

Abstract
Neurofibromatosis type 1 (NF1) is one of the most common brain tumor predisposition syndromes, in which affected children are prone to develop low-grade gliomas. While NF1-associated gliomas can be found in several brain regions, the majority arise in the optic nerves, chiasm, tracks, and radiations (optic pathway gliomas; OPGs). Owing to their location, 35–50% of affected children present with reduced visual acuity. Unfortunately, despite tumor stabilization following chemotherapy, most children have improved vision. For this reasons, more effective therapies are being sought that reflect a deeper understanding of the NF1 gene and the use of authenticated Nf1 genetically-engineered mouse strains. The implementation of these models for drug discovery and validation has galvanized molecularly-targeted clinical trials in children with NF1-OPG. Future research focused on defining the cellular and molecular factors that underlie optic glioma development and progression also has the potential to provide personalized risk assessment strategies for this pediatric population.
Keywords: astrocytoma, pilocytic, precision medicine, RAS, vision, low-grade glioma
Neurofibromatosis type 1 (NF1) is the most common inheritable tumor predisposition syndrome, occurring in approximately 1 in 2,500–3,000 people worldwide1, 2. NF1 affects nearly every organ system in the body with broad clinical ramifications, such that children and adults with this condition may exhibit pigmentary abnormalities (café-au-lait macules, skinfold freckling, Lisch nodules), tumors of the peripheral and central nervous system (neurofibromas and gliomas), learning and attention problems, autism spectrum symptomatology, bone abnormalities (long bone dysplasias, scoliosis), seizures, sleep disturbances, vasculopathies (moyamoya syndrome, renal artery stenosis), and non-nervous system cancers (breast cancer, pheochromocytoma). Of the tumors involving the nervous system, peripheral nerve sheath tumors (cutaneous and plexiform neurofibromas) predominate, and 8–13% of those individuals harboring a plexiform neurofibroma will develop a malignant peripheral nerve sheath tumor3.
Children and adults with NF1 are particularly prone to develop tumors of the central nervous system. In adults, high-grade gliomas may occur4, whereas in children, the most commonly-encountered brain tumor is a low-grade glioma, a World Health Organization grade I tumor (pilocytic astrocytoma) with low mitotic rates and low proliferative indices. While these pilocytic astrocytomas can occur anywhere in the brain, they are most frequently detected in the optic pathway and brainstem. In this respect, nearly two-thirds of gliomas are found in the optic pathway, with brainstem (15–20%), cerebellum (~5%), cerebral hemispheres (~5%) and subcortical structures (~5%) accounting for the remaining locations5.
Clinical Presentation and Natural History
Approximately 15–20% of children with NF1 will develop an optic pathway tumor6, 7; however, only 30–50% will be symptomatic from their glioma, and only one-third of affected children will require therapeutic intervention6–8. NF1-OPGs are most commonly seen in young children, with the majority occurring in children younger than seven years of age (mean 4.5 years)9. Rare cases of NF1-OPGs arising in adolescence or adulthood have also been reported10.
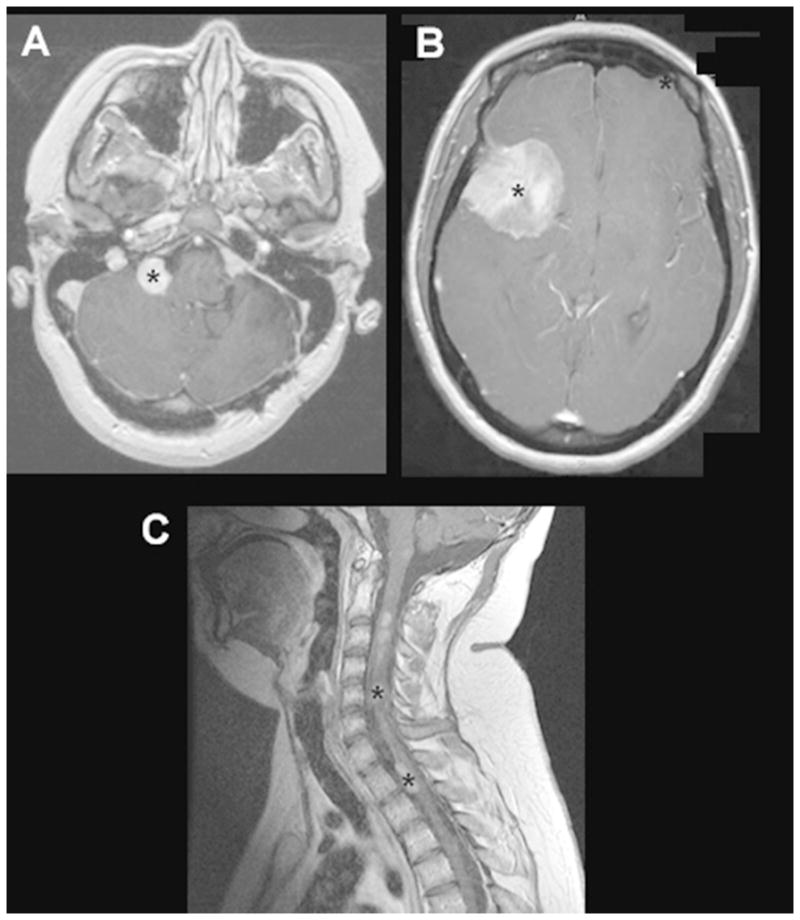
NF1-OPGs can occur anywhere along the optic pathway including the nerves, chiasm, post-chiasmatic tracts, and radiations (Figure 1)7, 11, 12. Tumor location largely dictates the presenting signs and symptoms, with optic nerve gliomas often resulting in unilateral proptosis, visual acuity loss, visual field defect, strabismus, relative afferent pupillary defect, and optic disc edema (papilledema) or atrophy13. With chiasmal involvement, precocious puberty can be the main presenting symptom, but visual acuity loss and visual field defects can also occur9, 14. Rarely, OPGs involving the hypothalamus can exert enough mass effect to cause obstructive hydrocephalus and resultant headache and vomiting. Those occurring in the optic tracks and radiations most often present with visual acuity deficits, but may result in other neurological signs depending on the involvement of adjacent structures.
Figure 1. OPGs in children with NF1.

Axial T2-weighted MR images of OPGs involving the (A) optic nerve, (B) optic chiasm, and (C) optic radiations. Asterisks denote the tumors.
The behavior of NF1-OPGs can be unpredictable, requiring that all children with NF1 undergo routine surveillance (discussed below). Currently, there are no clear prognostic features, although patient sex, tumor location, and age of the patient have each been associated with an increased risk of clinical progression. As such, girls are more likely to lose vision and require treatment for OPG than boys, and girls with optic nerve gliomas are 5–10 times more likely to experience visual decline than their male counterparts15, 16. In addition, NF1-OPGs occurring in the post-chiasmatic optic pathway tend to exhibit more aggressive clinical behavior than those involving the optic nerve or chiasm17. Finally, tumors presenting before the age of 2 years and after 8–10 years of age are typically more aggressive than those presenting in children between 2–8 years of age10, 17–19. Advanced neuroimaging techniques using diffusion tensor imaging, routinely included on most magnetic resonance imaging (MRI) studies, have revealed that a decrease in the integrity of the white matter tracts of the optic radiations, as measured by fractional anisotropy, is associated with abnormal visual acuity in NF1-related OPG, and may be predictive of future visual acuity loss20. Further prospective studies investigating this association are ongoing.
Screening and Surveillance
Recommended screening for children with NF1 entails annual eye exams in all children less than ten years of age, and at least every two years until 18 years of age9. Age-appropriate assessments of visual acuity are critical for NF1-OPG surveillance21, 22, since visual field testing is unreliable in young children and optic disc pallor does not predict vision outcome9. The most reliable information can be obtained when using Teller acuity cards (age 0–2 years), Lea figures (3–4 years of age), HOTV cards (4–6 years of age), and Snellen charts (≥6 years of age). Optic coherence tomography, a promising objective modality in NF1-associated OPG, measures retinal nerve fiber layer thickness (RNFL), a marker for visual loss in children with NF1. RNFL thickness correlates well with visual acuity, but sedation is required in young children to ensure full cooperation9. The use of this objective measure may circumvent some of the problems associated with accurately assessing vision, especially in children with NF1 and co-morbid attention and cognitive disabilities.
Recommended screening should be performed by an experienced pediatric ophthalmologist, and should include measurements of visual acuity, confrontational visual field evaluations, color vision testing, and assessments of pupils, eyelids, ocular motility, irises, and fundi9. All children with NF1 should undergo yearly measurements of weight and height plotted on standard growth charts to monitor for signs of precocious puberty9.
Screening baseline MRI evaluations are not indicated for NF1-OPG, as the detection of these tumors rarely changes management in the absence of clinical symptoms or signs23. There may be, however, a role for neuroimaging screening in children in whom reliable visual assessment cannot be performed.
Once an OPG has been identified, the frequency of neuroimaging and visual assessment depends on the site of the tumor, degree of visual impairment and associated symptoms as well as evidence of progressive disease9. There is no consensus on the specific interval of neuroimaging and visual assessments, but most centers experienced in treating patients with NF1-associated OPG perform eye examinations and vision testing every three months for the first year after diagnosis, with increasing intervals thereafter9. MRI examinations may be performed at similar or less frequent intervals.
Similarly, there is little consensus as to what constitutes sufficient clinical progression to warrant treatment, further complicating patient care. Most institutions rely on a combination of clinical deterioration and radiological progression24–27. However, clinical deterioration can include the onset of new neurological symptoms or endocrinologic changes24–27, a change in visual acuity18, or visual field loss combined with impaired visual acuity25. Radiologic progression can include an increase in tumor size, or further extension into the optic pathway or hypothalamus, but should not be claimed based on a change in the enhancement pattern alone9. While no evidence-based data exist to date, the following findings have been proposed as criteria for clinical progression: (1) a two-line change in Snellen, HOTV matching, or Lea matching visual acuity compared with the previous examination or (2) a two-line decline in Teller visual acuity9.
Pathophysiology
Like most solid tumors, NF1-OPGs are complex cellular ecosystems in which several different cell types participate in tumor initiation, evolution, and clinical progression (Figure 2). Among the critical cell types are neoplastic cellular elements (glioma stem cells and astrocytes) and non-neoplastic stromal cells (microglia, neurons and endothelial cells). The neoplastic cells are characterized by bi-allelic inactivation of the NF1 tumor suppressor gene, resulting in loss of NF1 protein (neurofibromin) expression. Whole genome sequencing of NF1-PA tumors confirmed genetic silencing of both NF1 alleles through mutation, methylation, or genomic loss. In addition, 35–50% of the cells in these tumors retain one normal NF1 gene (NF1 heterozygosity), and represent the non-neoplastic stromal cells28. The majority of these NF1 heterozygous mutant cells are immune system-like cells, called microglia, vitally important for normal brain function29, 30, but in this context, are key contributors to brain tumor pathogenesis31.
Figure 2. Ecosystem model for NF1-OPG therapeutic targeting.
The complex interactions between numerous cell types in the optic glioma determine tumor formation, maintenance, and vision loss. Neoplastic glia (glioma stem cells and tumor astrocytes) lacking NF1 gene expression produce chemokines that attract and activate microglia. These activated microglia elaborate growth factors that further promote tumor growth (e.g., CCL5), as well as secrete neurotoxins (e.g., IL-1β) that cause axonal injury, retinal ganglion cell death, and vision loss.
Since NF1-OPGs are rarely biopsied or surgically removed, human biological materials for mechanistic studies have been limited. Moreover, human low-grade glioma cells grow poorly in vitro and frequently undergo senescence32, and none have been successfully maintained as patient-derived xenografts33. For these reasons34, much of our understanding of the pathophysiology of NF1-associated low-grade gliomas derives from analyses of Nf1 genetically-engineered mouse models. Based on the genetics of their human counterparts, mice heterozygous for a targeted germline inactivating mutation in the Nf1 gene (Nf1+/− mice) have been engineered with a conditional Nf1 allele (Nf1flox) to enable somatic Nf1 loss in neuroglial progenitor cells during embryonic development35–37. Analysis of the resulting Nf1 mutant mice revealed that optic gliomas arise in >90% of mice by 3 months of age. Importantly, Nf1+/− cells are required for tumorigenesis38, further underscoring the contribution of stromal cells to glioma formation.
Analogous to the majority of pediatric NF1-OPG, the murine tumors are located in the prechiasmatic optic nerves and chiasm. Additionally, similar to their human counterparts, the resulting Nf1 optic gliomas have low proliferative indices (~1%), express glial fibrillary acidic protein (GFAP) and Olig2, and can be visualized by small-animal magnetic resonance imaging (MRI)39, 40. Relevant to NF1-OPG-associated vision loss in children, these tumors result in a time-dependent succession of events in the optic nerve, beginning with axonal injury at the site of the glioma, and then progressive retinal ganglion cell (RGC) apoptosis and loss, and culminating in reduced visual acuity15, 41–43.
Further analysis of these mice and their derivative neoplastic cells revealed that tumor cell proliferation results from loss of neurofibromin negative regulation of the RAS proto-oncogene (Figure 3). As such, neurofibromin is structurally and functionally similar to a family of proteins, termed GTPase activating proteins (GAPs), which function to accelerate the conversion of active, GTP-bound RAS to its inactive GDP-bound form. Neurofibromin inactivation of RAS abrogates the growth-promoting signal initiated by RAS at the plasma membrane. Consistent with this mechanism of tumor growth regulation, increased RAS activation has been observed in both human NF1-PAs44 and their murine counterparts45. Active RAS stimulates cell growth through a cascade of molecular intermediates whose successive phosphorylation results in their activation. These signaling intermediates include AKT and MEK, which increase cell growth through activation of the mechanistic target of rapamycin (mTOR) complex46 and ERK, respectively. Relevant to the design of human clinical trials, inhibition of AKT, mTOR, or MEK activity in Nf1 mutant mice results in reduced tumor growth in vivo47, 48, establishing the preclinical rationale for the use of these molecularly-targeted therapies in children with NF1-OPG (Table 1). However, it is worth noting that some of these therapies in mice require drug dosing that exceeds the maximal tolerated doses in children, and many do not result in durable stabilization of tumor growth47, 49. Moreover, cancer stem cells found in the murine tumors exhibit relative resistance to mTOR and MEK inhibition as a result of acquired adaptive responses50, which may further limit the use of these pharmacologic agents for the treatment of children with NF1-OPG. Taken together, it is essential that future targeted therapies consider pediatric dosing and potential glioma resistance.
Figure 3. Neurofibromin regulation of cell biology in the central nervous system.

The NF1 protein, neurofibromin, functions as a GTPase-activating protein (GAP) for p21-RAS, accelerating its conversion from an active RAS-GTP bound molecule to an inactive RAS-GDP bound form. RAS can be activated by G protein-coupled receptors (GPCRs), including chemokine receptors, and by receptor tyrosine kinase (RTK) binding of growth factors, like epidermal growth factor. Active RAS controls multiple downstream signaling pathways, engaging MEK and AKT through kinase intermediates, to activate ERK and the mechanistic target of rapamycin (mTOR) complex, respectively. In addition, RAS activation suppresses cyclic AMP (cAMP) generation, important for central nervous system neuron survival.
Table 1.
Treatments for NF1-OPG
| Trial | Study number | Status | Sponsor | Phase | Ages | Disease status | Primary Outcome |
|---|---|---|---|---|---|---|---|
| Lenalidomide | NCT01553149 | Active, not recruiting | NCI | II | 0 to 21 years | Recurrent, Refractory, Progressive | Response Rate |
| Selumetinib | NCT01089101 | Active, not recruiting | NCI | II | 3 to 21 years | Recurrent, refractory | Response Rate |
| Vinblastine +/− Bevacizumab | NCT02840409 | Active, recruiting | The Hospital for Sick Children | II | 6 months to 18 years | Unresectable or Progressive | Response Rate |
| MEK 162 | NCT02285439 | Active | Children’s Hospital Los Angeles | I/II | 1 to 18 years | Recurrent, Refractory, Progressive | Response Rate |
| MEK 162 | NCT01885195 | Active, not recruiting | Array BioPharma | II | ≥18 years | Recurrent or Progressive | Response Rate |
| RAD0001 (Everolimus) | NCT01158651 | Active, not recruiting | University of Alabama at Birmingham | II | 1 to 21 years | Chemotherapy refractory, radiologically progressive | Response Rate |
| Pomalidomide | NCT02415153 | Active, not recruiting | NCI | I | 3 to 20 years | Recurrent, Progressive, or Refractory | Maximum tolerated dose |
| Pegylated Interferon | NCT02343224 | Active, recruiting | Emory University | II | 3 to 18 years | Recurrent, refractory, Progressive | Response rate |
In addition to the neoplastic cells in the murine optic gliomas, a critical role for microglia in both tumor formation and maintenance has been established. Impairment of microglia infiltration by genetic reduction of an essential microglia chemotaxis receptor (CX3CR1) results in delayed tumor formation51, while either genetic or pharmacologic silencing of microglia function is sufficient to reduce optic glioma proliferation in vivo52–54. Using advanced RNA-sequencing, one of the relevant growth factors produced by these tumor-associated microglia was discovered55. The chemokine CCL5 was demonstrated to be important for glioma maintenance, such that inhibiting its function with neutralizing antibodies dramatically attenuated glioma growth in vivo.
One of the most common morbidities associated with NF1-OPG is progressive vision loss. Analysis of Nf1 mutant mice revealed that reduced neurofibromin expression in RGCs, the neurons that transmit visual information from the eye to the brain, results in increased programmed cell death by apoptosis. Neurofibromin regulation of RGC survival involves the generation of cyclic AMP (cAMP), such that Nf1+/− RGCs harbor reduced intracellular cAMP levels56. Elevating cAMP levels using an inhibitor of the enzyme responsible for cAMP degradation (phosphodiesterase-4 inhibitor; Rolipram) almost completely ameliorates the apoptosis and loss of RGCs associated with murine Nf1 optic glioma. Moreover, the progressive axonal injury that culminates in RGC death and visual impairment results from microglia production of neurotoxins, including interleukin-1β57. This finding suggests that there might exist a therapeutic window between the elaboration of neurotoxins and irreversible RGC loss during which time pharmacologic intervention might prevent vision loss. Using Nf1 optic glioma mice, the temporal course of tumorigenesis and retinal pathology was defined, identifying such a window before sufficient RGC apoptosis occurred (>50% RGC loss). Treatment of mice during this period resulted in preservation of RGC numbers two months following the cessation of therapy43, suggesting that vision stabilization might be possible.
Treatment
When clinical progression occurs, the mainstay of treatment is chemotherapy (Table 1). Other treatment modalities commonly used in brain tumors, namely surgery and radiation, are problematic in the setting of NF1-associated OPGs. Meaningful surgical resection of NF1-OPGs is usually impossible due to the location of these tumors. It is used, however, in the setting of large orbital tumors where there is no useful vision in the affected eye or to treat corneal exposure or proptosis. Hypothalamic and chiasmatic OPGs often require surgical debulking, and diagnostic biopsy should be considered in tumors arising in atypical locations9, 58. Radiation is not recommended in children with NF1, given the risk of secondary tumors (glioma and MPNSTs) in the setting of this tumor predisposition syndrome. In the NF1 population, there is an increased risk of moyamoya syndrome relative to the general population, a risk further heightened by radiation exposure to the large vessels of the Circle of Willis adjacent to the optic pathway59. Finally, the risk of late neurocognitive sequelae in children who have learning and attention differences should be considered before initiating radiation9.
While chemotherapy is often used in symptomatic OPG, few children ever regain normal visual acuity following treatment9 or experience improvements in vision60, 61. Some NF1 experts have raised the concern that chemotherapy may affect cognition in this vulnerable population62, 63; however, chemotherapy is usually effective at stabilizing disease or even shrinking NF1-OPGs. First-line OPG chemotherapy, initially proposed by Packer and colleagues, involves vincristine and carboplatin64. Second-line chemotherapy treatment options include vinblastine, which shows comparable efficacy to vincristine/carboplatin65, vinorelbine66, and temozolomide, an alkylating agent that should be used with caution in tumor-predisposition syndromes67. Bevacizumab has also been used to treat refractory NF1-OPG, and may improve visual outcome in some children, although the durability of these responses is unknown68. More recently, small molecule inhibitors have been used as investigational therapies for otherwise refractory tumors. Unfortunately, a phase II trial using sorafenib (a multi-kinase inhibitor) was stopped early due to an unexpected acceleration of tumor growth69. More promising is selumetinib, a MEK inhibitor that has shown growth inhibition in NF1-deficient GBM cell lines70 and in NF1-associated plexiform neurofibromas71. A phase II clinical trial in low-grade glioma, including NF1-associated OPG is ongoing, but results are not yet available (NCT01089101).
Future directions
Using Nf1 genetically-engineered mice, it now becomes possible to mechanistically define the factors that underlie tumor growth and associated vision loss. In this regard, the contribution of the germline NF1 gene mutation and patient sex have been evaluated as potential actionable risk factors relevant to gliomagenesis and clinical progression. While conflicting data exist in the literature regarding the existence of genotype-phenotype correlations in children with NF1-OPG72–74, proof-of-concept experiments were performed in which mice were generated with specific germline NF1 gene mutations reported in children with NF1-OPG (R681X) and adults with spinal neurofibromas (G848R). Mice harboring the G848R mutation as their germline Nf1 gene mutation did not develop optic gliomas, whereas those with the R681X mutation developed optic gliomas with greater volumes and proliferation indices than those harboring the engineered knockout allele as their germline NF1 gene mutation75. These exciting early-phase data support a model in which the particular germline NF1 gene mutation may have differential effects on neurofibromin expression and function in stromal cells (e.g., microglia) that serve to increase tumor growth and associated retinal pathology76. Studies are ongoing to examine other germline NF1 gene mutations, as well as create larger repositories of NF1 patient induced pluripotent stem cells for analysis.
Based on an analysis of children with NF1-OPG, girls with tumors located in the optic nerves were 5- to 10-fold more likely to require treatment for progressive vision loss15, 16. This interesting sexual dimorphism was further explored in Nf1 mutant mice, where only female mice were found to have reduced visual acuity from their optic glioma, despite equal tumor volumes and proliferative indices in male and female mice15. Sexually-dimorphic differences can result from chromosomal influences (organizational) or gonadal sex hormones (activational) through the effects on gene expression or hormonal receptors, respectively. In Nf1 mutant mice with optic glioma, there was no protective effect of male gonadal sex hormones. However, estrogen (estradiol) activation of microglia in female Nf1 mutant mice was responsible for the progressive loss of RGCs and thinning of the retinal nerve fiber layer57. These findings demonstrate that inhibition of gonadal sex hormone-mediated microglia activation might attenuate vision loss, suggesting new potential drug treatments to reduce glioma-associated vision loss in children with NF1- OPG.
Collectively, these findings indicate that the biological behavior of these tumors is heavily influenced by a myriad of factors, which each might be actionable. As such, the ability to incorporate sex and the germline NF1 gene mutation in combination with other factors, like asthma and genomic background77–80, into predicable risk assessment models is likely to improve our ability to manage these children. In addition, some of these factors may yield actionable outcomes, including new therapeutic approaches. In this regard, we are in a unique position to effectively translate basic laboratory research findings into improved management strategies for NF1-OPG. The availability of numerous authenticated preclinical models, human induced pluripotent stem cell reagents76, and a successful multi-institutional NF Clinical Trials Consortium81 offer unprecedented opportunities to apply precision medicine approaches to this common brain tumor in children with NF1.
Acknowledgments
Acknowledgements of research support: D.H.G. is supported by a Research Program Award from the National Institute of Neurological Disorders and Stroke (1-R35-NS097211-01).
We authors thank Dr. Corina Anastasaki for creating the expert illustrations.
Footnotes
Conflicts: None to disclose.
References
- 1.Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26(11):704–11. doi: 10.1136/jmg.26.11.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151(1):33–40. doi: 10.1093/oxfordjournals.aje.a010118. [DOI] [PubMed] [Google Scholar]
- 3.Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13(8):834–43. doi: 10.1016/S1474-4422(14)70063-8. [DOI] [PubMed] [Google Scholar]
- 4.Gutmann DH, Rasmussen SA, Wolkenstein P, et al. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1) Neurology. 2002;59(5):759–61. doi: 10.1212/wnl.59.5.759. [DOI] [PubMed] [Google Scholar]
- 5.Guillamo JS, Creange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003;126(Pt 1):152–60. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- 6.Lewis RA, Gerson LP, Axelson KA, Riccardi VM, Whitford RP. von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology. 1984;91(8):929–35. doi: 10.1016/s0161-6420(84)34217-8. [DOI] [PubMed] [Google Scholar]
- 7.Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63–6. doi: 10.1016/s0022-3476(94)70122-9. [DOI] [PubMed] [Google Scholar]
- 8.Fisher MJ, Loguidice M, Gutmann DH, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro-oncol. 2012;14(6):790–7. doi: 10.1093/neuonc/nos076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61(3):189–98. doi: 10.1002/ana.21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Listernick R, Ferner RE, Piersall L, Sharif S, Gutmann DH, Charrow J. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology. 2004;63(10):1944–6. doi: 10.1212/01.wnl.0000144341.16830.01. [DOI] [PubMed] [Google Scholar]
- 11.Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. 1989;114(5):788–92. doi: 10.1016/s0022-3476(89)80137-4. [DOI] [PubMed] [Google Scholar]
- 12.Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127(5):718–22. doi: 10.1016/s0022-3476(95)70159-1. [DOI] [PubMed] [Google Scholar]
- 13.Segal L, Darvish-Zargar M, Dilenge ME, Ortenberg J, Polomeno RC. Optic pathway gliomas in patients with neurofibromatosis type 1: follow-up of 44 patients. J AAPOS. 2010;14(2):155–8. doi: 10.1016/j.jaapos.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Habiby R, Silverman B, Listernick R, Charrow J. Precocious puberty in children with neurofibromatosis type 1. J Pediatr. 1995;126(3):364–7. doi: 10.1016/s0022-3476(95)70449-3. [DOI] [PubMed] [Google Scholar]
- 15.Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75(2):309–16. doi: 10.1002/ana.24093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher MJ, Loguidice M, Gutmann DH, et al. Gender as a disease modifier in neurofibromatosis type 1 optic pathway glioma. Ann Neurol. 2014;75(5):799–800. doi: 10.1002/ana.24157. [DOI] [PubMed] [Google Scholar]
- 17.Creange A, Zeller J, Rostaing-Rigattieri S, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999;122(Pt 3):473–81. doi: 10.1093/brain/122.3.473. [DOI] [PubMed] [Google Scholar]
- 18.Balcer LJ, Liu GT, Heller G, et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol. 2001;131(4):442–5. doi: 10.1016/s0002-9394(00)00852-7. [DOI] [PubMed] [Google Scholar]
- 19.Zeid JL, Charrow J, Sandu M, Goldman S, Listernick R. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J AAPOS. 2006;10(6):534–9. doi: 10.1016/j.jaapos.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 20.de Blank PM, Berman JI, Liu GT, Roberts TP, Fisher MJ. Fractional anisotropy of the optic radiations is associated with visual acuity loss in optic pathway gliomas of neurofibromatosis type 1. Neuro-oncol. 2013;15(8):1088–95. doi: 10.1093/neuonc/not068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher MJ, Avery RA, Allen JC, et al. Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology. 2013;81(21 Suppl 1):S15–24. doi: 10.1212/01.wnl.0000435745.95155.b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avery RA, Ferner RE, Listernick R, Fisher MJ, Gutmann DH, Liu GT. Visual acuity in children with low grade gliomas of the visual pathway: implications for patient care and clinical research. J Neurooncol. 2012;110(1):1–7. doi: 10.1007/s11060-012-0944-y. [DOI] [PubMed] [Google Scholar]
- 23.King A, Listernick R, Charrow J, Piersall L, Gutmann DH. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet A. 2003;122A(2):95–9. doi: 10.1002/ajmg.a.20211. [DOI] [PubMed] [Google Scholar]
- 24.Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143–9. doi: 10.1002/ana.410410204. [DOI] [PubMed] [Google Scholar]
- 25.Thiagalingam S, Flaherty M, Billson F, North K. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology. 2004;111(3):568–77. doi: 10.1016/j.ophtha.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Liu GT. Optic gliomas of the anterior visual pathway. Curr Opin Ophthalmol. 2006;17(5):427–31. doi: 10.1097/01.icu.0000243016.90004.12. [DOI] [PubMed] [Google Scholar]
- 27.Astrup J. Natural history and clinical management of optic pathway glioma. Br J Neurosurg. 2003;17(4):327–35. doi: 10.1080/02688690310001601216. [DOI] [PubMed] [Google Scholar]
- 28.Gutmann DH, McLellan MD, Hussain I, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013;23(3):431–9. doi: 10.1101/gr.142604.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reemst K, Noctor SC, Lucassen PJ, Hol EM. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front Hum Neurosci. 2016;10:566. doi: 10.3389/fnhum.2016.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–34. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–7. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raabe EH, Lim KS, Kim JM, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–9. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23(43):7267–73. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 34.Ricker CA, Pan Y, Gutmann DH, Keller C. Challenges in Drug Discovery for Neurofibromatosis Type 1-Associated Low-Grade Glioma. Front Oncol. 2016;6:259. doi: 10.3389/fonc.2016.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63(24):8573–7. [PubMed] [Google Scholar]
- 36.Zhu Y, Harada T, Liu L, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132(24):5577–88. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee DY, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22(1):131–8. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22(14):5100–13. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol. 2005;57(1):119–27. doi: 10.1002/ana.20337. [DOI] [PubMed] [Google Scholar]
- 40.Banerjee D, Hegedus B, Gutmann DH, Garbow JR. Detection and measurement of neurofibromatosis-1 mouse optic glioma in vivo. Neuroimage. 2007;35(4):1434–7. doi: 10.1016/j.neuroimage.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim KY, Ju WK, Hegedus B, Gutmann DH, Ellisman MH. Ultrastructural characterization of the optic pathway in a mouse model of neurofibromatosis-1 optic glioma. Neuroscience. 2010;170(1):178–88. doi: 10.1016/j.neuroscience.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegedus B, Hughes FW, Garbow JR, et al. Optic nerve dysfunction in a mouse model of neurofibromatosis-1 optic glioma. J Neuropathol Exp Neurol. 2009;68(5):542–51. doi: 10.1097/NEN.0b013e3181a3240b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toonen JA, Ma Y, Gutmann DH. Defining the temporal course of murine neurofibromatosis-1 optic gliomagenesis reveals a therapeutic window to attenuate retinal dysfunction. Neuro Oncol. 2016 doi: 10.1093/neuonc/now267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gutmann DH, Giordano MJ, Mahadeo DK, Lau N, Silbergeld D, Guha A. Increased neurofibromatosis 1 gene expression in astrocytic tumors: positive regulation by p21-ras. Oncogene. 1996;12(10):2121–7. [PubMed] [Google Scholar]
- 45.Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005;65(1):236–45. [PubMed] [Google Scholar]
- 46.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65(7):2755–60. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 47.Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68(5):1520–8. doi: 10.1158/0008-5472.CAN-07-5916. [DOI] [PubMed] [Google Scholar]
- 48.Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol. 2015;17(6):843–53. doi: 10.1093/neuonc/nou329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banerjee S, Gianino SM, Gao F, Christians U, Gutmann DH. Interpreting mammalian target of rapamycin and cell growth inhibition in a genetically engineered mouse model of Nf1-deficient astrocytes. Mol Cancer Ther. 2011;10(2):279–91. doi: 10.1158/1535-7163.MCT-10-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen YH, McGowan LD, Cimino PJ, et al. Mouse low-grade gliomas contain cancer stem cells with unique molecular and functional properties. Cell Rep. 2015;10(11):1899–912. doi: 10.1016/j.celrep.2015.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol. 2013;73(2):303–8. doi: 10.1002/ana.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16(9):1098–112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 53.Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res. 2008;68(24):10358–66. doi: 10.1158/0008-5472.CAN-08-2506. [DOI] [PubMed] [Google Scholar]
- 54.Simmons GW, Pong WW, Emnett RJ, et al. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol. 2011;70(1):51–62. doi: 10.1097/NEN.0b013e3182032d37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solga AC, Pong WW, Kim KY, et al. RNA Sequencing of Tumor-Associated Microglia Reveals Ccl5 as a Stromal Chemokine Critical for Neurofibromatosis-1 Glioma Growth. Neoplasia. 2015;17(10):776–88. doi: 10.1016/j.neo.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown JA, Gianino SM, Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci. 2010;30(16):5579–89. doi: 10.1523/JNEUROSCI.3994-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toonen JA, Solga AC, Ma Y, Gutmann DH. Estrogen activation of microglia underlies the sexually dimorphic differences in Nf1 optic glioma-induced retinal pathology. J Exp Med. 2016 doi: 10.1084/jem.20160447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolin G, Parkin P, Mabbott D, et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer. 2009;53(7):1231–7. doi: 10.1002/pbc.22198. [DOI] [PubMed] [Google Scholar]
- 59.Ullrich NJ, Robertson R, Kinnamon DD, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology. 2007;68(12):932–8. doi: 10.1212/01.wnl.0000257095.33125.48. [DOI] [PubMed] [Google Scholar]
- 60.Dalla Via P, Opocher E, Pinello ML, et al. Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol. 2007;9(4):430–7. doi: 10.1215/15228517-2007-031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalin-Hajdu E, Decarie JC, Marzouki M, Carret AS, Ospina LH. Visual acuity of children treated with chemotherapy for optic pathway gliomas. Pediatr Blood Cancer. 2014;61(2):223–7. doi: 10.1002/pbc.24726. [DOI] [PubMed] [Google Scholar]
- 62.Larizza L, Gervasini C, Natacci F, Riva P. Developmental abnormalities and cancer predisposition in neurofibromatosis type 1. Curr Mol Med. 2009;9(5):634–53. doi: 10.2174/156652409788488801. [DOI] [PubMed] [Google Scholar]
- 63.de Blank PM, Berman JI, Fisher MJ. Systemic Chemotherapy and White Matter Integrity in Tracts Associated with Cognition Among Children With Neurofibromatosis Type 1. Pediatr Blood Cancer. 2016;63(5):818–24. doi: 10.1002/pbc.25896. [DOI] [PubMed] [Google Scholar]
- 64.Packer RJ, Lange B, Ater J, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. 1993;11(5):850–6. doi: 10.1200/JCO.1993.11.5.850. [DOI] [PubMed] [Google Scholar]
- 65.Bouffet E, Jakacki R, Goldman S, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol. 2012;30(12):1358–63. doi: 10.1200/JCO.2011.34.5843. [DOI] [PubMed] [Google Scholar]
- 66.Cappellano AM, Petrilli AS, da Silva NS, et al. Single agent vinorelbine in pediatric patients with progressive optic pathway glioma. J Neurooncol. 2015;121(2):405–12. doi: 10.1007/s11060-014-1652-6. [DOI] [PubMed] [Google Scholar]
- 67.Gururangan S, Fisher MJ, Allen JC, et al. Temozolomide in children with progressive low-grade glioma. Neuro Oncol. 2007;9(2):161–8. doi: 10.1215/15228517-2006-030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Avery RA, Hwang EI, Jakacki RI, Packer RJ. Marked recovery of vision in children with optic pathway gliomas treated with bevacizumab. JAMA Ophthalmol. 2014;132(1):111–4. doi: 10.1001/jamaophthalmol.2013.5819. [DOI] [PubMed] [Google Scholar]
- 69.Karajannis MA, Legault G, Fisher MJ, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol. 2014;16(10):1408–16. doi: 10.1093/neuonc/nou059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.See WL, Tan IL, Mukherjee J, Nicolaides T, Pieper RO. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012;72(13):3350–9. doi: 10.1158/0008-5472.CAN-12-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dombi E, Baldwin A, Marcus LJ, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550–60. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hutter S, Piro RM, Waszak SM, et al. No correlation between NF1 mutation position and risk of optic pathway glioma in 77 unrelated NF1 patients. Human genetics. 2016;135(5):469–75. doi: 10.1007/s00439-016-1646-x. [DOI] [PubMed] [Google Scholar]
- 73.Bolcekova A, Nemethova M, Zatkova A, et al. Clustering of mutations in the 5′ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma. 2013;60(6):655–65. doi: 10.4149/neo_2013_084. [DOI] [PubMed] [Google Scholar]
- 74.Sharif S, Upadhyaya M, Ferner R, et al. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J Med Genet. 2011;48(4):256–60. doi: 10.1136/jmg.2010.081760. [DOI] [PubMed] [Google Scholar]
- 75.Toonen JA, Anastasaki C, Smithson LJ, et al. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum Mol Genet. 2016;25(9):1703–13. doi: 10.1093/hmg/ddw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anastasaki C, Woo AS, Messiaen LM, Gutmann DH. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum Mol Genet. 2015;24(12):3518–28. doi: 10.1093/hmg/ddv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Porcelli B, Zoellner NL, Abadin SS, Gutmann DH, Johnson KJ. Associations between allergic conditions and pediatric brain tumors in Neurofibromatosis type 1. Fam Cancer. 2016;15(2):301–8. doi: 10.1007/s10689-015-9855-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abadin SS, Zoellner NL, Schaeffer M, Porcelli B, Gutmann DH, Johnson KJ. Racial/Ethnic Differences in Pediatric Brain Tumor Diagnoses in Patients with Neurofibromatosis Type 1. J Pediatr. 2015;167(3):613–20. e1–2. doi: 10.1016/j.jpeds.2015.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reilly KM. Using the Collaborative Cross to Study the Role of Genetic Diversity in Cancer-Related Phenotypes. Cold Spring Harb Protoc. 2016;2016(3) doi: 10.1101/pdb.prot079178. pdb prot079178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warrington NM, Sun T, Luo J, et al. The cyclic AMP pathway is a sex-specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res. 2015;75(1):16–21. doi: 10.1158/0008-5472.CAN-14-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gutmann DH, Blakeley JO, Korf BR, Packer RJ. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs. 2013;22(4):443–62. doi: 10.1517/13543784.2013.772979. [DOI] [PMC free article] [PubMed] [Google Scholar]