macrophages are responsible for the increase in TNF‐α, but additional studies are needed to confirm this.
By counting the number of microvessels with green fluorescence of BSA‐FITC in 200 X frames, we were able to assess the density of perfused microvessels. In diabetic rats, the density of perfused microvessels in the cortex, hippocampus and thalamus were significantly lower (P < 0.05) than that in control rats. This reduction in the density of microvessel perfusion would create hypoxic environments that will compromise the metabolic needs of neurons and other cell types. As such, these regions will also be more susceptible to ischaemia‐reperfusion injury (Natarajan et al., 2008), a likely reason for the exacerbated cerebral infarct in diabetic rats following transient MCA occlusion.
Increasing expression of Glo‐I in cSMCs of diabetic rats
AAV serotypes exhibit selective tropism for certain cell types. In the brain, AAV2/9 shows preferential tropism for SMCs and neurons (Rahim et al., 2011). In the vasculature, endothelin‐1 is synthesized by SMCs and ECs. By cloning the endothelin‐1 promoter in front of Glo‐I cDNA, we were able to selectively target expression of Glo‐I protein in cSMCs of the cerebral vasculature (Figure 2A). Agrawal and colleagues (Pankajakshan et al., 2012) also reported successful transfection of GFP into the SMCs of swine arteries using AAV2/9. Using this custom‐designed virus, we found that restoring Glo‐I protein in cSMCs of T1D rats (25% higher than that in Con rats) was sufficient to blunt EC dysfunction and its downstream consequences. At this time it is not clear what if any would be the consequence of higher levels of Glo‐I expression in cSMCs of T1D rats.
In the brain, endothelin‐1 is also expressed in selected hippocampal and cortical neurons (Petrov et al., 2002). It is therefore likely that our virus could also be increasing expression of Glo‐I in selected neuron populations as well. An increase in expression of Glo‐I in neurons would protect neurovascular units from methylglyoxal, as well as improve the ability of neurons to regulate the reactivity of the cerebral microvasculature. These benefits may also be responsible, at least in part for the reduction in ischaemia‐reperfusion injury seen in AAV2/9‐Endo‐Glo‐I‐treated Dia rats.
In summary, the present study is the first to show that dysregulation of the VAP‐1 – methylglyoxal – Glo‐I axis in cSMCs with a consequent increase in methylglyoxal production, is responsible for EC dysfunction, increased transcytosis, reduction in density of microvessels perfused, inflammation and exacerbated ischaemia‐reperfusion injury following transient occlusion of a MCA in the brains of rats with diabetes induced by STZ (Figure 10). The data further suggest that lowering methylglyoxal in cSMCs could be therapeutically beneficial to prevent cerebral vascular defects in T1D and possibly Type 2 diabetes.
Figure 10.
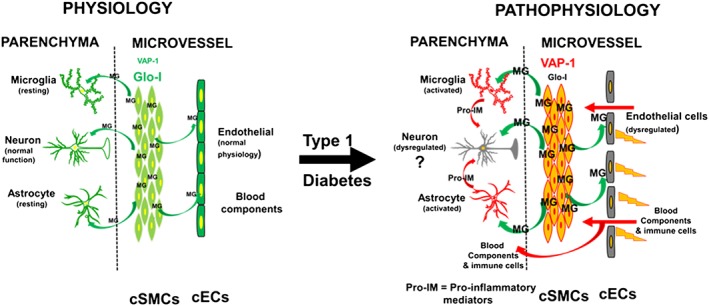
A schematic model summarizing data obtained from this study and a proposed mechanism by which cSMCs impair the function of endothelial cells in T1D. While others have shown that pro‐inflammatory data can impair neuronal function, at this time, we do not have any data of neuronal function in the Dia and Dia‐Endo‐Glo‐I groups of animals; hence, the question marks.
Author contributions
K.R.B. proposed the hypothesis and designed the study. F.A., J.S., W.G.M, H‐S.J., C‐H.S., B.P., G.J.R. and K.R.B. performed experiments, analysed data and wrote and edited the manuscript. The authors thank Dr. Caronda J. Moore for helpful suggestions. We also thank Janice A. Taylor and James R. Talaska (Confocal Laser Scanning Microscope Core Facility, University of Nebraska Medical Center). We thank Drs. Matthew Zimmerman, University of Nebraska Medical Center, for assisting with measuring ROS levels using EPR, Tsuneya Ikezu (University of Boston) for pZac 2.1 and Howard Fox for the use of ApoTome for screening slides.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgments
This work was supported in part by the Gene Therapy Research Program, GTRP 1053 and a pilot project from the Nebraska Redox Biology Center, Lincoln NE (P20‐RR 17675, P30GM103335).
Alomar, F. , Singh, J. , Jang, H. ‐S. , Rozanzki, G. J. , Shao, C. H. , Padanilam, B. J. , Mayhan, W. G. , and Bidasee, K. R. (2016) Smooth muscle‐generated methylglyoxal impairs endothelial cell‐mediated vasodilatation of cerebral microvessels in type 1 diabetic rats. British Journal of Pharmacology, 173: 3307–3326. doi: 10.1111/bph.13617.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alomar F (2014). Glyoxalase‐1 gene transfer to prevent cerebral microvascular and neuronal dysfunctions in type 1 diabetes mellitus PhD thesis, University of Nebraska Medical Center.
- American Diabetes Association (2010). Diagnosis and classification of diabetes mellitus. Dia Care 33 (Suppl. 1): S62–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrick DM, Sun H, Patel KP, Mayhan WG (2011). Chronic resveratrol treatment restores vascular responsiveness of cerebral arterioles in type 1 diabetic rats. Am J Physiol Heart Circ Physiol 301: H696–H703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrick DM, Sun H, Mayhan WG (2012). Influence of exercise training on ischaemic brain injury in type 1 diabetic rats. J Appl Physiol 113: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvilommi AM, Salmi M, Jalkanen S (1997). Organ‐selective regulation of vascular adhesion protein‐1 expression in man. Eur J Immunol 27: 1794–1800. [DOI] [PubMed] [Google Scholar]
- Baynes JW, Thorpe SR (1999). Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48: 1–9. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Reijmer YD (2014). Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 63: 2244–2252. [DOI] [PubMed] [Google Scholar]
- Bono P, Salmi M, Smith DJ, Leppänen I, Horelli‐Kuitunen N, Palotie A et al. (1998). Isolation, structural characterization, and chromosomal mapping of the mouse vascular adhesion protein‐1 gene and promoter. J Immunol 161: 2953–2960. [PubMed] [Google Scholar]
- Bono P, Jalkanen S, Salmi M (1999). Mouse vascular adhesion protein 1 is a sialoglycoprotein with enzymatic activity and is induced in diabetic insulitis. Am J Pathol 155: 1613–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ (2004). Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood‐glucose levels. Eur J Pharmacol 490: 159–168. [DOI] [PubMed] [Google Scholar]
- Carlberg I, Mannervik B (1995). Glutathione reductase. Methods Enzymol 113: 484–490. [DOI] [PubMed] [Google Scholar]
- Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS et al. (2005). 6‐Hydroxydopamine‐induced alterations in blood–brain barrier permeability. Eur J Neurosci 22: 1158–1168. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Gorantla S, Gendelman HE, Knibbe J, Casale GP, Makarov E et al. (2011). Loss of neuronal integrity during progressive HIV‐1 infection of humanized mice. J Neurosci 31: 3148–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon R (2012). Assessing burrowing, nest construction, and hoarding in mice. J Vis Exp: e2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar A, Dhar I, Desai KM, Wu L (2010). Methylglyoxal scavengers attenuate endothelial dysfunction induced by methylglyoxal and high concentrations of glucose. Br J Pharmacol 161: 1843–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler MG, Plant LD, Sokoloff G, Hawk AJ, Aneas I, Wuenschell GE et al. (2012). Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J Clin Invest 122: 2306–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler MG, Gorfinkle N, Papale LA, Wuenschell GE, Termini J, Escayg A et al. (2013). Glyoxalase 1 and its substrate methylglyoxal are novel regulators of seizure susceptibility. Epilepsia 54: 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez JM, Yorek MA, Grant MB (2015). Combination therapies prevent the neuropathic, proinflammatory characteristics of bone marrow in streptozotocin‐induced diabetic rats. Diabetes 64: 643–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Heistad DD, Faraci FM (1992). Effect of diabetes mellitus on flow‐mediated and endothelium‐dependent dilatation of the rat basilar artery. Stroke 23: 1494–1498. [DOI] [PubMed] [Google Scholar]
- Gilman S (2006). Time course and outcome of recovery from stroke: relevance to stem cell treatment. Exp Neurol 199: 37–41. [DOI] [PubMed] [Google Scholar]
- Han Y, Randell E, Vasdev S, Gill V, Gadag V, Newhook LA et al. (2007). Plasma methylglyoxal and glyoxal are elevated and related to early membrane alteration in young, complication‐free patients with Type 1 diabetes. Mol Cell Biochem 305: 123–131. [DOI] [PubMed] [Google Scholar]
- Hasim S, Hussin NA, Alomar F, Bidasee KR, Nickerson KW, Wilson MA (2014). A glutathione‐independent glyoxalase of the DJ‐1 superfamily plays an important role in managing metabolically generated methylglyoxal in Candida albicans . J Biol Chem 289: 1662–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM et al. (2005). Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438: 662–666. [DOI] [PubMed] [Google Scholar]
- Jaakkola K, Kaunismaki K, Tohka S, Yegutkin G, Vanttinen E, Havia T et al. (1999). Human vascular adhesion protein‐1 in smooth muscle cells. Am J Pathol 155: 1953–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman K, Iadecola C (2015). Neurovascular regulation in the ischaemic brain. Antioxid Redox Signal 22: 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubcakova V, Curzi ML, Flachskamm C, Hambsch B, Landgraf R, Kimura M (2013). The glycolytic metabolite methylglyoxal induces changes in vigilance by generating low‐amplitude non‐REM sleep. J Psychopharmacol 27: 1070–1075. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodl CT, Franc DT, Rao JP, Anderson FS, Thomas WM, Mueller BA et al. (2008). Diffusion tensor imaging identifies deficits in white matter microstructure in subjects with type 1 diabetes that correlate with reduced neurocognitive function. Diabetes 57: 3083–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaloff H, Gavras H, Brecher P (1980). Reninlike enzymatic activity in the cerebral microvessels of the rat. Am J Physiol 238: H384–H388. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang R, Desai K, Wu L (2011). Upregulation of aldolase B and overproduction of methylglyoxal in vascular tissues from rats with metabolic syndrome. Cardiovasc Res 92: 494–503. [DOI] [PubMed] [Google Scholar]
- Ma Y, Wang J, Wang Y, Yang GY (2016). The biphasic function of microglia in ischaemic stroke. Prog Neurobiol. doi: 10.1016/j.pneurobio.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Mathys KC, Ponnampalam SN, Padival S, Nagaraj RH (2002). Semicarbazide‐sensitive amine oxidase in aortic smooth muscle cells mediates synthesis of a methylglyoxal‐AGE: implications for vascular complications in diabetes. Biochem Biophys Res Commun 297: 863–869. [DOI] [PubMed] [Google Scholar]
- Mayhan WG (1989). Impairment of endothelium‐dependent dilatation of cerebral arterioles during diabetes mellitus. Am J Physiol 256: H621–H625. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan AC, Thornalley PJ (1989). Glyoxalase activity in human red blood cells fractioned by age. Mech Ageing Dev 48: 63–71. [DOI] [PubMed] [Google Scholar]
- Mohanty JG, Nagababu E, Rifkind JM (2014). Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol 5: 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moheet A, Mangia S, Seaquist ER (2015). Impact of diabetes on cognitive function and brain structure. Ann New York Acad Sci 1353: 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler AA (2008). Hypoxia inducible factor‐1 upregulates adiponectin in diabetic mouse hearts and attenuates post‐ischaemic injury. J Cardiovasc Pharmacol 51: 178–187. [DOI] [PubMed] [Google Scholar]
- Nemet I, Varga‐Defterdarovic L, Turk Z (2004). Preparation and quantification of methylglyoxal in human plasma using reverse‐phase high‐performance liquid chromatography. Clin Biochem 37: 875–881. [DOI] [PubMed] [Google Scholar]
- Pankajakshan D, Makinde TO, Gaurav R, Del Core M, Hatzoudis G, Pipinos I et al. (2012). Successful transfection of genes using AAV‐2/9 vector in swine coronary and peripheral arteries. J Surg Res 175: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitti DB, Klingensmith GJ, Bell RA, Andrews JS, Dabelea D, Imperatore G et al. (2009). Glycaemic control in youth with diabetes: the SEARCH for diabetes in Youth Study. J Pediatr 155: 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov T, Steiner J, Braun B, Rafols JA (2002). Sources of endothelin‐1 in hippocampus and cortex following traumatic brain injury. Neuroscience 115: 275–283. [DOI] [PubMed] [Google Scholar]
- Pickup JC (2015). Banting Memorial Lecture 2014, Technology and diabetes care: appropriate and personalized. Dia Med 32: 3–13. [DOI] [PubMed] [Google Scholar]
- Pun PB, Murphy MP (2012). Pathological significance of mitochondrial glycation. Int J Cell Biol 2012: 843505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim AA, Wong AM, Hoefer K, Buckley SM, Mattar CN, Cheng SH et al. (2011). Intravenous administration of AAV2/9 to the fetal and neonatal mouse leads to differential targeting of CNS cell types and extensive transduction of the nervous system. FASEB J 25: 3505–3518. [DOI] [PubMed] [Google Scholar]
- Rama‐Rao KV, Kielian T (2015). Neuron‐astrocyte interactions in neurodegenerative diseases: role of neuroinflammation. Clin Exp Neuroimmunol 6: 245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhi AK, Berg JP, Berg TJ (2013). Glyoxalase 1 enzyme activity in erythrocytes and Ala111Glu polymorphism in type 1‐diabetes patients. Scand J Clin Lab Invest 73: 175–181. [DOI] [PubMed] [Google Scholar]
- Salmi M, Jalkanen S (2001). VAP‐1: an adhesin and an enzyme. Trends Immunol 22: 211–216. [DOI] [PubMed] [Google Scholar]
- Seelig GF, Meister A (1995). Glutathione synthesis: γ‐glutamylcysteine synthetase from rat kidney. Methods Enzymol 113: 379–390. [DOI] [PubMed] [Google Scholar]
- Shao CH, Capek HL, Patel KP, Wang M, Tang K, DeSouza C et al. (2011). Carbonylation contributes to SERCA2a activity loss and diastolic dysfunction in a rat model of type 1 diabetes. Diabetes 60: 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao CH, Tian C, Ouyang S, Moore CJ, Alomar F, Nemet I et al. (2012). Carbonylation induces heterogeneity in cardiac ryanodine receptor function in diabetes mellitus. Mol Pharmacol 82: 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Vanhoutte PM (2009). Reactive oxygen‐derived free radicals are key to the endothelial dysfunction of diabetes. J Diabetes 1: 151–162. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomanek L (2015). Proteomic responses to environmentally induced oxidative stress. J Exp Biol 218: 1867–1879. [DOI] [PubMed] [Google Scholar]
- Uruno A, Yagishita Y, Yamamoto M (2015). The Keap1‐Nrf2 system and diabetes mellitus. Arch Biochem Biophys 566: 76–84. [DOI] [PubMed] [Google Scholar]
- Vander Jagt DL, Hunsaker LA (2003). Methylglyoxal metabolism and diabetic complications: roles of aldose reductase, glyoxalase‐I, betaine aldehyde dehydrogenase and 2‐oxoaldehyde dehydrogenase. Chem Biol Interact 143‐144: 341–351. [DOI] [PubMed] [Google Scholar]
- Wessels AM, Simsek S, Remijnse PL, Veltman DJ, Biessels GJ, Barkhof F et al. (2006). Voxel‐based morphometry demonstrates reduced grey matter density on brain MRI in patients with diabetic retinopathy. Diabetologia 49: 2474–2480. [DOI] [PubMed] [Google Scholar]
- Woerdeman J, van Duinkerken E, Wattjes MP, Barkhof F, Snoek FJ, Moll AC et al. (2014). Proliferative retinopathy in type 1 diabetes is associated with cerebral microbleeds, which is part of generalized microangiopathy. Dia Care 37: 1165–1168. [DOI] [PubMed] [Google Scholar]
- Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N et al. (2012). Transcriptional control of glyoxalase 1 by Nrf2 provides a stress‐responsive defence against dicarbonyl glycation. Biochem J 443: 213–222. [DOI] [PubMed] [Google Scholar]
- Zhang D, Hu X, Qian L, O'Callaghan JP, Hong JS (2010). Astrogliosis in CNS pathologies: is there a role for microglia? Mol Neurobiol 41: 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Mayhan WG, Sun H (2008). A modified suture technique produces consistent cerebral infarction in rats. Brain Res 1246: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]