

Abstract
Background & Aims
Chronic gastrointestinal inflammation increases the risk of cancer by mechanisms that are not well understood. Indoleamine-2,3-dioxygenase 1 (IDO1) is a heme-binding enzyme that regulates the immune response via catabolization and regulation of tryptophan availability for immune cell uptake. IDO1 expression is increased during the transition from chronic inflammation to gastric metaplasia. We investigated whether IDO1 contributes to the inflammatory response that mediates loss of parietal cells leading to metaplasia.
Methods
Chronic gastric inflammation was induced in Ido1−/− and CB57BL/6 (control) mice by gavage with Helicobacter felis or overexpression of interferon gamma in gastric parietal cells. We also performed studies in Jh−/− mice, which are devoid of B cells. Gastric tissues were collected and analyzed by flow cytometry, immunostaining, and reverse transcription quantitative PCR. Plasma samples were analyzed by ELISA. Gastric tissues were obtained from 20 patients with gastric metaplasia and 20 patients without gastric metaplasia (controls) and analyzed by reverse transcription quantitative PCR; gastric tissue arrays were analyzed by immunohistochemistry. We collected genetic information on gastric cancers from The Cancer Genome Atlas database.
Results
H felis gavage induced significantly lower levels of pseudopyloric metaplasia in Ido1−/− mice, which had lower frequencies of gastric B cells, than in control mice. Blood plasma from H felis-infected control mice had increased levels of autoantibodies against parietal cells, compared to uninfected control mice, but this increase was lower in Ido1−/− mice. Chronically inflamed stomachs of Ido1−/− mice had significantly lower frequencies of natural killer cells in contact with parietal cells, compared with stomachs of control mice. Jh−/− mice had lower levels of pseudopyloric metaplasia than control mice in response to H felis infection. Human gastric pre-neoplasia and carcinoma specimens had increased levels of IDO1 mRNA compared with control gastric tissues, and IDO1 protein co-localized with B cells. Co-clustering of IDO1 mRNA with B-cell markers was corroborated by the TCGA database.
Conclusions
IDO1 mediates gastric metaplasia by regulating the B-cell compartment. This process appears to be associated with type II hypersensitivity/autoimmunity. The role of autoimmunity in the progression of pseudopyloric metaplasia warrants further investigation.
Keywords: kynurenine, gastritis, SPEM, humoral immunity
INTRODUCTION
It is well established that a prolonged latency period of chronic inflammation (~8–10 years) precedes the development of gastrointestinal cancers1, 2, including gastric cancer3, 4. Therefore, the quality of inflammation can change during this long-lived process: in the later stages of chronic inflammation, immune cells should display a pathological phenotype that can trigger carcinogenesis. This “switch” in the inflammatory milieu has not been characterized. We have recently identified a mouse model (the Gli1−/− model) in which the progression from chronic gastric inflammation to metaplasia does not occur5. This model led to the identification of several metaplasia-associated genes, several of which played a role in immunity. One of these differentially induced genes was indoleamine-2,3-dioxygenase 1 (IDO1). We therefore sought to assess the contribution of IDO1 to gastric metaplasia and determine its mechanism.
IDO1 is traditionally known to suppress T cell immunity6. It functions by metabolizing tryptophan into kynurenine7. In doing so this enzyme restricts the tryptophan pool in tumor microenvironments, therefore reducing T cell numbers6. The enzyme also increases kynurenine levels in the microenvironment, which stimulates regulatory T cell (Treg) differentiation8. However recently, IDO1 has been described to regulate other populations, e.g., B cells9–11 and epithelial cells12. In addition, we recently reported that IDO1 regulates neutrophil abundance (rather than T cells) and their IFN-γ production during cecal Clostridium difficile infection13. Hence IDO1 function is likely variable within different pathological contexts.
Given the role of IDO1 in immunity and its association with gastric metaplasia, we sought to determine its function and mechanism in this disease. We hypothesized that IDO1 is a critical component involved in the transition from chronic inflammation to gastric metaplasia. The elucidation of IDO1 function would therefore shed some light on the immune components involved in this transition. To address this hypothesis, we analyzed chronically inflamed gastric microenvironments in IDO1-deficient versus proficient mice, and compared our findings to molecular pathways of human gastric cancer.
METHODS
Human Gastric Samples
Human gastric samples were obtained during surgical procedures according to standard tissue procurement mechanisms managed by the Department of Pathology of the University of Hong Kong, under IRB-approved protocol number UW-140611. The samples were de-identified and private information such as names, dates of birth, or medical record numbers was not provided. The samples were collected from the lesser gastric curvature of patients with intestinal metaplasia versus normal patients. The increase in marker expression (TFF-2 and CD44) indicating spasmolytic peptide-expressing metaplasia (SPEM) was confirmed in the metaplastic samples. A total of 20 normal and 20 metaplastic gastric samples were used. For paraffin immunohistochemistry of human gastric tissue, a tissue microarray of human inflamed, cancerous and normal stomach was used (US Biomax, catalog # IC00011c).
Animals
Ido1−/− and CB57BL/6 (generated by Baban et al.14) were obtained from Jackson Labs – stock #005867. Jh−/− mice on a C57BL/6 background were provided by Dr. Cascalho (University of Michigan)15. All the animals used in our studies were male, except in in Suppl. Fig. 12 where both males and females were used for the analysis. All animal models used in this study including WT, Ido1−/−, IFN-γ, IFN-γIdo1−/−, and Jh−/− were on a CB57BL/6 background. Animals were housed in groups (3–5 animals per cage) in microisolator cages under specific pathogen-free conditions. Animals were infected at 8 weeks of age for 6 months. Prior to infection, cage bedding was replaced between cages repetitively for 14 days to normalize the microflora between cages. The H+/K+-ATPase-IFN-γ model was provided by Dr. Dlugosz (University of Michigan)16.
Fluorescence Activated Cell Sorting (FACS) analysis and sorting
FACS was performed as described previously13. Live cells were gated using LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (cat. #L34957, Life Technologies, Grand Island, NY). The following antibodies were used for B cells: B220-PE-Cy7 (clone RA3-6B2, cat. #103221, Biolegend), and IgM-PE (clone eB121-15F9, cat. #12-5890-81, eBiosciences, San Diego, CA). The following antibodies were used for T cells and myeloid cells: CD4-FITC (clone GK1.5, cat. #11-0041-85; eBioscience), CD25–PE–Cy7 (clone PC61.5, cat. #25-0251-81; eBioscience), CD11b–eFluor 450 (clone M1/70, cat. #48-0112-82; eBioscience), and Ly6G-PE (clone 1A8, cat. #127607; BioLegend). RNA extraction of isolated cells for microarray analysis was performed using the RNEasy Microkit (Qiagen).
Statistics
Data were tested for normality using the Shapiro-Wilk W test (Prism, GraphPad Software, La Jolla, CA). Data were compared using one-way analysis of variance (ANOVA) with Dunnet’s (parametric) or Dunn’s (non-parametric) multiple comparison tests (Prism). P values less than 0.05 were considered significant.
Study Approval
All studies were approved by the University of Michigan Institutional Animal Care and Use Committee (PRO00005890). The human data were obtained by analyzing de-identified samples collected during surgical procedures performed by the Department of Pathology of the University of Hong Kong, under IRB-approved protocol number UW-140611. The TCGA human data were obtained by analysing de-identified databases previously generated by the TCGA study17, which did not require additional human sample collection17. Hence, IRB approval for the TCGA data was described in the previous study for which the samples were originally collected17. The human tissue array was obtained from US Biomax and had been previously de-identified by the company.
Supplementary Methods
Further information about the Methods utilized in this paper can be accessed in the Supplementary Methods section.
RESULTS
IDO1 contributes to gastric metaplasia
We identified IDO1 mRNA to be induced in human gastric metaplastic tissue relative to normal (Fig. 1A, left panel, and Suppl. Fig. 1). Moreover, using The Cancer Genome Atlas (TCGA) database, IDO1 mRNA was also induced in gastric cancers relative to normal stomach (Fig. 1A, right panel). To model the role of IDO1 in gastric immunopathology, we chronically infected WT and Ido1−/− mice with H. felis. We previously established that gastric metaplasia develops at 6 month, but not at 2 months following H. felis infection5. Consistent with this previous observation5, we found IDO1 mRNA to be induced at 6 months – but not 2 months – following H. felis infection (Fig. 1B). The induction of gastric IDO1 mRNA was abrogated in Ido1−/− mice (Fig. 1B). As IDO1 metabolizes tryptophan into kynurenine (Suppl. Fig. 2A), we confirmed the levels of kynurenine to be induced in the chronically infected stomach, which was abolished in the Ido1−/− mice (Suppl. Fig. 2B). The generation of downstream metabolites of kynurenine was not substantial in the inflamed stomach (Suppl. Fig. 2B). These experiments confirmed the induction of IDO1 mRNA, and stimulation of IDO1 activity, during chronic (6 month) H. felis infection, which was abrogated in Ido1−/− mice.
Figure 1. IDO1 deletion reduces gastric hypertrophy, metaplastic lesions and parietal cell loss.
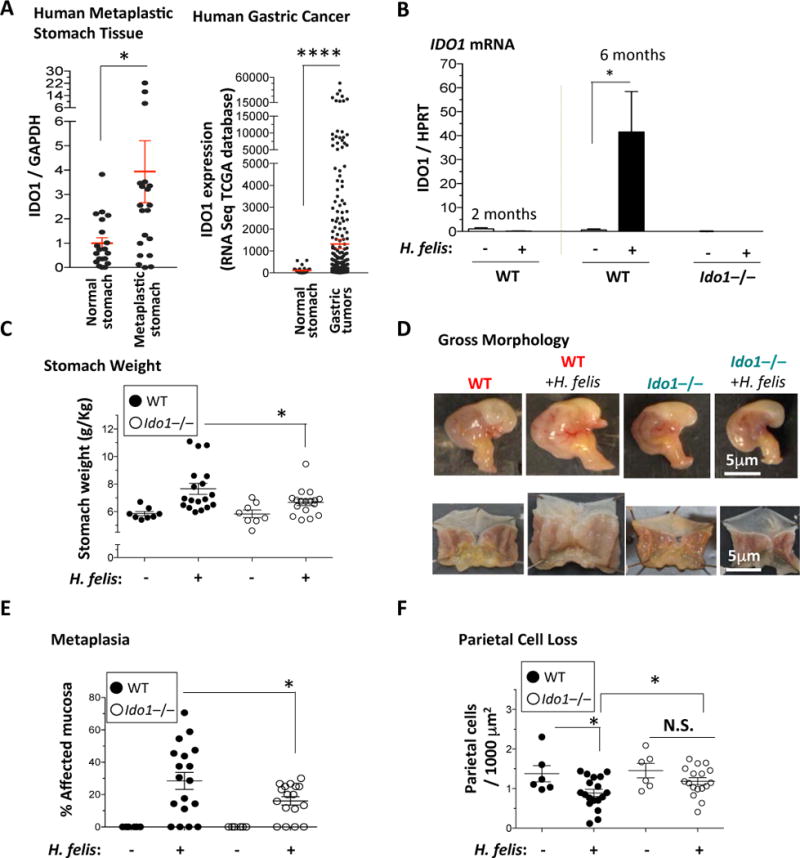
A) Left Panel, RT-qPCR of IDO1 mRNA expression (relative to GAPDH) in human gastric metaplasia samples relative to normal stomach. Right Panel, IDO1 mRNA expression levels by RNASeq in gastric tumor versus normal stomach tissue obtained from TCGA. Each dot represents tissue from one patient. B) RT-qPCR analysis of IDO1 mRNA in 2 months and 6 months H. felis-infected WT stomachs versus uninfected, and 6 months H. felis-infected Ido1−/− stomachs versus uninfected. C) Stomach weight normalized to total body weight in WT versus Ido1−/− mice +/− 6-month H. felis infection. D) Representative images showing the gross morphological appearance of WT versus Ido1−/− mouse stomachs +/− H. felis. E) Scatterplot showing the percentage area of metaplastic gastric mucosa in WT versus Ido1−/− mice +/− 6-month H. felis. Metaplasia was assessed and quantified using trefoil factor 2 (TFF-2) and intrinsic factor (IF) as shown in Suppl. Fig. 3A. F) Scatterplot showing the number of parietal cells per 1,000 μm2 of glandular tissue in WT versus Ido1−/− mice +/− 6-month H. felis. Parietal cells were quantified using Fiji (ImageJ) and the fluorescent staining described in Suppl. Fig. 3B. N = 5 mice per uninfected group, 18 mice in the infected WT group, and 17 mice in the infected Ido1−/− group. Error bars represent the means and standard error of the means. Data were compared using one-way analysis of variance (ANOVA) with Dunnet’s (parametric) or Dunn’s (non-parametric) for multiple comparison tests (Prism), and unpaired t-test (parametric) or Mann-Whitney test (non-parametric) for single comparisons (Prism). Each data point represents one mouse. * P < 0.05. ** P < 0.01. **** P < 0.0001. N.S. = not significant.
We then investigated the role of IDO1 in the generation of gastric metaplasia. As stomach hypertrophy, assessed by measuring stomach weight, is directly associated with metaplasia5, we measured the stomach weight normalized to total body weight of the mouse. We observed an increase in relative stomach weight in 6-month H. felis-infected WT mice relative to uninfected WT mice (Fig. 1C). However, this increase was significantly reduced in Ido1−/− mice (Fig. 1C). This is corroborated by the macroscopic appearance of the stomachs in WT versus Ido1−/− mice (Fig. 1D). The reduction in stomach weight increase of infected Ido1−/− mice correlated with a significant reduction in metaplastic lesions (Fig. 1E; quantification criteria in Suppl. Fig. 3A). Hence Ido1−/− mice exhibited a significant reduction of gastric metaplastic lesions.
Since parietal cell loss triggers metaplastic development18, we assessed the degree of parietal cell loss in the Ido1−/− mice by staining for H+/K+-ATPase-β as shown in Suppl. Fig. 3B. While infected WT mice showed a significant reduction in parietal cell number (per area), this significant reduction was not observed in Ido1−/− stomachs (Figure 1F). We conclude that IDO1 contributes to the development of parietal cell loss and gastric metaplasia.
IDO1 regulates B cell abundance during chronic gastric inflammation
To investigate the mechanism of IDO1 in gastric inflammation, we used a mouse model that overexpresses IFN-γ in the stomach (H+/K+-ATPase-IFN-γ; will be referred to as IFN-γ model from here onwards)16. IFN-γ is a known inducer of IDO119, and this model exhibited a robust induction of IDO1 mRNA in the stomach (Suppl. Fig. 4A). We have previously shown the IFN-γ mouse model to develop chronic gastric inflammation. To study the function of IDO1, we bred the IFN-γ model onto an IDO1-deficient background to generate H+/K+-ATPase-IFN-γ/Ido1−/− (will be referred to as IFN-γIdo1−/−). The IFN-γIdo1−/− model lacked gastric IDO1 expression and induction (Suppl. Fig. 4A). We performed microarray analysis to determine the effect of IDO1 loss. The microarray showed that the IDO1-dependent genes in the inflamed stomach almost exclusively comprised immunoglobulin light and heavy chain cassettes, CD79A, and Pou2af1, which are B cell markers (Fig. 2A). These genes were upregulated in the IFN-γ model versus non-transgenic, but were not induced in IFN-γIdo1−/− (Fig. 2A). The expression patterns of CD79A, IDO1 and Igkv1-133 from this array were confirmed by RT-qPCR (Fig. 2B and Suppl. Fig. 4). This indicated that IDO1 deficiency was affecting the B cell compartment in the inflamed gastric mucosa.
Figure 2. Ido1−/− stomachs display a B cell-deficient phenotype.
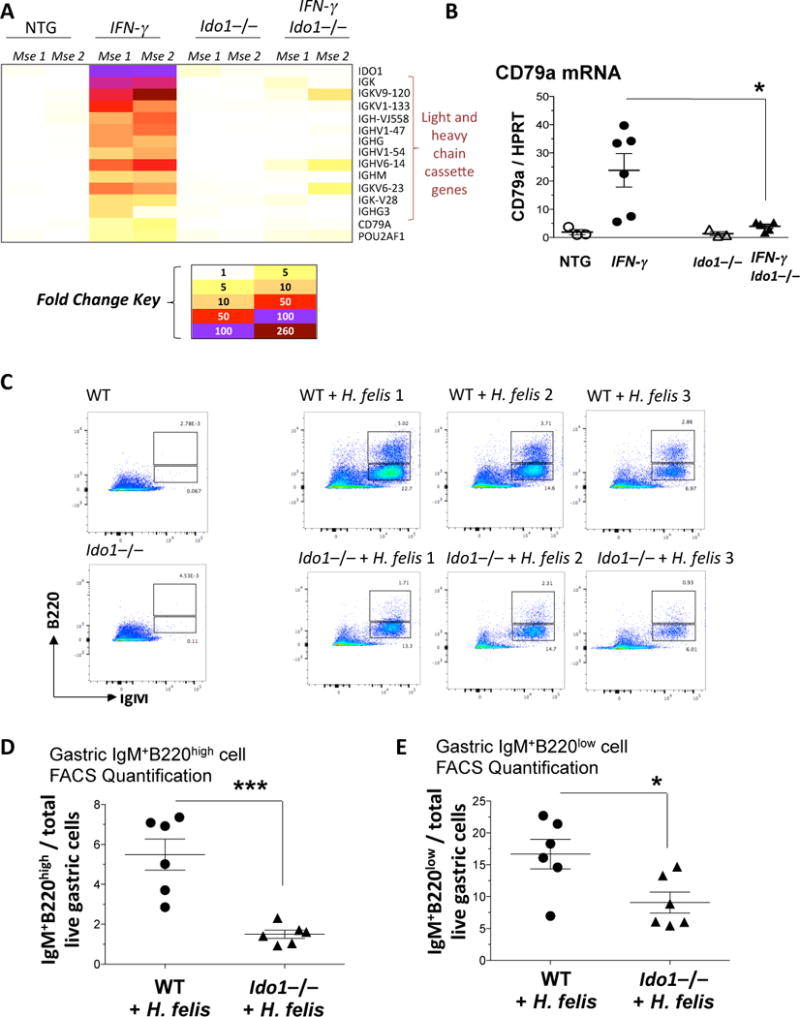
A) Fold change heatmap of the differentially regulated genes induced in 2 month old IFN-γ but not in IFN-γIdo1−/− stomachs. B) RT-qPCR of the B cell marker, CD79a, in IFN-γ versus IFN-γIdo1−/− stomachs (2 month old), relative to non-transgenic controls. C) FACS analysis of immature naïve (IgM+B220low) and mature naïve (IgM+B220high) B cells in WT versus Ido1−/− stomachs +/− H. felis following 6 month infection. D) Graphical representation of FACS mature naïve (IgM+B220high) B cell percentages in H. felis-infected WT versus Ido1−/− stomachs. E) Graphical representation of FACS immature naïve (IgM+B220low) B cell percentages in H. felis-infected WT versus Ido1−/− stomachs. Error bars represent the standard error of the mean. Each data point represents one mouse. * P < 0.05. *** P < 0.001.
To investigate the effect of IDO1 deficiency on B cells, we performed B cell FACS analysis in Ido1−/− versus WT H. felis-infected stomachs. We used the classification from previous studies, which characterized i) immature naïve B cells as B220lowIgM+, and ii) mature naïve B cells as B220highIgM+20, 21. We observed a robust increase in both immature and mature naïve gastric B cell populations during chronic H. felis infection (Fig. 2C). However, there was a significant decrease in mature B220highIgM+ and immature B220lowIgM+ B cells in infected Ido1−/− stomachs (Fig. 2C–E). This indicated that mature naïve B cells were: 1) either depleted in inflamed Ido1−/− stomachs, or otherwise 2) exhibited an increased rate of class switch recombination. To determine the mechanism, we assessed class switch recombination of B cells in chronically inflamed Ido1−/− versus WT stomachs.
Our identification of class-switched B cells began with the observation of the expression of immunoglobulin γ heavy chain and variable κ light chain cassettes (Suppl. Fig. 5A). We used a B cell-deficient model lacking the joining chain (Jh−/− mice) and observed the effect of B cell deficiency on the expression of heavy and light chain cassettes (Suppl. Fig. 5A, left panel). H. felis infection (6 months) increased the expression of Ighm, Ighg, Igl and Igk genes (for μ heavy chain, γ heavy chain, λ light chain, and κ light chain, respectively), which were abolished in the infected Jh−/− stomachs (Suppl. Fig. 5A, left panel). However, the majority of these cassette genes – except for Ighm (μ) and Iglv (λ variable) – were not expressed by B220+IgM+ B cells (Suppl. Fig. 5A, middle panel). Instead, they were expressed by CD11b−Ly6G+ immune cells (Suppl. Fig. 5A, right panel). Analysis of these CD11b−Ly6G+ immune cells – after being negatively gated for CD4−CD8− – showed that they coincided with B220+IgM− B cells (Suppl. Fig. 5B–H). This data correlated with a previous report showing that “class-switched B cells” expressed the B220+IgMlow markers (as opposed to B220+IgMhigh for naïve B cells)22. Moreover, these CD11b−Ly6G+ immune cells expressed the Ly6c gene (Suppl. Fig. 5A, right panel, third gene from the top). This also correlated with a previous study that showed that Ly6c is a marker of a differentiation of B cells into plasma cells23. We therefore conclude that the CD11b−Ly6G+ population (after negatively gating T cells) represents the class switched population producing IgG (Ighg gene). To determine the effect of IDO1, we measured the frequency of this CD11b−Ly6G+ population, and showed that it was reduced – rather than increased – in chronically infected Ido1−/− stomachs (Suppl. Fig. 5I). This demonstrated that the reduction of naïve B cell population in inflamed Ido1−/− stomachs (Fig. 2C) was not due to an increase in class switch recombination of these cells.
IDO1 does not regulate gastric CD4+ T cell, myeloid, or MDSC cell abundance
IDO1 has traditionally been defined as an enzyme that metabolizes tryptophan into kynurenine7. In doing so, the enzyme diminishes the tryptophan pool required for T cell proliferation6, and generates kynurenine that stimulates Treg differentiation8. We therefore measured the abundance of CD4+ T cells and CD4+CD25+ Tregs in infected WT and Ido1−/− stomachs. We first observed a clear increase in T cell and Treg numbers following 6-month H. felis infection (Suppl. Fig. 6A). However, contrary to the commonly described role of IDO1, we did not observe a difference in CD4+ T cell and CD4+CD25+ Treg cell number between infected WT and Ido1−/− stomachs (Suppl. Fig. 6A–C). In addition to the lack of difference in CD4+ T cell numbers, we did not observe a significant difference in Th1 cytokine expression (IFN-γ, TNF-α, and IL-1β) between WT and Ido1−/− stomach tissue (Suppl. Fig. 6D). We conclude that Ido1−/− did not alter the T cell response in the context of chronic gastric inflammation.
Since a recent study showed that IDO1 indirectly regulates MDSC recruitment24, we quantified myeloid cells and MDSCs in the gastric microenvironment of WT and Ido1−/− stomachs. We observed a robust increase in gastric MDSCs following 6 months of H. felis infection, but no difference was observed between infected WT and Ido1−/− stomachs (Suppl. Fig. 7A–C). We conclude that IDO1 does not regulate MDSC or myeloid cell frequency in the chronically inflamed stomach.
IDO1 does not affect H. felis bacterial DNA abundance
H. felis flagellar filament B (Fla-B) is used as an indicator of bacterial abundance5, 25. Even though Ido1−/− showed a trend for increased H. felis DNA in gastric tissue (which correlates with reduced gastric immunopathology), the difference in H. felis DNA was not significant between WT and Ido1−/− (Suppl. Fig. 8). This indicated that IDO1 did not have a significant effect on H. felis bacterial abundance.
IDO1 deficiency correlates with reduced anti-parietal cell autoantibody levels and natural killer cell-to-cell contact with parietal cells
The reduction of gastric B cells in Ido1−/− suggested that IDO1 might regulate the autoimmune response against parietal cells in the inflamed stomach. The presence of parietal cell autoantibody is a common occurrence in Helicobacter pylori (H. pylori)-infected patients26, although the pathological mechanism for these autoantibodies is not clear27. Autoimmune reactions, although responding to internal “self” antigen – as opposed to external antigen in the case of hypersensitivity – are believed to share overlapping mechanisms with hypersensitivity reactions28. There are 4 types of hypersensitivity reactions according to the Coombs and Gell classification29. Amongst the 4 types, only type II is dependent on antibody-mediated binding of target cells29. The latter mechanism can be mediated by antigen-dependent cell-mediated cytotoxicity (ADCC), in which natural killer (NK) cells bind antibody-coated target cells and initiate cell death29. We therefore investigated whether hallmarks of type II hypersensitivity/autoimmunity were present and regulated by IDO1. In the following paragraph, we will only use the term “type II autoimmunity” as it involves the targeting of internal/“self” (parietal cell) antigen – rather than external antigen. We therefore assayed two hallmarks of type II autoimmunity: 1) autoantibody binding to parietal cells, and 2) NK cell association with parietal cells.
We first measured anti-parietal cell autoantibody in infected WT versus Ido1−/− mouse plasma. While chronic H. felis infection significantly increased anti-parietal cell antibodies measured by ELISA, Ido1−/− mice did not exhibit such a robust significant increase (Fig. 3A). We observed a significant increase of natural killer cell/parietal cell contact points in the chronically infected WT gastric mucosa, but not in the Ido1−/− gastric mucosa (Fig. 3B–D). Low power images and the negative control are also shown in Suppl. Fig. 9. The latter images additionally suggest that the total number of NK cells might have been reduced in the infected Ido1−/− stomach (Suppl. Fig. 9). Hence, the reduction in NK/parietal contact might have also been attributed to lower abundance of NK cells. Irrespective of either scenario, the data provide supportive evidence that the hallmarks of type II autoimmunity (parietal cell autoantibody and NK/parietal cell interaction) were reduced in inflamed Ido1−/− stomachs. Hence, a reduction of hallmarks of type II autoimmunity is associated with IDO1 deficiency in the chronically inflamed stomach.
Figure 3. IDO1 deficiency associated with reduced autoantibody production and NK cell-to-cell contact with parietal cells.
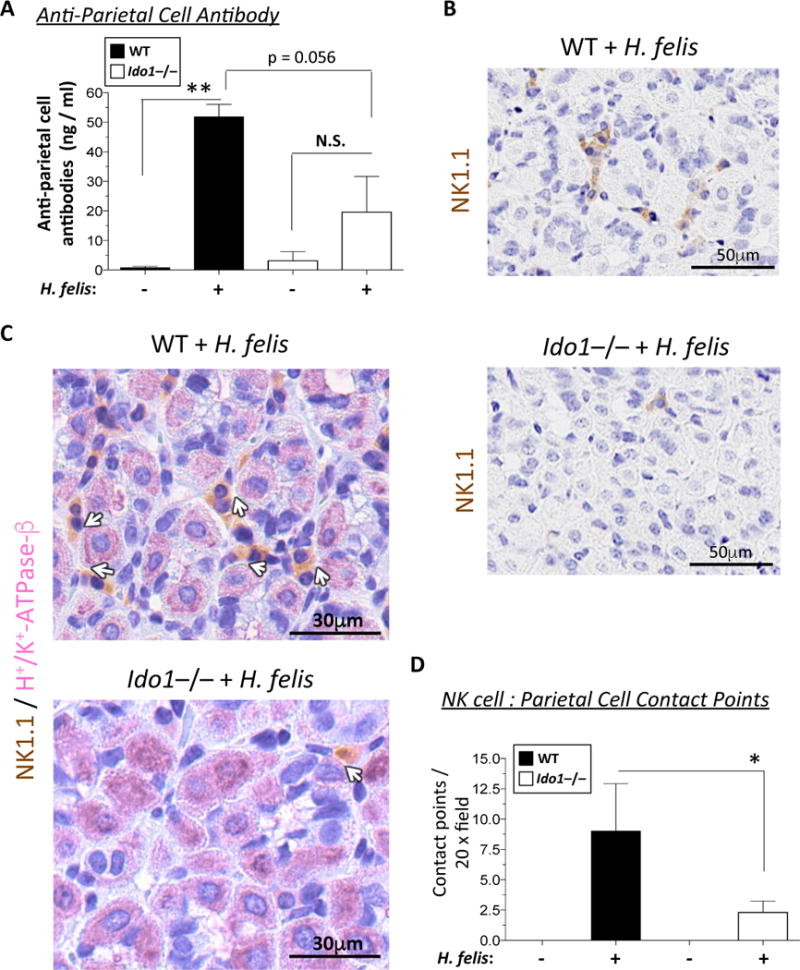
A) Bar graph representation of plasma anti-parietal cell antibody levels in 6-month infected WT versus Ido1−/− mice compared to uninfected controls. B) Immunohistochemical staining of NK1.1-HRP antibody (brown) in 6-month infected WT versus Ido1−/− stomachs. These insets are obtained from the low power images shown in Suppl. Fig. 9A. C) Immunohistochemical double staining of NK1.1-HRP (brown) and H+/K+-ATPase-β (pink) of parietal cells in 6-month infected WT versus Ido1−/− stomachs. White arrows indicate the contact points between the NK cells and the parietal cells. D) Bar graph representation of morphometric quantification of NK cell/parietal cell contact points per 20x microscopic field. Data were compared using one-way analysis of variance (ANOVA) with Dunnet’s (parametric) or Dunn’s (non-parametric) for multiple comparison tests (Prism), Error bars represent the standard error of the mean. * P < 0.05. ** P < 0.01.
IDO1 alters the transcriptional profile of gastric B cells
To investigate the mechanism of how IDO1 regulates B cells, we first determined the expression pattern of IDO1. IDO1 was expressed by both gastric epithelial cells and B cells in metaplastic mouse stomach (Suppl. Fig. 10A), and inflamed human stomach (Suppl. Fig. 10B). Moreover, both stromal and epithelial expression of IDO1 was detected in human gastric cancer (Suppl. Fig. 10C). Since B cells expressed IDO1 endogenously, we determined the effect of IDO1 deficiency on gastric B cells’ transcriptional profile. We FACS sorted B cells from 6-month infected WT versus Ido1−/− gastric mucosa and performed microarray analysis (Fig. 4). First, the microarray corroborated the enrichment of B cell-specific genes in these isolated cells (Suppl. Fig. 11 and Suppl. Dataset 1; for raw data, please refer to Suppl. Dataset 2). Second, the microarray identified differences in the transcriptional profile between WT and Ido1−/− gastric B cells (Fig. 4 and Suppl. Dataset 3). Although the majority of the identified genes have not been previously characterized in B cell function, the microarray still identified some known markers (Fig. 4). These included: 1) the upregulation of BMP receptor 2 (Bmpr2) in gastric Ido1−/− B cells. Bmpr2 is not normally detected in WT gastric B cells after chronic infection with H. felis (Fig. 4), but is induced in IDO1-deficient gastric B cells (Fig. 4). BMP signaling can play a role in modulating B cell activation in certain contexts such as bone marrow and peripheral blood, in which BMP signaling can repress B cell proliferation30–32 and antibody production33; 2) the upregulation of granzyme B (Gzmb) in gastric Ido1−/− B cells. Gzmb upregulation is indicative of incomplete T cell help34; and 3) downregulation of VH11 (Igh-V11) in Ido1−/− gastric B cells. VH11 is a marker of an autoreactive subset of B1 cells35 (Fig. 4). We did not observe an effect of IDO1 on the expression of maturation factors Prdm1 (Blimp1), Xbp-1, Bcl6 or Pax5 in gastric B cells (data not shown). Hence, although the mechanism of IDO1 regulation of B cells remains unclear, the data demonstrates an altered transcriptional profile – indicative of altered functionality – of Ido1−/− gastric B cells relative to WT.
Figure 4. IDO1-deficient gastric B cells exhibit an altered transcriptional profile.
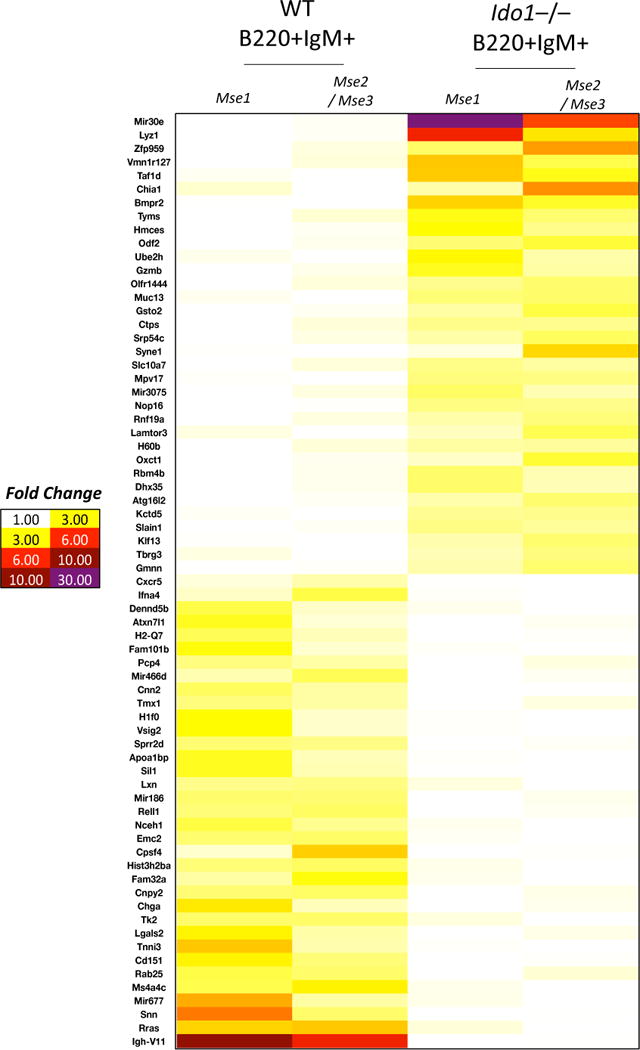
Microarray heatmap of FACS isolated gastric B220+IgM+ B cells from 6-month infected WT versus Ido1−/− stomachs. The microarray illustrates the altered transcription profile of Ido1−/− versus WT gastric B cells isolated from 6-month infected stomachs. “Mse 1” refers to isolated RNA from gastric B cells of one mouse, whereas “Mse 2/Mse 3” represents pooled isolated RNA from gastric B cells of two mice to serve as a replicate.
B cells are necessary for metaplastic development
Since a previous study showed that the cμ gene knockout of the constant immunoglobulin heavy chain locus (μMT mice) did not affect gastric metaplastic development36, we aimed to address this inconsistency. Several studies have shown that the μMT model does not exhibit total B cell deficiency, and in fact is capable of producing antibodies37–40. This is not surprising since heavy chain genes are normally deleted from the chromosome during B cell class switching. Hence other downstream genes (e.g., δ, γ, ε, or α) can be similarly utilized when the μ gene is genetically deleted in the μMT model. To avoid this redundancy, we used the Jh−/− mouse model41, which contains a disrupted J segment gene, and therefore exhibits complete B cell and immunoglobulin deficiency38. While the chronically infected WT mice showed an expanded gastric B220+IgM+ B cell populations compared to uninfected WT, the Jh−/− mice lost their gastric B cells (Fig. 5A and quantification in Fig. 5B). These mice did not exhibit a significant change in H. felis Fla-B bacterial DNA (Suppl. Fig. 12A). The loss of B cells was validated by RT-qPCR for CD79a and Igk v1-133 (Suppl. Fig. 12B, C). These Jh−/− mice exhibited significantly reduced metaplastic lesions in the stomach (Fig. 5C, D). Hence, the data suggest that B cells contribute to metaplasia, contrary to the previous interpretation based on the μMT model36.
Figure 5. Mice with B cell deficiency (lacking the JH locus) show reduced metaplasia.
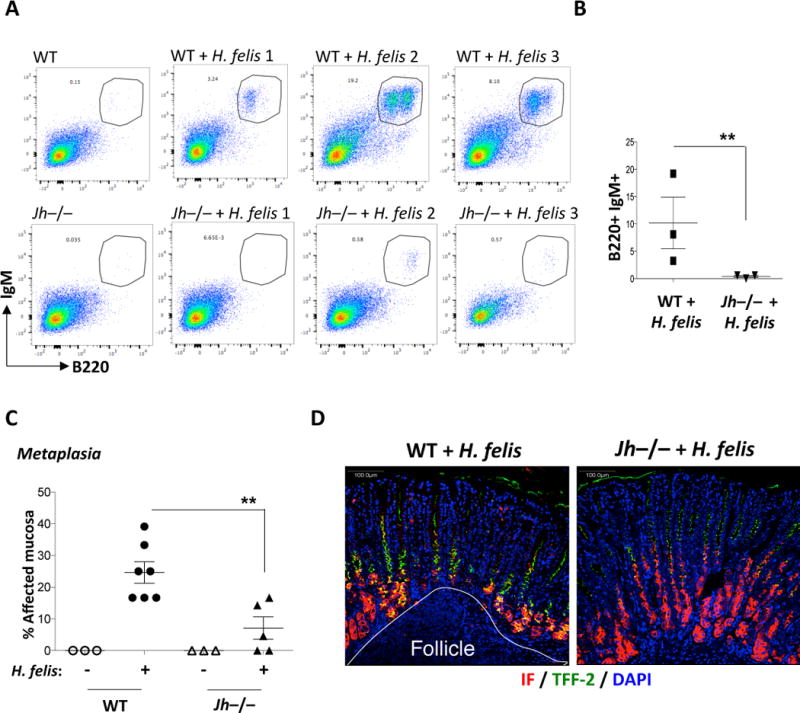
A) FACS analysis of mature B220+IgM+ B cells in 6-month H. felis-infected WT versus Jh−/− stomachs. B) Graphical representation of FACS percentages of B cells in H. felis-infected WT versus Jh−/− stomachs. C) Scatterplot showing the percentage of metaplastic gastric areas in WT versus Jh−/− mice +/− 6-month H. felis. D) Representative images of metaplastic versus non-metaplastic areas from H. felis-infected WT versus Jh−/− stomachs respectively. Intrinsic factor (IF) = red; Trefoil factor 2 (TFF-2) = green; DAPI = blue. Error bars represent the standard error of the mean. Each data point represents one mouse. ** P < 0.01.
IDO1 and B cell markers are amongst the highest-regulated genes that cluster with a human gastric adenocarcinoma subtype
To correlate our finding to human gastric cancer, we used TCGA17 to determine whether IDO1 and B cell regulation are involved. IDO1 and five B cell markers (IDO1, CD79A, IGHG1, IGLL5, IGHC, IGVK-A2) were amongst the 10 highest differentially-induced gene, and clustered with one molecular subtype of human gastric adenocarcinoma called “expression cluster 2”17 (Fig. 6). The high expression of these markers in “expression cluster 2” does not preclude the induction of IDO1 in other gastric cancer subtypes, as induction is generally detected in pooled gastric cancer samples in Fig. 1A. However, as “expression cluster 2” is associated with Epstein Barr Virus (EBV) infection17, this implicates stronger induction of IDO1/B cell signaling in heightened inflammatory immunopathology caused by EBV.
Figure 6. List of the top cluster-specific genes for each human expression cluster.
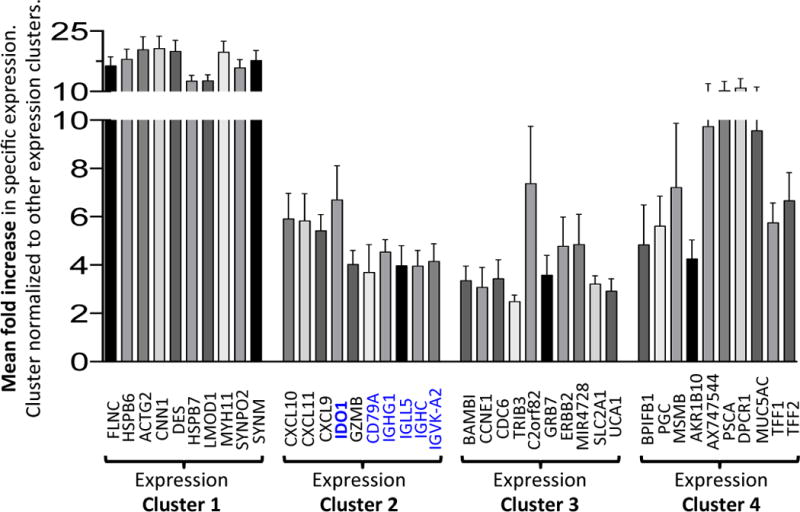
Bar graph showing the top 10 most up-regulated genes in each expression cluster defined by the TCGA study17. IDO1 and B cell-specific genes (CD79A, IGHG1, IGLL5, IGHC and IGVK-A2) are highlighted in blue font. For each gene, the values represent the mean fold change in expression across the samples in that cluster. Error bars = mean +/− SEM.
Mouse models of gastric pre-neoplasia exhibit some similarity in gene induction patterns to human “expression cluster 2”
Since mouse models of gastric metaplasia are limited in that they fail to progress to gastric cancer, it was important to assess the relevance of our mouse studies to human cancer. Even though the pre-neoplastic models do no replicate the pathology of fully developed gastric cancer, we evaluated the suitability of our analyses by comparing gene expression patterns that were induced in both mouse models and human cancer subtypes from the TCGA17. We observed considerable overlap between the expression profiles corresponding to the H. felis model and human “expression cluster 2” (Suppl. Fig. 13). IDO1 was the highest-expressed common gene in these two sets (Suppl. Fig. 13). Other commonly induced genes in cluster 2 included B cell-specific genes, CD79a, IgJ and Pou2af1, and the natural killer cell gene NKG7 (Suppl. Fig. 13). A similar result was obtained using the IFN-γ-overexpressing model, with highest similarity to “expression cluster 2” (Suppl. Fig. 14A). These comparisons were also performed against the general TCGA classification of gastric cancer subtypes (EBV, MSI, GS, and CIN), and showed the highest similarity between the mouse pre-neoplastic models and the EBV subtype (Suppl. Fig. 14B, C). This is not surprising since “expression cluster 2” is enriched with EBV-positive cancers17. Therefore, some similarity exists in the patterns of differentially induced genes between mouse models of gastric pre-neoplasia and human adenocarcinoma.
DISCUSSION
The major conclusion of this study is that IDO1 contributes to pseudopyloric metaplasia in the stomach. The mechanism appears to be mediated by IDO1 regulation of the B cell compartment, which associates with hallmarks of type II autoimmunity. Our proposed mechanism is summarized in Suppl. Diagram 1. This conclusion is based on the following experimental evidence. First, we showed that IDO1-deficient mice develop reduced metaplasia and gastric B cell frequency. The effect on B cells was not due to increased class switching. In addition, no apparent impact on T cell or myeloid cell abundance was detected. Second, we showed that this phenotype associates with a reduced autoantibody response against parietal cells, and reduced NK cell-to-cell contact with parietal cells, both of which represent hallmarks of reduced type II autoimmunity. Third, we showed that IDO1-deficient gastric B cells exhibit an altered transcriptional profile from WT gastric B cells, and that B cell-deficient mice develop reduced metaplastic lesions. Finally, we showed that the IDO1 expression is strongly increased in human gastric metaplasia and cancer, and associates with B cell markers. Collectively, this experimental evidence suggests that IDO1-mediated B cell regulation contributes to pseudopyloric metaplasia, and associates with hallmarks of type II autoimmunity during gastric carcinogenesis.
Previous studies have shown associations between parietal cell autoantibodies, H. pylori infection, and human gastric cancer42–48, although the pathological mechanisms for these autoantibodies remained unclear. IDO1 has also been shown to be associated with human gastric cancer17, 49, 50. First, Strong et al. detected high IDO1 induction in EBV-associated gastric cancer49. Second, this was recently confirmed by an independent patient sample of EBV-associated gastric cancer50. Third, the TCGA identified IDO1 in one of the four types of patient expression clusters, called “expression cluster 2”, which was associated with EBV-positive gastric cancer17. In this study, we showed that the H. felis and IFN-γ-overexpressing mouse models were similar in their expression patterns to human “expression cluster 2” and the EBV subtype. The similar induction of IDO1 in EBV and H. felis is possibly a manifestation of the heightened immune response and immunopathology in both infectious contexts. Furthermore, we also showed an induction of IDO1 expression in human gastric metaplasia. Hence, the modelled pathways showed relevance to the gastric pre-neoplastic and neoplastic microenvironment. However, despite these observations, further work is required to determine the differences in the applicability of these mechanisms to human gastric pre-neoplastic etiology versus cancer.
One consideration in this study is that, despite our previous finding that IFN-γ overexpression sufficiently induces metaplasia16, we did not observe a difference in IFN-γ expression between infected WT and Ido1−/− stomachs. This observation is not surprising since IFN-γ functions upstream of IDO1 induction, and inflamed Ido1−/− stomachs do not exhibit a difference in CD4+ T cell frequency. Thus, we proposed that IDO1 functions downstream of IFN-γ, which is illustrated in Suppl. Diagram 1.
The finding that IDO1 did not alter T cell numbers or cytokine expression is not entirely surprising given the recent findings by Thaker et al in colorectal tumor models12. In that study, the authors showed IDO1 deficiency to reduce colorectal polyps in the AOM + DSS model12. This parallels our finding that IDO1 deficiency reduces pre-neoplastic lesions in the stomach. However, their study also demonstrated that the reduction of polyps did not appear to be mediated by a T cell mechanism12. However, they proposed IDO1 to act directly on neoplastic epithelial cell proliferation via β-catenin12. Another study showed – in line with our findings – that IDO1 is necessary for B cell and autoantibody production in rheumatoid arthritis51. This contrasts with a recent study, which shows that B cell-intrinsic IDO1 suppresses T cell-independent humoral immunity10. The difference between our study and the latter study is perhaps not surprising since H. felis-mediated immunopathology requires T cell-dependent adaptive immunity and B cell-T cell interactions36, whereas the latter study focussed on thymic-independent B cell activation10. However, our results shed new light on B cell-mediated gastric metaplasia development, which is contrary to a previous report using the μMT model36. This is because the μMT model is not fully B cell deficient as it is capable of producing antibodies and generating viable B cells37–40. Hence, it is clear from this study, and from our previous work where IDO1 restricts neutrophil infiltration13, that the IDO1 mechanism is not limited to the traditionally described pathway of restricting T cell proliferation. Moreover, the effect of IDO1 on B cells might also be subject to variability due to the heterogeneity of B cell biology. This could depend on the extent of involvement of thymic-dependent versus independent responses, or B2 versus B1 contribution to the pathological context under study. Future studies will explore these aspects as outlined in the following paragraph.
It is a novel and paradigm-shifting concept to consider B cell involvement and autoreactive ADCC in gastric carcinogenesis. However our observations are not surprising, since the development of lymphoid follicles, which contain B cell compartments, has been extensively studied previously in a process called “lymphoid neogenesis” (as reviewed by Pitzalis et al.52). Lymphoid neogenesis in the stomach comprises the development of B and T cell compartments in an extensive process that enables in situ antigen presentation and B cell/T cell interactions. Therefore, the biology of B cells in the stomach – as part of these structures – is very complex and uncharacterized. The resulting follicles from lymphoid neogenesis are considered tertiary lymphoid organs (also called ectopic lymphoid structures), as they contain structures that resemble those of the lymph node. The tertiary lymphoid organs contain high endothelial venule (HEV), which regulate extravasation and trafficking, follicular dendritic cells, T follicular helper cells, and B cells and T cells that mature into effector and memory cells. The study of tertiary lymphoid organs has not been sufficiently utilized in the study of gastric carcinogenesis. Hence, future studies can begin to dissect out the cellular components of these tertiary lymphoid structures (follicles) in hopes of identifying novel therapeutic strategies against gastric metaplasia and carcinoma.
We conclude that IDO1 mediates pseudopyloric metaplasia in the stomach and associates with human gastric metaplasia and cancer. Our study provides evidence that IDO1 contributes to B cell regulation in the inflamed gastric microenvironment, which associates with antibody production and autoimmunity. These findings prompt further investigation of autoimmunity in gastric carcinogenesis.
Supplementary Material
Acknowledgments
We would like to acknowledge the American Gastroenterological Association (AGA)/Gastric Cancer Foundation (GCF) for providing Grant N017489 (El-Zaatari) that supported this study. Additionally, we would like to acknowledge DoD grant CA160431 (El-Zaatari) for supporting the final stages of this study. We would also like to acknowledge the NIH for Public Health Service Grants NIDDK R01 DK087708-01 (Kao); 5P30DK034933 (Owyang); University of Michigan Comprehensive Cancer John S. and Suzanne C. Munn Cancer Fund (Todisco); DK062041 and CA118875 (Dlugosz), which also supported this study.
Abbreviations
- IDO1
indoleamine-2,3-dioxygenase-1
- H. felis
Helicobacter felis
- ADCC
Antigen-dependent cell-mediated cytotoxicity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have nothing to disclose.
Author contributions:
Mohamad El-Zaatari: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; statistical analysis; obtained funding; study supervision.
Adam J. Bass: study concept and design; acquisition of data; analysis and interpretation of data; technical and material support; critical revision of the manuscript for important intellectual content.
Reanne Bowlby: acquisition of data; analysis and interpretation of data; statistical analysis; technical and material support.
Min Zhang: acquisition of data; technical support.
Li-Jyun Syu: acquisition of data; analysis and interpretation of data; material support.
Yitian Yang: acquisition of data; analysis and interpretation of data; material support.
Helmut Grasberger: study concept and design; analysis and interpretation of data; critical revision of the manuscript for important intellectual content.
Andrew Shreiner: study concept and design; analysis and interpretation of data; critical revision of the manuscript for important intellectual content.
Bei Tan: acquisition of data; analysis and interpretation of data.
Shrinivas Bishu: study concept and design; analysis and interpretation of data; critical revision of the manuscript for important intellectual content.
Andrea Todisco: acquisition of data; analysis and interpretation of data; material support.
Wai K. Leung: analysis and interpretation of data; material support.
Nobuhiko Kamada: study concept and design; analysis and interpretation of data; critical revision of the manuscript for important intellectual content.
Marilia Cascalho: study concept and design; analysis and interpretation of data; material support; critical revision of the manuscript for important intellectual content.
Andrzej A. Dlugosz: study concept and design; material support; critical revision of the manuscript for important intellectual content.
John Y. Kao: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; statistical analysis; obtained funding; study supervision.
References
- 1.Farraye FA, Odze RD, Eaden J, et al. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:746–74. 774 e1–4. doi: 10.1053/j.gastro.2009.12.035. quiz e12–3. [DOI] [PubMed] [Google Scholar]
- 2.Farraye FA, Odze RD, Eaden J, et al. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:738–45. doi: 10.1053/j.gastro.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 3.Correa P, Haenszel W, Cuello C, et al. A model for gastric cancer epidemiology. Lancet. 1975;2:58–60. doi: 10.1016/s0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 4.Kim N, Park RY, Cho SI, et al. Helicobacter pylori infection and development of gastric cancer in Korea: long-term follow-up. J Clin Gastroenterol. 2008;42:448–54. doi: 10.1097/MCG.0b013e318046eac3. [DOI] [PubMed] [Google Scholar]
- 5.El-Zaatari M, Kao JY, Tessier A, et al. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS One. 2013;8:e58935. doi: 10.1371/journal.pone.0058935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 7.Takikawa O, Yoshida R, Kido R, et al. Tryptophan degradation in mice initiated by indoleamine 2,3-dioxygenase. J Biol Chem. 1986;261:3648–53. [PubMed] [Google Scholar]
- 8.Mezrich JD, Fechner JH, Zhang X, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nouel A, Pochard P, Simon Q, et al. B-Cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. J Autoimmun. 2015;59:53–60. doi: 10.1016/j.jaut.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Shinde R, Shimoda M, Chaudhary K, et al. B Cell-Intrinsic IDO1 Regulates Humoral Immunity to T Cell-Independent Antigens. J Immunol. 2015;195:2374–82. doi: 10.4049/jimmunol.1402854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godin-Ethier J, Hanafi LA, Duvignaud JB, et al. IDO expression by human B lymphocytes in response to T lymphocyte stimuli and TLR engagement is biologically inactive. Mol Immunol. 2011;49:253–9. doi: 10.1016/j.molimm.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Thaker AI, Rao MS, Bishnupuri KS, et al. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology. 2013;145:416–25. e1–4. doi: 10.1053/j.gastro.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Zaatari M, Chang YM, Zhang M, et al. Tryptophan catabolism restricts IFN-gamma-expressing neutrophils and Clostridium difficile immunopathology. J Immunol. 2014;193:807–16. doi: 10.4049/jimmunol.1302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baban B, Chandler P, McCool D, et al. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 15.AbuAttieh M, Rebrovich M, Wettstein PJ, et al. Fitness of cell-mediated immunity independent of repertoire diversity. J Immunol. 2007;178:2950–60. doi: 10.4049/jimmunol.178.5.2950. [DOI] [PubMed] [Google Scholar]
- 16.Syu LJ, El-Zaatari M, Eaton KA, et al. Transgenic expression of interferon-gamma in mouse stomach leads to inflammation, metaplasia, and dysplasia. Am J Pathol. 2012;181:2114–25. doi: 10.1016/j.ajpath.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weis VG, Sousa JF, LaFleur BJ, et al. Heterogeneity in mouse spasmolytic polypeptide-expressing metaplasia lineages identifies markers of metaplastic progression. Gut. 2013;62:1270–9. doi: 10.1136/gutjnl-2012-302401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yasui H, Takai K, Yoshida R, et al. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: its possible occurrence in cancer patients. Proc Natl Acad Sci USA. 1986;83:6622–6. doi: 10.1073/pnas.83.17.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones MA, DeWolf S, Vacharathit V, et al. Investigating B Cell Development, Natural and Primary Antibody Responses in Ly-6A/Sca-1 Deficient Mice. PLoS One. 2016;11:e0157271. doi: 10.1371/journal.pone.0157271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crouch EE, Li Z, Takizawa M, et al. Regulation of AID expression in the immune response. J Exp Med. 2007;204:1145–56. doi: 10.1084/jem.20061952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han JH, Akira S, Calame K, et al. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity. 2007;27:64–75. doi: 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrammert J, Kallberg E, Agace WW, et al. Ly6C expression differentiates plasma cells from other B cell subsets in mice. Eur J Immunol. 2002;32:97–103. doi: 10.1002/1521-4141(200201)32:1<97::AID-IMMU97>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 24.Holmgaard RB, Zamarin D, Li Y, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015;13:412–24. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoicov C, Fan X, Liu JH, et al. T-bet knockout prevents Helicobacter felis-induced gastric cancer. J Immunol. 2009;183:642–9. doi: 10.4049/jimmunol.0900511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claeys D, Faller G, Appelmelk BJ, et al. The gastric H+,K+-ATPase is a major autoantigen in chronic Helicobacter pylori gastritis with body mucosa atrophy. Gastroenterology. 1998;115:340–7. doi: 10.1016/s0016-5085(98)70200-8. [DOI] [PubMed] [Google Scholar]
- 27.Rusak E, Chobot A, Krzywicka A, et al. Anti-parietal cell antibodies - diagnostic significance. Adv Med Sci. 2016;61:175–179. doi: 10.1016/j.advms.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 28.Bartunkova J, Kayserova J, Shoenfeld Y. Allergy and autoimmunity: parallels and dissimilarity: the yin and yang of immunopathology. Autoimmun Rev. 2009;8:302–8. doi: 10.1016/j.autrev.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Gell PGH, Coombs RRA. The classification of allergic reactions underlying disease. In: Coombs RRA, Gells PGH, editors. Clinical Aspects of Immunology. Oxford, England: Blackwell Science; 1963. [Google Scholar]
- 30.Kersten C, Sivertsen EA, Hystad ME, et al. BMP-6 inhibits growth of mature human B cells; induction of Smad phosphorylation and upregulation of Id1. BMC Immunol. 2005;6:9. doi: 10.1186/1471-2172-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kersten C, Dosen G, Myklebust JH, et al. BMP-6 inhibits human bone marrow B lymphopoiesis–upregulation of Id1 and Id3. Exp Hematol. 2006;34:72–81. doi: 10.1016/j.exphem.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Seckinger A, Meissner T, Moreaux J, et al. Bone morphogenic protein 6: a member of a novel class of prognostic factors expressed by normal and malignant plasma cells inhibiting proliferation and angiogenesis. Oncogene. 2009;28:3866–79. doi: 10.1038/onc.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huse K, Bakkebo M, Oksvold MP, et al. Bone morphogenetic proteins inhibit CD40L/IL-21-induced Ig production in human B cells: differential effects of BMP-6 and BMP-7. Eur J Immunol. 2011;41:3135–45. doi: 10.1002/eji.201141558. [DOI] [PubMed] [Google Scholar]
- 34.Hagn M, Sontheimer K, Dahlke K, et al. Human B cells differentiate into granzyme B-secreting cytotoxic B lymphocytes upon incomplete T-cell help. Immunol Cell Biol. 2012;90:457–67. doi: 10.1038/icb.2011.64. [DOI] [PubMed] [Google Scholar]
- 35.Rowley B, Tang L, Shinton S, et al. Autoreactive B-1 B cells: constraints on natural autoantibody B cell antigen receptors. J Autoimmun. 2007;29:236–45. doi: 10.1016/j.jaut.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roth KA, Kapadia SB, Martin SM, et al. Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J Immunol. 1999;163:1490–7. [PubMed] [Google Scholar]
- 37.Ghosh S, Hoselton SA, Schuh JM. mu-chain-deficient mice possess B-1 cells and produce IgG and IgE, but not IgA, following systemic sensitization and inhalational challenge in a fungal asthma model. J Immunol. 2012;189:1322–9. doi: 10.4049/jimmunol.1200138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macpherson AJ, Lamarre A, McCoy K, et al. IgA production without mu or delta chain expression in developing B cells. Nat Immunol. 2001;2:625–31. doi: 10.1038/89775. [DOI] [PubMed] [Google Scholar]
- 39.Perona-Wright G, Mohrs K, Taylor J, et al. Cutting edge: Helminth infection induces IgE in the absence of mu- or delta-chain expression. J Immunol. 2008;181:6697–701. doi: 10.4049/jimmunol.181.10.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orinska Z, Osiak A, Lohler J, et al. Novel B cell population producing functional IgG in the absence of membrane IgM expression. Eur J Immunol. 2002;32:3472–80. doi: 10.1002/1521-4141(200212)32:12<3472::AID-IMMU3472>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Trounstine M, Alt FW, et al. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol. 1993;5:647–56. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Weck MN, Schottker B, et al. Gastric parietal cell antibodies, Helicobacter pylori infection, and chronic atrophic gastritis: evidence from a large population-based study in Germany. Cancer Epidemiol Biomarkers Prev. 2013;22:821–6. doi: 10.1158/1055-9965.EPI-12-1343. [DOI] [PubMed] [Google Scholar]
- 43.Ayesh MH, Jadalah K, Al Awadi E, et al. Association between vitamin B12 level and anti-parietal cells and anti-intrinsic factor antibodies among adult Jordanian patients with Helicobacter pylori infection. Braz J Infect Dis. 2013;17:629–32. doi: 10.1016/j.bjid.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergman MP, Faller G, D’Elios MM, et al. Gastric Autoimmunity. In: Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington (DC): 2001. [PubMed] [Google Scholar]
- 45.Sterzl I, Hrda P, Matucha P, et al. Anti-Helicobacter Pylori, anti-thyroid peroxidase, anti-thyroglobulin and anti-gastric parietal cells antibodies in Czech population. Physiol Res. 2008;57(Suppl 1):S135–41. doi: 10.33549/physiolres.931498. [DOI] [PubMed] [Google Scholar]
- 46.Sugiu K, Kamada T, Ito M, et al. Anti-parietal cell antibody and serum pepsinogen assessment in screening for gastric carcinoma. Dig Liver Dis. 2006;38:303–7. doi: 10.1016/j.dld.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 47.Lo CC, Hsu PI, Lo GH, et al. Implications of anti-parietal cell antibodies and anti-Helicobacter pylori antibodies in histological gastritis and patient outcome. World J Gastroenterol. 2005;11:4715–20. doi: 10.3748/wjg.v11.i30.4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonilla Palacios JJ, Miyazaki Y, Kanayuma S, et al. Serum gastrin, pepsinogens, parietal cell and Helicobacter pylori antibodies in patients with gastric polyps. Acta Gastroenterol Latinoam. 1994;24:77–82. [PubMed] [Google Scholar]
- 49.Strong MJ, Xu G, Coco J, et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog. 2013;9:e1003341. doi: 10.1371/journal.ppat.1003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SY, Park C, Kim HJ, et al. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology. 2015;148:137–147 e9. doi: 10.1053/j.gastro.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 51.Scott GN, DuHadaway J, Pigott E, et al. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J Immunol. 2009;182:7509–17. doi: 10.4049/jimmunol.0804328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitzalis C, Jones GW, Bombardieri M, et al. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14:447–62. doi: 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.