

In primary sclerosing cholangitis (PSC), annular fibrosis around intrahepatic and extrahepatic bile ducts leads to progressive liver disease for which there is no effective therapy except liver transplantation.1 The concentric accumulation of connective tissue around bile ducts suggests that the cholangiocyte plays an integral role in PSC pathogenesis. However, what initiates changes in cholangiocyte phenotype and how cholangiocytes interact with cells in the peribiliary extracellular matrix like immune cells and stromal components is largely unknown. Over the last 10 years, genome-wide association studies in PSC have revealed >20 risk genes.2–5 A significant observation that can be derived from these data, which also applies to other non-Mendelian phenotypes, is that the predominant risk contribution is likely to come from one or more environmental sources, rather than the genetic aberrations. Indeed, in PSC, we estimate that <10% of the overall liability is accounted for by the genetic findings; with extrapolations into hypothetical, larger study populations, it is unlikely to exceed 30%–40%.6 7 Research dissecting the remaining environmental contribution to PSC and other complex diseases is methodologically challenging. The exposures of an organism throughout life as a whole have recently been referred to as the exposome8 and include a myriad of components of both the external (eg, xenobiotics) and internal (eg, gut microbes) milieu.
There is a long tradition and effective means for detecting infectious exposures in medicine. Robert Koch in the late 19th century proposed criteria to identify a disease as infectious.9 These included (i) the organism is regularly associated with the disease, (ii) the organism can be isolated from the diseased host and grown in culture and (iii) the disease can be reproduced when the organism is introduced into a healthy susceptible host. With the development of nucleic acid sequence-based identification of microbes, as well as the recognition that the behaviour of a particular microbe can be influenced by the community in which it resides, Koch’s original postulates have evolved.10 11 More specifically, the concept has developed to incorporate culture-independent techniques that can identify putative pathogen nucleic acid sequences or even microbe-derived products (eg, short-chain fatty acids) in association with a diseased tissue that decrease with resolution of disease and increase with clinical relapse.11 These ideas suggest that ‘the scientific community should consider infectious disease causation in a broader systems biology context …’ and ‘… as technology advances and new scientific discoveries are made, there must be dynamic adaptation of Koch’s postulates …’.10 Recent examples of an expanded paradigm thus incorporate the transfer of body weight-associated, IgA-associated or liver injury-associated gut microbiota in causation of obesity, IBD or alcoholic hepatitis, respectively.12–14
There is less of a tradition and only preliminary tools to detect non-infectious environmental exposures in medicine. In light of the strong environmental component in PSC liability determined by the genetic studies, a reconsideration as to how PSC may fulfil the revised Koch’s criteria is however timely. A rationale for this proposition can be made on several sets of considerations, each of which will be addressed in this commentary. These include (i) the evolution of Koch’s postulates to account for complex exposure-driven disease,10 11 for example, obesity and IBD12 15; (ii) similarities in cholangiocyte phenotype and downstream signalling pathways between secondary sclerosing cholangitis (SSC) due to a direct microbial infection (eg, Cryptosporidium parvum)16 and PSC; (iii) accumulating evidence supporting a critical role for the intestinal microbiome in the pathogenesis of PSC17; (iv) a similar genetic architecture in PSC as in a prototypical exposure-driven autoimmune disease, coeliac disease18; and (v) a similar genetic architecture in PSC as in cholestatic drug-induced injury.19 20
We began studying the cholangiopathy in HIV-infected patients (a form of SSC due to biliary tract infection by opportunistic pathogens, including C parvum)21 both because it was a significant clinical problem and as a result of our own clinical experience with C parvum-induced SSC.22 To summarise key points from multiple in vitro and in vivo studies,16 23 we found that C parvum, presumably via retrograde invasion of the biliary tree from the small intestine, resulted in a pro-inflammatory, activated cholangiocyte phenotype and that the process involved a number of key molecules (eg, toll-like receptors, nuclear factor KB, C/EBP Beta and NRAS).24 25 Moreover, the infection was associated with altered cholangiocyte cytokine and miRNA expression26 27 as well as inflammation and fibrosis.16 More recently, and relevant to this commentary, we noted that virtually all of the changes described as a result of cholangiocyte invasion by C parvum also occurred when microbial-derived products (eg, lipopolysaccharide, flagellin) were used as an alternative to the microbe itself.25 28 Additionally, cholangiocytes in vitro develop a senescent phenotype (ie, proliferative arrest), likely involving these same pathways and processes, when chronically exposed to microbial-derived-products29 (figure 1). Importantly, we discovered that these pathways and processes are likely operative in human PSC.
Figure 1.

Model of cholangiocyte activation. (A) Bile ducts are lined by cholangiocytes, specialised epithelia that under normal physiological conditions modify bile through the transport of water, ions and solutes. Portal myofibroblasts are adjacent to bile duct epithelia within the portal tract and are distinct from hepatic stellate cells which line the hepatic sinusoids; both can differentiate to matrix depositing myofibroblasts under injurious conditions. (B) Cholangiocytes exist in a harsh environment and are exposed to a variety of insults such as microbes, pathogen-associated molecular patterns, danger-associated molecular patterns, xenobiotics and bile acid-induced damage during cholestasis (from a variety of potential mechanisms, eg, oxidative stress). Recognition of these insults, for example, via pathogen recognition receptors or damage-associated molecular pattern receptors, promotes an activated cholangiocyte phenotype characterised by increased proliferation and secretion of profibrotic (eg, connective tissue growth factor) and proinflammatory (eg, interleukin 6 and 8) mediators. In this model, the activated cholangiocyte promotes hepatobiliary repair processes and recruits a variety of innate (eg, macrophages) and professional (eg, T cell) immune cells. On persistent insult, some injured cholangiocytes enter the cellular state of senescence, characterised by withdrawal from the cell cycle, and transition to a hypersecretory proinflammatory state, that is, senescence-associated secretory phenotype. On persistence, such an inflammatory/fibrotic environment will lead to sclerosing cholangitis. Reproduced with permission of Kari C. Toverud.
A series of divergent published and unpublished data are evolving that independently suggest, from a different perspective, microbial contributions to PSC. These data in patients with PSC include (i) expression of microbial receptors on cholangiocytes,30 and the occurrence of bacterobilia,31 32 (iii) bacteraemia of the portal venous system,33 (iv) promising results of antibiotics in uncontrolled studies34–37 and (v) genomic associations with loci implicated in host/microbiome interactions.38 The clinical relationship between the gut (including potentially those microbes that reside therein) and the liver in PSC was established 50 years ago with the observation that PSC frequently occurred in the setting of IBD.39 40 A series of studies recently published in Gut have demonstrated that the gut microbiome in PSC is distinct from those microbial communities observed in IBD patients without PSC and healthy controls.41–44 The relationship between the gut microbial communities and the liver is bidirectional, meaning that there is an impact from host factors onto the gut microbiota by bile and intestinal secretions and the immune system, as much as from microbial metabolites and constituents provided via the gut mucosa and the portal circulation (figure 2).17 Support for a role of intestinal microbiota in PSC also comes from studies performed in germ-free mice. In the biliary bile acid toxicity model (Abcb4-/-), an aggravation of bile duct disease was observed in the germ-free animals compared with conventionally raised mice.45 In contrast, in the immune-driven NOD.c3c4 model, an amelioration of bile duct disease was observed in the germ-free animals.46 These differences highlight the complexity of the relationship between gut-derived exposures and the liver and the bile ducts, and suggest that various components of PSC pathogenesis may be affected differently. Further studies are now urgently needed to link the microbial community alterations of PSC and their metabolic and immunological consequences for hepatobiliary physiology and disease.
Figure 2.

The gut–liver relationship in primary sclerosing cholangitis (PSC). There is bidirectional relationship between the gut and the liver in terms of delivery of a number of endogenous metabolites and bioactive compounds to the gut (eg, bile acids). Reversely, there is an ongoing delivery of compounds from the intestinal environment via portal blood to the liver. In this bilateral concept, the gut microbiota and the liver comprise an integrated physiological machinery under the influence of endogenous as well as external factors, in which the role of cholangiocytes warrants further attention. Reproduced with permission from Ref. 17.
Given the above considerations supporting the importance of the environment in the pathogenesis of PSC, many key questions remain largely unanswered. For example, what is the nature of a potential environmental exposure in PSC? Is it a small molecule or a peptide-derived substance? Is it dietary or microbial in origin (or both, meaning xenobiotic transformed by microbial metabolism)? Is it singular and specific in nature (like gluten in coeliac disease) or is it a compound microbial impact (like most likely the case in Crohn’s disease)? Are there different factors responsible for triggering and driving disease, respectively? At this time, answers to these questions can only be provided based on circumstantial evidence. From the genetic perspective, the strong human leucocyte antigen (HLA) class II associations (DRB1) are suggestive of at least one singular causative compound of key importance, similar to the situation in coeliac disease or drug-induced injury (figure 3).47 In comparison, the genetic architecture of non-autoimmune, inflammatory diseases like Crohn’s disease is largely different (figure 3), possibly due to a broader spectrum of environmental factors being involved. Further to this thinking, in coeliac disease and drug-induced liver injury, the site of metabolism is crucial for disease localisation (in coeliac disease by transglutaminase 2 in the proximal intestine, in drug-induced liver injury in the liver). In PSC, disease distribution stretches across the distal ileum throughout most of the colon (with a right-sided predominance) into the entire surface of the intrahepatic and extrahepatic bile ducts. Our knowledge of the metabolic machinery of the epithelium in the biliary fraction of this surface (cholangiocytes) is still relatively rudimentary compared with hepatocytes and enterocytes. This also makes assessments as to what could be the components involved with causative factors very difficult. Possibly, as shown in figure 4, working from the currently known genetic co-variables of PSC development, HLA class II in particular, is a more direct way of determining the identity of causative factors than unbiased –omics technologies providing broad, correlative data.
Figure 3.

Genome-wide association study outcomes shown as Manhattan plots. In primary sclerosing cholangitis (PSC) and prototypical autoimmune diseases, there is a strong human leucocyte antigen association (chromosome 6). A similar genetic architecture is also seen in diseases elicited by specific environmental exposures (exemplified by drug-induced liver injury and coeliac disease), this contrasts the situation in diseases where a compound environmental insult is involved, exemplified by Crohn’s disease. The figure shows Manhattan plots with results of genome-wide association studies in (A) PSC, (B) flucloxacillin-induced liver injury, (C) coeliac disease and (D) Crohn’s disease. The X axis shows the chromosomal location, the Y axis the -log10 p values of the association statistics. Panel (B) is reproduced with permission from Ref. 19. Panels (A), (C) and (D) plotted from data in Ref. 20.
Figure 4.
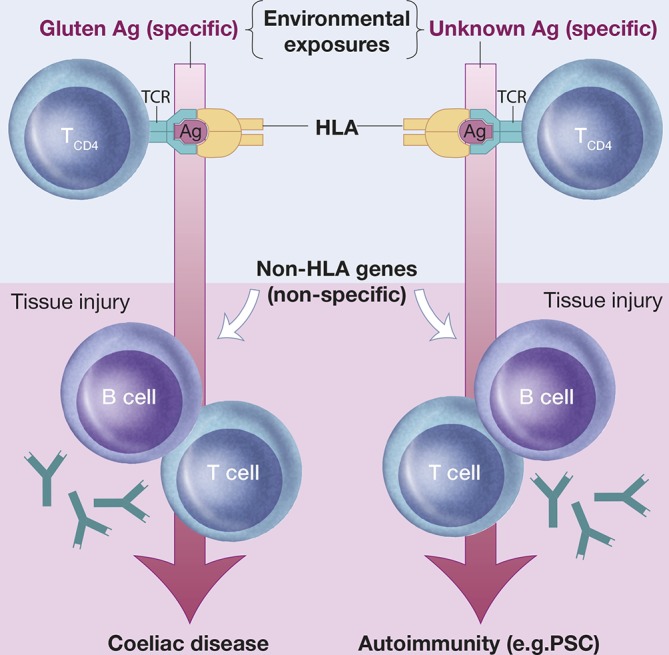
A coeliac disease model of primary sclerosing cholangitis (PSC) susceptibility. In coeliac disease, the disease-associated human leucocyte antigen (HLA) variants direct the adaptive immune response to gluten. Exposure to gluten, as well as the resulting gluten-specific adaptive immunity, respectively, is required to maintain autoantibody production and immunopathology in coeliac disease. Such observations challenge the concept that autoimmunity requires immune activation towards self-antigens. It may thus be hypothesized that the strong genetic HLA associations in PSC (and other autoimmune diseases) point to specific causal environmental exposures determined by the HLA/antigen (Ag)/T-cell receptor (TCR) interactions. Within this concept, genetic and environmental factors are co-dependent in disease causation with the implication that genetic risk factors may hold clues as to the identity of pathogenic environmental factors. For further reading, see Ref. 47. Reproduced with permission of Kari C. Toverud.
In summary, the evolution of Koch’s postulates based on the availability of new technologies, the similarities in the intracellular signalling pathways, processes and pathophysiological outcomes of SSC versus PSC, as well as recent data strongly implicating an important role for the intestinal microbiome in PSC, all justify serious consideration and further experiments to test the possibility that PSC is caused by an environmental exposure. Genetic and environmental factors are inseparable and co-dependent in the causation of PSC as in any disease, and specific genetic findings made over the last decade, HLA related in particular, may for this reason guide the way forward in a methodologically challenging research space. The molecular machinery of the cholangiocyte is likely to play a key role at this gene–environment intersection, and basic research to enhance our understanding of normal and activated cholangiocyte function is urgently needed to test for their involvement with putative pathogenic factors.
Footnotes
Contributors: All authors have contributed equally to the article.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med 2016;375:1161–70. 10.1056/NEJMra1506330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji SG, Juran BD, Mucha S, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269–73. 10.1038/ng.3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellinghaus D, Jostins L, Spain SL, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet 2016;48:510–8. 10.1038/ng.3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu JZ, Hov JR, Folseraas T, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 2013;45:670–5. 10.1038/ng.2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melum E, Franke A, Schramm C, et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat Genet 2011;43:17–19. 10.1038/ng.728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014;46:1173–86. 10.1038/ng.3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stahl EA, Wegmann D, Trynka G, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet 2012;44:483–9. 10.1038/ng.2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogler G, Vavricka S. Exposome in IBD: recent insights in environmental factors that influence the onset and course of IBD. Inflamm Bowel Dis 2015;21:400–8. 10.1097/MIB.0000000000000229 [DOI] [PubMed] [Google Scholar]
- 9.Koch R. Ueber bakteriologische Forschung Verh X Int Med Congr 1890. Berlin, 1892. [Google Scholar]
- 10.Byrd AL, Segre JA. Infectious disease. Adapting Koch’s postulates. Science 2016;351:224–6. 10.1126/science.aad6753 [DOI] [PubMed] [Google Scholar]
- 11.Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev 1996;9:18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–131. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- 13.Palm NW, de Zoete MR, Cullen TW, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014;158:1000–10. 10.1016/j.cell.2014.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrere G, Wrzosek L, Cailleux F, et al. Fecal Microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J Hepatol 2017;66:806–15. 10.1016/j.jhep.2016.11.008 [DOI] [PubMed] [Google Scholar]
- 15.Kaser A, Zeissig S, Blumberg RS. Genes and environment: how will our concepts on the pathophysiology of IBD develop in the future? Dig Dis 2010;28:395–405. 10.1159/000320393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Hara SP, Bogert PS, Trussoni CE, et al. TLR4 promotes Cryptosporidium parvum clearance in a mouse model of biliary cryptosporidiosis. J Parasitol 2011;97:813–21. 10.1645/GE-2703.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlsen TH. Primary sclerosing cholangitis: 50 years of a gut-liver relationship and still no love? Gut 2016;65:1579–81. 10.1136/gutjnl-2016-312137 [DOI] [PubMed] [Google Scholar]
- 18.Karlsen TH, Chung BK. Genetic risk and the development of autoimmune liver disease. Dig Dis 2015;33(Suppl 2):13–24. 10.1159/000440706 [DOI] [PubMed] [Google Scholar]
- 19.Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a Major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 2009;41:816–9. 10.1038/ng.379 [DOI] [PubMed] [Google Scholar]
- 20.Liu JZ, Hov JR, Folseraas T, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 2013;45:670–5. 10.1038/ng.2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen XM, LaRusso NF. Cryptosporidiosis and the pathogenesis of AIDS-cholangiopathy. Semin Liver Dis 2002;22:277–90. 10.1055/s-2002-34505 [DOI] [PubMed] [Google Scholar]
- 22.Cockerill FR, Hurley DV, Malagelada JR, et al. Polymicrobial cholangitis and Kaposi’s sarcoma in blood product transfusion-related acquired immune deficiency syndrome. Am J Med 1986;80:1237–41. 10.1016/0002-9343(86)90695-9 [DOI] [PubMed] [Google Scholar]
- 23.O’Hara SP, Chen XM. The cell biology of Cryptosporidium infection. Microbes Infect 2011;13:721–30. 10.1016/j.micinf.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen XM, O’Hara SP, LaRusso NF. The immunobiology of cholangiocytes. Immunol Cell Biol 2008;86:497–505. 10.1038/icb.2008.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Hara SP, Splinter PL, Gajdos GB, et al. NFkappaB p50-CCAAT/enhancer-binding protein beta (C/EBPbeta)-mediated transcriptional repression of microRNA let-7i following microbial infection. J Biol Chem 2010;285:216–25. 10.1074/jbc.M109.041640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen XM, Splinter PL, O’Hara SP, et al. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem 2007;282:28929–38. 10.1074/jbc.M702633200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou R, O’Hara SP, Chen XM. MicroRNA regulation of innate immune responses in epithelial cells. Cell Mol Immunol 2011;8:371–9. 10.1038/cmi.2011.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Hara SP, Splinter PL, Trussoni CE, et al. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J Biol Chem 2011;286:30352–60. 10.1074/jbc.M111.269464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabibian JH, O’Hara SP, Splinter PL, et al. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014;59:2263–75. 10.1002/hep.26993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen XM, O’Hara SP, Nelson JB, et al. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J Immunol 2005;175:7447–56. 10.4049/jimmunol.175.11.7447 [DOI] [PubMed] [Google Scholar]
- 31.Olsson R, Björnsson E, Bäckman L, et al. Bile duct bacterial isolates in primary sclerosing cholangitis: a study of explanted livers. J Hepatol 1998;28:426–32. 10.1016/S0168-8278(98)80316-4 [DOI] [PubMed] [Google Scholar]
- 32.Pohl J, Ring A, Stremmel W, et al. The role of dominant stenoses in bacterial infections of bile ducts in primary sclerosing cholangitis. Eur J Gastroenterol Hepatol 2006;18:69–74. 10.1097/00042737-200601000-00012 [DOI] [PubMed] [Google Scholar]
- 33.O’Toole A, Alakkari A, Keegan D, et al. Primary sclerosing cholangitis and disease distribution in inflammatory bowel disease. Clin Gastroenterol Hepatol 2012;10:439–41. 10.1016/j.cgh.2011.11.010 [DOI] [PubMed] [Google Scholar]
- 34.Elfaki DA, Lindor KD. Antibiotics for the treatment of primary sclerosing cholangitis. Am J Ther 2011;18:261–5. 10.1097/MJT.0b013e3181b7b8c0 [DOI] [PubMed] [Google Scholar]
- 35.Färkkilä M, Karvonen AL, Nurmi H, et al. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial. Hepatology 2004;40:1379–86. 10.1002/hep.20457 [DOI] [PubMed] [Google Scholar]
- 36.Silveira MG, Torok NJ, Gossard AA, et al. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. Am J Gastroenterol 2009;104:83–8. 10.1038/ajg.2008.14 [DOI] [PubMed] [Google Scholar]
- 37.Tabibian JH, Weeding E, Jorgensen RA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis - a pilot study. Aliment Pharmacol Ther 2013;37:604–12. 10.1111/apt.12232 [DOI] [PubMed] [Google Scholar]
- 38.Maroni L, Hohenester SD, van de Graaf SFJ, et al. Knockout of the primary sclerosing cholangitis-risk gene Fut2 causes liver disease in mice. Hepatology 2017. 10.1002/hep.29029 [DOI] [PubMed] [Google Scholar]
- 39.Thorpe ME, Scheuer PJ, Sherlock S. Primary sclerosing cholangitis, the biliary tree, and ulcerative colitis. Gut 1967;8:435–48. 10.1136/gut.8.5.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warren KW, Athanassiades S, Monge JI. Primary sclerosing cholangitis. A study of forty-two cases. Am J Surg 1966;111:23–38. [DOI] [PubMed] [Google Scholar]
- 41.Kummen M, Holm K, Anmarkrud JA, et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017;66:611–9. 10.1136/gutjnl-2015-310500 [DOI] [PubMed] [Google Scholar]
- 42.Quraishi MN, Sergeant M, Kay G, et al. The gut-adherent Microbiota of PSC-IBD is distinct to that of IBD. Gut 2017;66:386–8. 10.1136/gutjnl-2016-311915 [DOI] [PubMed] [Google Scholar]
- 43.Rühlemann MC, Heinsen FA, Zenouzi R, et al. Faecal Microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis. Gut 2017;66:753–4. 10.1136/gutjnl-2016-312180 [DOI] [PubMed] [Google Scholar]
- 44.Sabino J, Vieira-Silva S, Machiels K, et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016;65:1681–9. 10.1136/gutjnl-2015-311004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tabibian JH, O’Hara SP, Trussoni CE, et al. Absence of the intestinal Microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016;63:185–96. 10.1002/hep.27927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schrumpf E, Kummen M, Valestrand L, et al. The gut Microbiota contributes to a mouse model of spontaneous bile duct inflammation. J Hepatol 2017;66:382–9. 10.1016/j.jhep.2016.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol 2013;13:294–302. 10.1038/nri3407 [DOI] [PMC free article] [PubMed] [Google Scholar]