

Abstract
Mensacarcin is a highly oxygenated polyketide that was first isolated from soil-dwelling Streptomyces bacteria. It exhibits potent cytostatic properties (mean of 50% growth inhibition = 0.2 μm) in almost all cell lines of the National Cancer Institute (NCI)-60 cell line screen and relatively selective cytotoxicity against melanoma cells. Moreover, its low COMPARE correlations with known standard antitumor agents indicate a unique mechanism of action. Effective therapies for managing melanoma are limited, so we sought to investigate mensacarcin's unique cytostatic and cytotoxic effects and its mode of action. By assessing morphological and biochemical features, we demonstrated that mensacarcin activates caspase-3/7–dependent apoptotic pathways and induces cell death in melanoma cells. Upon mensacarcin exposure, SK-Mel-28 and SK-Mel-5 melanoma cells, which have the BRAFV600E mutation associated with drug resistance, showed characteristic chromatin condensation as well as distinct poly(ADP-ribose)polymerase-1 cleavage. Flow cytometry identified a large population of apoptotic melanoma cells, and single-cell electrophoresis indicated that mensacarcin causes genetic instability, a hallmark of early apoptosis. To visualize mensacarcin's subcellular localization, we synthesized a fluorescent mensacarcin probe that retained activity. The natural product probe was localized to mitochondria within 20 min of treatment. Live-cell bioenergetic flux analysis confirmed that mensacarcin disturbs energy production and mitochondrial function rapidly. The subcellular localization of the fluorescently labeled mensacarcin together with its unusual metabolic effects in melanoma cells provide evidence that mensacarcin targets mitochondria. Mensacarcin's unique mode of action suggests that it may be a useful probe for examining energy metabolism, particularly in BRAF-mutant melanoma, and represent a promising lead for the development of new anticancer drugs.
Keywords: apoptosis, bioenergetics, flow cytometry, melanoma, mitochondrial metabolism
Introduction
Historically, natural product discoveries have directly or indirectly contributed to a large number of new drug leads. A recent analysis of new medicines approved by the United States Food and Drug Administration between 1981 and 2014 revealed that ∼50% of all small molecule pharmaceuticals were based on natural products or derivatives thereof (1). Similarly, numerous anticancer agents in current clinical use are based on genetically encoded small molecules, including anticancer drugs with DNA interaction properties like doxorubicin and bleomycin and tubulin-acting metabolites like vincristine and epothilone. Natural products with their unique structures and properties continue to serve as an invaluable inspiration for new drug entities.
Mensacarcin (1; see Fig. 2) is a highly oxidized and stereogenic complex molecule with potent antitumor activity.3 The polyketide can be obtained in high yields from its producing organism Streptomyces bottropensis (unoptimized yield of 50 mg/liter) and was named after the location where the soil sample originated, next to the university's cafeteria (“mensa” in German). Its structure is related to the bioactive metabolite cervicarcin isolated from Streptomyces ogaensis (3). Initial cytotoxic evaluation of mensacarcin revealed potent antitumor activity comparable with that of doxorubicin, a clinically used anticancer drug for the treatment of a broad spectrum of cancer (4, 5). No total synthesis of mensacarcin has been published thus far; however, related synthetic programs toward the highly functionalized hexahydroanthracene backbone indicate the importance of the epoxide moieties within mensacarcin for antitumor activity (6–8). Indeed, semi-synthetic modifications targeting the side chain epoxide revealed a correlation of cytotoxicity with the degree of oxidation in the side chain (9). Detailed studies on mensacarcin's biosynthesis by Bechthold and co-workers (10) enabled the heterologous expression of mensacarcin's biosynthetic gene cluster to yield 1 and analogues. Its biogenesis entails several unusual enzyme activities, among them a new mechanism of epoxide formation in polyketides (9, 11). Mensacarcin was submitted to the NCI-60 human tumor cell line screen and showed strong anti-proliferative effects in all tested cell lines and low COMPARE correlations to known anticancer agents (12). Given the encouraging cytostatic and cytotoxic responses induced by mensacarcin in the NCI in vitro cell assay, the present study aims to examine mensacarcin's cellular mode of action. In 2017, it is estimated that there will be 87,100 new cases of melanoma in the United States and 9,730 deaths from the disease (13). Classical chemotherapy regimens confer only very low success rates with a median survival rate of 8 ± 2 months for patients with stage IV melanoma (14, 15). Melanoma genetics revealed that 50% of fast progressing melanomas contain a mutation in the gene that encodes B-Raf, which leads to constitutive activation of downstream signaling in the mitogen-activated protein kinase pathway (16). The BRAF V600E mutation is a hallmark for high-risk melanoma associated with shortened patient survival rates and tumor drug resistance (17, 18), and B-Raf has emerged as a validated target for melanoma intervention. B-Raf inhibitors like vemurafenib and dabrafenib show immense short-term tumor repression. However, chemoresistance is quickly acquired by the tumor, and disease relapse within several months is commonly observed. These limited treatment options indicate a need for new anti-melanoma drug leads with alternative targets, which could potentially be used in combination therapies to overcome intrinsic or acquired resistance to combat BRAF-mutant melanoma (18, 19). Mensacarcin's unique response pattern in the NCI-60 screen and pronounced selective cytotoxicity against the melanoma cell line panel motivated us to evaluate and characterize the biological effects in selected cell lines and explore its mode of action further. Considering the limited availability of effective therapies for melanoma, we are seeking to investigate mensacarcin's potential as an antitumor drug lead. In this study, we use a combination of molecular and cell-based assays to provide insights into the mechanism of mensacarcin-induced growth inhibition and cell death. We demonstrate that mensacarcin activates caspase-dependent apoptotic pathways and induces cell death with relative selectivity against melanoma cells. In addition, our results indicate that the target of mensacarcin is located in mitochondria and affects mitochondrial function in an atypical manner.
Figure 2.

Evaluation of cytostatic versus cytotoxic effects of mensacarcin in melanoma and colon cancer cells. A, chemical structure of mensacarcin (1). B, morphology of cells after 24-h exposure to 1 μm mensacarcin examined by phase-contrast microscopy. C, dose- and time-dependent growth inhibition and cytotoxicity of mensacarcin in SK-Mel-28 and SK-Mel-5 melanoma cells and HCT-116 colon cancer cells. Cell viability was determined by an MTT assay and LDH release assay as described under “Experimental Procedures.” Results are presented as mean ± S.D. (error bars) of three replicates (n = 3).
Results
Analysis of the NCI-60 cancer cell line screen
The growth inhibitory and cytotoxic activity patterns of mensacarcin in the NCI-60 cell line screen were evaluated using mean graphs illustrating the mean of 50% growth inhibition (GI50)4 and 50% lethal concentration (LC50) of all tested cancer cell lines versus the response of the individual cell line (Fig. 1). Mensacarcin exhibits uniformly strong growth inhibition in all tested cell lines with a mean GI50 of 0.2 μm (Fig. 1A) with renal cell line SN12C being especially sensitive. COMPARE analysis of the GI50 data set revealed no correlation between mensacarcin and compounds from the NCI Developmental Therapeutics Program standard agent database of 170 compounds (correlation < 0.3). The COMPARE algorithm ranks compounds by similarity of response patterns, which can give an indication of a similar mechanisms of action (12, 20). Interestingly, mensacarcin displays a unique pattern of growth inhibition with no known similarity to other agents, which has been referred to as “COMPARE-negative.” Besides its strong universal anti-proliferative effect, most cell lines treated with mensacarcin did not reach the LC50 with the highest concentration tested (100 μm) (Fig. 1B). The strongest responding cell lines in the mean log LC50 analysis were the melanoma panel with an average LC50 of 10 μm, RPMI-8226 (leukemia), NCI-H522 (non-small lung cell), COLO205 (colon), SNB-75 (CNS), A498 (renal), and MDA-N (breast). Given mensacarcin's relative selective cytotoxicity to melanoma cells in the NCI-60 screen, subsequent experiments were focused on two melanoma cell lines (SK-Mel-5 and SK-Mel-28) that gave distinct responses. We also selected a colon cancer carcinoma (HCT-116) as a representative non-responsive cell line.
Figure 1.

Growth inhibition and lethality pattern of mensacarcin in the NCI-60 human tumor cell line screen. Presented are the mean graphs that show the difference in response to mensacarcin (NSC 718798) of the individual cell line to the mean response of all cell lines for the log GI50 (A) and the log LC50 (B). The NCI-60 screen data were retrieved from the publically available NCI Developmental Therapeutics Program database (https://dtp.cancer.gov/).5
Mensacarcin exhibits general cytostatic but cell type–specific cytotoxic effects
Lethality of compounds in the NCI cancer screen is indirectly examined by measurement of total cellular protein at the beginning of the assay and at the end of the 48-h treatment using sulforhodamine B (https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm)5 (21). The present study sought to examine growth inhibition and cell fate induced by mensacarcin in selected cancer cell lines in a direct and comprehensive manner. We chose two representatives from the NCI melanoma cell line panel (SK-Mel-28 and SK-Mel-5 cells) to characterize mensacarcin's efficacy against malignant melanomas and compared its activity against HCT-116 colon carcinoma cells. SK-Mel-5 and SK-Mel-28 are both well-characterized melanoma cell line models with a BRAF V600E mutation. Eight of the nine NCI-60 melanoma cell lines carry the BRAF mutation, with SK-Mel-2 being the exception. All melanomas were responsive in the NCI LC50 assay, with SK-Mel-28 being the least responsive cell line in the panel. As illustrated in Fig. 2B, both melanoma cells and HCT-116 colon cancer cells treated with mensacarcin show strong reduction in cell density and morphological changes. Cell viability was monitored and quantitatively assessed via measuring reduction of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) by viable cells to a purple formazan. Mensacarcin concentrations as low as 0.5 μm induced strong, concentration-dependent growth inhibition after 24 h in melanoma cells and in HCT-116 colon cancer cells (Fig. 2C). Next, we investigated the effect of mensacarcin treatment on membrane integrity of cells. The loss of membrane integrity is a distinct sign of necrotic cell death and results in release of LDH into the extracellular matrix. Our results demonstrated that mensacarcin induced concentration- and time-dependent cell death in the two tested melanoma cell lines, concurrent with growth inhibition but only moderate LDH release. Interestingly, HCT-116 colon carcinoma cells are strongly inhibited by mensacarcin in the cell viability assay but do not show signs of cell death in the LDH cytotoxicity assays when treated with mensacarcin. In accordance to the initial NCI-60 cancer screen, our data demonstrate that mensacarcin induced universal strong cytostasis independent of cancer type, but cell death was only observed in melanoma cells.
Initial evidence for apoptosis in SK-Mel-28 cells that were exposed to mensacarcin was observed by confocal microscopy. Hoechst staining was used to visualize nuclei and their morphological changes via fluorescence microscopy in cells upon exposure to mensacarcin (Fig. 3A). Changes in nuclear morphology included chromatin condensation. We also observed various characteristic stages of apoptotic nuclear disassembly, including formation of spherical aggregates along the nuclear lamina, formation of necklace-shaped rings, shrinking of the nucleus, and nuclear collapse (22) (Fig. 3A, c and d). Moreover, the use of ER-Tracker revealed that mensacarcin-treated melanoma cells showed cytotoxic responses, including detachment and cell shrinkage. In contrast to the melanoma cells, no nuclear alterations or other signs of apoptosis were observed in HCT-116 cells treated with mensacarcin (Fig. 3A, f).
Figure 3.

Mensacarcin induces rapid apoptotic cell death in melanoma cells. A, changes in nuclear morphology with chromatin condensation as a hallmark for apoptosis were examined by confocal fluorescence microscopy. Presented are images of SK-Mel-28 or HCT-116 cells that were grown in the presence of 0.5% (v/v) DMSO (a and e) or 50 μm mensacarcin (b and f) for 15 h and analyzed after staining with Hoechst 33342 and ER-Tracker Green. Insets show examples for cells with chromatin condensation in a single focal plane, which visualizes the necklace-shaped rings characteristic of apoptosis (c), or in a maximum intensity projection (d). B, apoptotic cell death was evaluated by immunoblot analysis of caspase 3 activation and PARP-1 cleavage. Shown are immunoblots of cell lysates of cells grown in the presence of various concentrations of mensacarcin or 0.25% (v/v) DMSO for 15 h (SK-Mel-28, SK-Mel-5) or 48 h (HCT-116). Analyzed lysates were pools of duplicates. β-Actin levels served as loading control. C, DNA damage associated with early apoptosis in SK-Mel-5 cells induced by mensacarcin was determined by an alkaline comet assay. Cells were grown in the presence of various amounts of mensacarcin or 0.5% (v/v) DMSO for 6 h or 15 h. At least 100 nuclei were randomly selected and scored for DNA damage. Results are presented as mean ± S.E. (error bars) (n = 100; two slides, 50 cells/slide). *, p < 0.1; ***, p < 0.001 versus DMSO control. B, representative images of mensacarcin-induced comets after 15-h treatment. Doxorubicin (5 μm) was used as a positive control.
Given the observed cytotoxicity, we then tested the ability of mensacarcin to induce activation of apoptotic signaling pathways. A characteristic event in apoptosis is the proteolytic activation of the caspase cascade, eventually resulting in the initiation of downstream apoptosis-executing processes. Effector caspases like caspase-3 and -7 cleave the nuclear enzyme poly(ADP-ribose)polymerase-1 (PARP-1) into specific 89- and 24-kDa fragments. PARP-1 is generally activated during DNA damage and initiates DNA repair; however, during apoptosis, caspase-mediated cleavage of PARP-1 inhibits DNA repair mechanisms, eventually leading to DNA fragmentation (23, 24). The proteolytic activation of procaspases and the presence of the specific 89-kDa PARP fragment are therefore considered hallmarks of apoptosis. Although not well understood, caspase-independent as well as PARP-1–independent programmed cell death has been described (23, 25). Melanoma cells were exposed to increasing mensacarcin concentrations for 6–48 h, and activation of caspase-3 and subsequent cleavage of PARP-1 was investigated by immunoblot analysis. Mensacarcin concentrations of 0.5–1 μm or greater induced the formation of 89-kDa PARP-1 fragments as well as caspase-3 activation in SK-Mel-28 and SK-Mel-5 beginning between 6 and 15 h after exposure (Fig. 3B). Cleavage of the pro-apoptotic proteins could not be observed in HCT-116 cells after 48 h of treatment.
Next, we examined SK-Mel-5 cells for mensacarcin-triggered DNA damage as a sign of early apoptosis by an alkaline comet assay. This single-cell gel electrophoresis technique allows detection of DNA-altering lesions, including DNA single-strand breaks or double-strand breaks, or indicates incomplete excision repair (26). Mensacarcin treatment for 6 or 15 h induced a significant dose-dependent increase of DNA in the comet tail (Fig. 3C). The early onset of DNA damage is surprising, and the amount of DNA fragmentation increases in cells with a longer incubation time of 15 h. The comet assay verifies that mensacarcin treatment leads to genomic instability in SK-Mel-5 cells, a classic hallmark of early apoptotic onset (27).
Verification of caspase-dependent apoptosis in melanoma cells and release of cytochrome c
To provide some insight into the potential mechanism of mensacarcin-induced cell death, we tested the ability of mensacarcin to activate the downstream effector caspases of the major apoptotic signaling pathways: caspase-3 and caspase-7. Mensacarcin induced activation of caspase-3 and -7 in SK-Mel-28 and SK-Mel-5 cells over a time frame consistent with loss of cell viability (12 and 24 h) but did not induce caspase-dependent cell death in HCT-116 colon carcinoma cells (Fig. 4). In control experiments, we found that treatment with the broad-spectrum caspase inhibitor Z-VAD-FMK (28) (10 μm) alone produced no significant change in the viability of either SK-Mel-28, SK-Mel-5, or HCT-116 cells as determined by caspase activation assays (Fig. 4). Co-treatment of cells with mensacarcin and Z-VAD-FMK attenuated mensacarcin-induced caspase activation at all tested mensacarcin concentrations (Fig. 4). These results indicate that mensacarcin activates caspase-dependent apoptotic pathways in melanoma cells. Taken together, we found that mensacarcin induces caspase and PARP activation, DNA damage, and changes in nuclear morphology, which strongly suggests that mensacarcin induces cell death through apoptotic pathways.
Figure 4.

Mensacarcin induces activation of caspase-3 and -7 in a cell type–specific manner in melanoma. Shown is the concentration–response relationship for mensacarcin-induced caspase-3/7 activation in SK-Mel-28 and SK-Mel-5 cells and HCT-116 after 12-h (A) and 24-h (B) exposure to mensacarcin (0.5, 2, 10, and 50 μm), vehicle (DMSO), or the positive control staurosporine (10 μm). We also tested the broad-spectrum caspase inhibitor Z-VAD-FMK alone (10 μm) and in combination with mensacarcin (0.5, 2, 10, and 50 μm), respectively. In all cases, the caspase-activating activity of mensacarcin was attenuated by Z-VAD-FMK. Caspase-3/7 activity was measured at the end of each treatment using a luminescence-based assay coupled with automated luminescence detection. Scatter plots represent mean ± S.D. (error bars) of triplicates (n = 3). *, p < 0.1; **, p < 0.01; ***, p < 0.001 mensacarcin versus DMSO treatment.
Mitochondrially mediated programmed cell death can be monitored by the release of the apoptogenic factor cytochrome c from the mitochondria into the cytosol (29). We treated SK-Mel-5 cells for 6 h with 50 μm mensacarcin and 10 μm staurosporine, a known inducer of mitochondrial apoptosis. Cells were lysed and fractionated using differential centrifugation to obtain a cytosolic fraction for immunoblot analysis. The Western blot result indicated that cytochrome c was released from the mitochondria into the cytosol for the mensacarcin and staurosporine treatment (Fig. 5). Notably, in melanoma cells, mensacarcin was an order of magnitude more potent in rapid induction of apoptotic pathways than the anticancer agent doxorubicin (data not shown).
Figure 5.

Mensacarcin treatment triggers cytochrome c release from mitochondria into the cytoplasm of melanoma cells. A, the cytosolic fractions of SK-Mel-5 cells non-treated or treated with mensacarcin (50 μm) and staurosporine (10 μm) for 6 h were analyzed for cytochrome c content by Western blot analysis. GAPDH levels served as loading control. B, densitometric measurement of Western blotting. Staurosporine control was set to 100% cytochrome c release. Results are presented as mean ± S.D. (error bars) of triplicates (n = 3). *, p < 0.1; ***, p < 0.001 versus DMSO control.
Mensacarcin represents a unique chemical scaffold with nine stereogenic centers and two epoxide groups that could be prone to nucleophilic attack by nucleobases (30). Various conditions to covalently bind mensacarcin with DNA were tested, but no DNA-mensacarcin adducts formed with either calf thymus DNA or single nucleobases or in isolated DNA from treated melanoma cells could be detected via LC-MS analysis. Direct binding to immobilized double-stranded DNA on a biosensor platform via biolayer interferometry (31) was successful for other DNA-acting molecules (ethidium bromide, doxorubicin, Hoechst 33342), but no specific binding event could be detected for mensacarcin (data not shown). Additionally, we investigated mensacarcin's ability to intercalate DNA, but mensacarcin was not able to replace known intercalators from isolated DNA (32). We conclude that mensacarcin does not intercalate or alkylate DNA.
Effects on the cell cycle by mensacarcin-treated melanoma cells
To further examine the mechanism of action of mensacarcin, the cell cycle distribution was investigated by flow cytometric analysis of propidium iodide (PI)-stained cells. Consistent with our immunoblot analysis and fluorescence microscopy results, increasing mensacarcin concentrations resulted in a dose-dependent increase in apoptotic cells with hypodiploid DNA content for SK-Mel-28 and SK-Mel-5 cells (sub-G1 population) but produced only a minor increase of the sub-G1 population in HCT-116 cells (Fig. 6A). Interestingly, the phase distribution of the remaining cells did not change significantly in the presence of mensacarcin in melanoma cells, whereas in HCT-116 cells, a moderate S-/G2-phase arrest could be observed (Fig. 6A). Depending on their mechanism of action, many known anticancer agents induce a phase-specific cell cycle arrest. For example, the DNA-acting reagents doxorubicin and etoposide both arrest cells mainly in G2 phase (33, 34). Mensacarcin, in contrast, induced only a modest shift in the cell cycle distribution. We examined the effect of mensacarcin on synchronized SK-Mel-5 cells; however, we found that mensacarcin does not arrest cells in a specific phase but found induction of apoptosis in melanoma cells independent of cell cycle phase (data not shown).
Figure 6.

Cell cycle analysis of mensacarcin-treated cells. A, cell cycle distribution was analyzed by flow cytometric measurements after PI staining. B and C, cell cycle analysis (B) and quantification of apoptotic cells (C) appearing in sub-G1 fraction upon mensacarcin treatment. SK-Mel-28, SK-Mel-5 cells, and HCT-116 were grown in the presence of various concentrations of mensacarcin or 0.5% (v/v) DMSO for 24 h. The apoptotic cell fraction was calculated by including and gating on the sub-G1 peak. Results are presented as mean ± S.D. (error bars) of duplicates (n = 2). *, p < 0.05; ***, p < 0.001 versus DMSO control.
Rhodamine-labeled mensacarcin induces apoptotic cell death in melanoma cells
We next sought to determine the subcellular compartment targeted by mensacarcin. To monitor its localization in live cells via fluorescence microscopy, we synthesized a rhodamine-labeled mensacarcin probe. Rhodamine was linked to mensacarcin using tetramethylrhodamine-5-carbonyl azide (TMRCA) following established protocols (35). Upon heating TMRCA to 80 °C, the azide group undergoes a Curtius rearrangement to yield the isocyanide group, which instantly reacts with mensacarcin's hydroxyl groups to form the carbamate-linked conjugate (TMR-mensa) (36, 37) (Fig. 7A). After 1 h, three predominant products with combined rhodamine- and mensacarcin-specific spectroscopic features (absorbance at 545 and 330 nm, respectively) were found in the reaction mixture, indicating that all three secondary hydroxyl groups of 1 were utilized. The rhodamine-mensacarcin conjugates were purified via HPLC separation and analyzed by high-resolution mass spectrometry and UV properties. Only singly labeled mensacarcin probes were observed and combined for application in microscopy studies. TMR-mensa also showed growth inhibition and cytotoxicity properties that were similar to unlabeled mensacarcin. In SK-Mel-5 cells, growth was inhibited starting at 1.0 μm TMR-mensa (versus 0.5 μm mensacarcin), and LDH release was induced with 1.5 μm TMR-mensa (versus 0.5 μm mensacarcin) (Fig. 7B). Caspase-mediated cleavage of PARP-1 and induction of apoptosis could be detected after 15 h with 5 μm TMR-mensa (versus 1 μm with mensacarcin) (Fig. 7C).
Figure 7.

Preparation of a fluorescently labeled mensacarcin and activity validation. A, synthesis of the rhodamine-mensacarcin conjugate. B, evaluation of the cytotoxic and apoptotic activity of the rhodamine-mensacarcin conjugate. Shown are dose-dependent growth inhibition and cytotoxicity of the rhodamine-mensacarcin conjugate in SK-Mel-5 cells (48-h treatment) compared with treatment with unlabeled mensacarcin determined by MTT assay and LDH assay. Vehicle-treated cells were grown in the presence of 1% (v/v) DMSO. Results are presented as mean ± S.D. (error bars) (n = 3). C, apoptotic cell death in SK-Mel-5 cells induced by TMR-mensa was evaluated by immunoblot analysis of caspase-3 activation and PARP-1 cleavage. Cells were grown in the presence of various concentrations of TMR-mensa or 0.5% (v/v) DMSO for 15 h. Analyzed lysates were pools of duplicates. β-Actin levels served as loading control.
Rhodamine-labeled mensacarcin localizes in mitochondria
To study the subcellular distribution of TMR-mensa, SK-Mel-5 cells were incubated with the fluorescent probe for 20 min and co-stained with markers for the nucleus, endoplasmic reticulum, and mitochondria. Confocal fluorescence microscopy confirmed a quick uptake of TMR-mensa into the cells. A general hazy background of rhodamine fluorescence throughout the cell indicated some nonspecific binding and/or excess of fluorescence probe. SK-Mel-5 cells adopt a heterogeneous cell population ranging from smaller cells with tightly packed organelles to widespread cells with defined structures that enable the distribution pattern for TMR-mensa to be readily visualized. Interestingly, TMR-mensa is strictly excluded from the nucleus and does not co-localize with the nuclear Hoechst stain (Pearson correlation r = −0.30) (Fig. 8, A and B). This stands in strong contrast to TMRCA, the uncoupled rhodamine dye, which does accumulate in the nucleus (Fig. 8C). This result supports our previous findings that mensacarcin does not directly interact with nuclear DNA. In addition, the fluorescence imaging studies indicated that TMR-mensa substantially co-localizes with the mitochondrial marker. This is particularly evident in cell areas with well-defined cellular compartments (Fig. 8A). A Pearson correlation of r = 0.61 (r = 0.73 in well-defined areas; Fig. 8A (inset)) provides statistical evidence for a linear relationship between TMR-mensa and the mitochondrial stain. In addition to this distinct mitochondrial localization, a less defined non-mitochondrial perinuclear region with vesicle-like structures also showed enhanced TMR-mensa fluorescence. It is possible that these fluorescent features correspond to activity-independent accumulation of TMR-mensa in endosomal/lysosomal compartments, which are known to trap tetramethylrhodamine in the acidic lysosomal lumen. Additionally, rhodamine dyes are known to show enhanced fluorescence at lower pH values such that acidic compartments are stained more brightly (38). The mitochondrial localization of TMR-mensa observed through imaging was also supported by subcellular fractionation studies. Using differential centrifugation, we obtained an enriched mitochondrial fraction, a crude light membrane fraction, and a cytosolic fraction from TMR-mensa–treated cells. As shown in Fig. 8D, rhodamine fluorescence is enriched in the mitochondrial fraction of SK-Mel-5 cells after 15 h of mensacarcin treatment and less abundant in the light membrane fraction (lysosomes, peroxisomes, and microsomes) and cytosolic fraction. Similar trends were already observed after a 30-min incubation with mensacarcin (data not shown). The fluorescence of the TMR-mensa probe is not exclusively found in mitochondria; therefore, we cannot exclude the possibility that non-mitochondrial compartments may also contribute to the overall activity of mensacarcin. The present study does show that TMR-mensa is significantly enriched in mitochondria, suggesting that mensacarcin's principal site of action may be located in this compartment.
Figure 8.
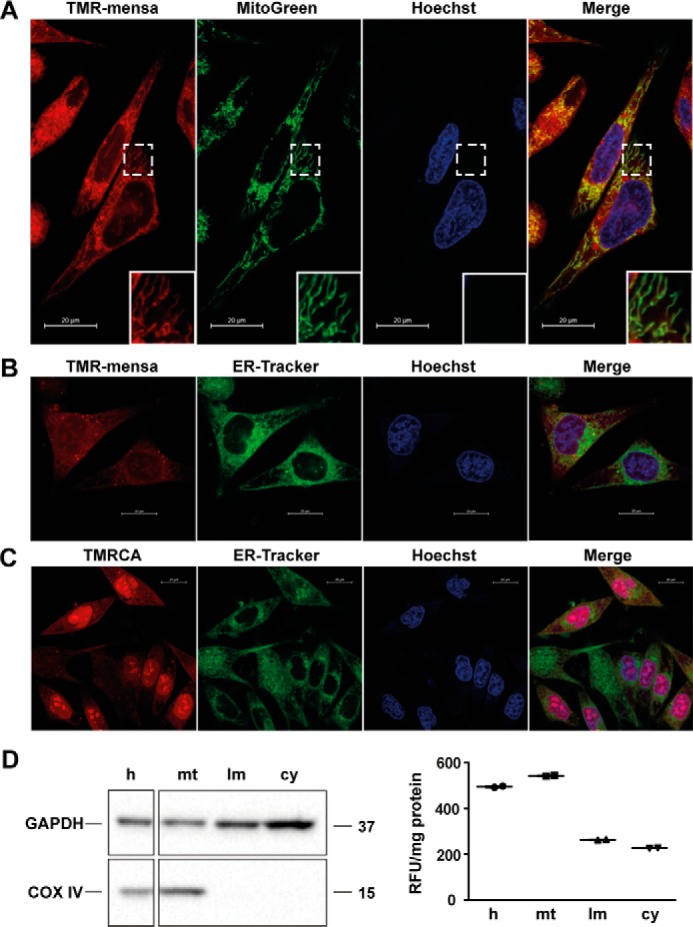
Cell localization studies in melanoma cells with rhodamine-mensacarcin probe. A, SK-Mel-5 cells were stained with TMR-mensa, Hoechst 33342 (nucleus), and ER-Tracker Green (endoplasmic reticulum) or MitoGreen (mitochondria) for 20 min. Images were captured by inverted confocal fluorescence microscopy and merged to examine co-localization. TMR-mensa does co-localize with the MitoGreen with well-defined mitochondrial staining (inset). B, TMR-mensa does not co-localize with the ER-Tracker or Hoechst. C, control cells were stained with uncoupled rhodamine dye (TMRCA) and present an overall diffuse background fluorescence with dye accumulation in nuclei. D, cell subfractionation of TMR-mensacarcin–treated SK-Mel-5 cells (15-h exposure) into homogenate (h), mitochondrial fraction (mt), light membrane fraction (lm), and cytosolic fraction (cy). Fluorescence of each cellular subfraction was analyzed and normalized to protein content (excitation, 530 nm; emission, 590 nm). The enrichment of mitochondria in the mitochondrial fraction and the absence of mitochondria in the light membrane and cytosolic fractions was analyzed by immunoblot analysis using cytochrome c oxidase subunit IV (COX IV) as mitochondrial marker. The enrichment of cytosolic proteins in the cytosolic fraction was analyzed using GAPDH as marker protein.
Mensacarcin causes mitochondrial stress in melanoma cells
Based on the co-localization studies, we identified mitochondria as the major compartment labeled by our fluorescent-mensacarcin probe. This indicates that mensacarcin's universal cytostatic and potent cell type–specific cytotoxic properties might be derived from interference with mitochondrial activity. To investigate the effect of mensacarcin on mitochondrial function and cellular bioenergetics, we monitored cellular oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) in real time as measures of mitochondrial respiration and glycolysis, respectively. Using a Seahorse extracellular flux analyzer, we assessed the energy metabolism of SK-Mel-28, SK-Mel-5, and HCT-116 cells before and after exposure to mensacarcin. On melanoma cells, mensacarcin had a pronounced effect. The addition of mensacarcin induced a concentration-dependent reduction of basal OCR in both melanoma cell lines. With exposure to 50 μm mensacarcin, oxidative consumption decreased rapidly in the first 5 min of exposure and stabilized at 48% of the baseline in SK-Mel-28 and 74% in SK-Mel-5 after 60 min. The addition of the control, ATP-synthase inhibitor oligomycin (1 μm in well), which suppresses ATP production and reduces oxidative phosphorylation to the minimum necessary to balance proton leak, lowered OCR to ∼30% of the baseline value. In contrast, mensacarcin had a minimal effect on oxygen consumption in HCT-116 cells, reducing the OCR only to 85% of control values at the highest dose tested (Fig. 9A). This is consistent with previous assays that showed that this colon carcinoma cell line is relatively insensitive to mensacarcin. For comparison, colon cancer cells were responsive to the ATP-synthase inhibitor oligomycin (1 μm in well), which lowered OCR to ∼18% of the baseline.
Figure 9.

Mensacarcin impairs mitochondrial function in melanoma cells. A, OCR and ECAR were measured using the Seahorse analyzer XF24. After basal measurements, mensacarcin was injected at different concentrations (black arrows) (n = 3). B, OCR was measured after mensacarcin was injected (black arrow) in different concentrations, followed by consecutive injections of oligomycin (1 μm), FCCP (0.5 μm), and antimycin A (0.5 μm)/rotenone (0.5 μm) (n = 3). C, basal bioenergetics state of SK-Mel-28, SK-Mel-5, and HCT-116 cells (n = 25). Error bars, S.D. ***, p < 0.001 versus DMSO control.
In addition to measuring the OCR, we also measured the effects of mensacarcin and oligomycin on media acidification (ECAR). In agreement with numerous prior studies, oligomycin produced an increase in ECAR that represents a compensatory increase in glycolysis (39). Consistent with our previous assays and OCR measurements, mensacarcin had a modest effect on ECAR in HCT-116 cells. However, interestingly, mensacarcin had distinct but variable effects on ECAR in melanoma cells. In contrast to oligomycin, mensacarcin did not induce a compensatory increase in glycolysis. Low concentrations of mensacarcin (2 μm) yielded ECAR responses similar to control, whereas higher concentrations (50 μm) produced slight decreases in the acidification rate in melanoma cells. These results suggest complex concentration-dependent effects on cell glycolysis and lactate formation.
To gain further insight into the mensacarcin-induced mitochondrial impairment in melanoma cells, we assessed key parameters of mitochondrial function by consecutively exposing cells to well-described mitochondria-perturbing reagents in a so-called mitochondria stress test. Following the addition of mensacarcin, we sequentially added oligomycin, FCCP, and, last, rotenone and antimycin A. This well-established protocol provides information on basal respiration, ATP-linked respiration, proton leak, maximal respiration capacity, and non-mitochondrial respiration. As shown in Fig. 9B, basal respiration is lowered dose-dependently by treatment with mensacarcin, suggesting a decrease in oxidative phosphorylation. Oligomycin inhibits ATP synthase and reduces OCR, FCCP uncouples oxygen consumption from ATP production and raises OCR to a maximal value, and antimycin A and rotenone target the electron transport chain and reduce OCR to a minimal value. Mensacarcin affects the mitochondrial function of cells with a distinct bioenergetic signature. The rapid and substantial decrease in OCR upon the addition of mensacarcin suggests that mensacarcin targets a pathway that is critical to oxidative phosphorylation (Fig. 9B). However, because the addition of oligomycin produced a further decrease in OCR in all cases, it appears that mensacarcin has either an incomplete effect or that it targets a modulatory pathway. Furthermore, because FCCP addition caused OCR to return to near control levels (the difference between post-FCCP and post-rotenone/antimycin A OCR is unaltered), mensacarcin does not appear to directly target a protein that is necessary for the electron transport chain. Consistent with this interpretation, known electron transport chain inhibitors (e.g. BAY 87-2243 (40), apoptolodins (41), and rotenone/antimycin A (42)) produce a decrease in OCR that cannot be rescued by the addition of FCCP. The effects of mensacarcin on ECAR also provide valuable insights into its consequences for mitochondrial function. Small molecules that inhibit ATP synthase, like oligomycin, produce a characteristic, compensatory increase in glycolysis that is observable as an increase in ECAR. In contrast, mensacarcin produced no such increase in ECAR. At high concentrations, mensacarcin appeared to have the opposite effect, producing a decrease in ECAR. Such a decrease in ECAR has been observed in response to compounds that inhibit glycolysis (e.g. phloretin, deoxyglucose, and dichloroacetate) (43). These results may suggest that mensacarcin does not directly target the electron transport chain or ATP-synthase but instead targets a pathway that produces precursors for oxidative phosphorylation (e.g. glycolysis), targets proteins that support mitochondrial health, or targets components of a regulatory pathway. In addition to their role in cellular respiration, mitochondria also play a key role in regulating apoptotic cell death. Therefore, alteration of the equilibrium between pro-apoptotic and anti-apoptotic proteins of the Bcl-2 family not only can prompt the mitochondrial apoptotic pathway but also can induce mitochondrial failure (44).
Therefore, it will be interesting to investigate the impact of mensacarcin on direct mitochondrial acting pro-apoptotic factors in the future. Given its effects on mitochondrial function, we considered whether the selectivity of mensacarcin for different cell lines might correlate with metabolic traits of those lines. Cancer cells often show substantial alterations in their metabolic pathways. Many cancer cells have elevated glucose uptake rates and increased aerobic glycolysis, first described by Otto Warburg (45), whereas other tumors demonstrate an increase in mitochondrial energy metabolism (46). Some of these differences can be captured by the OCR/ECAR ratio, which indicates the rate of oxidative respiration relative to glycolysis. It is interesting to note that melanoma cells are both relatively sensitive to mensacarcin and that these same cell lines have relatively low OCR/ECAR ratios (Fig. 9C). The increased dependence on glycolysis is especially prominent in BRAF-driven tumorigenesis (39, 47). Consistent with this trend, we found that melanoma cells are more sensitive to mensacarcin and exhibit lower OCR/ECAR ratios compared with HCT-116 (Fig. 9C). Although only correlative, this may suggest a mechanism for the cell type selectivity of mensacarcin.
Discussion
In summary, mensacarcin (1), a highly complex polyketide isolated from a soil-dwelling Streptomyces, showed potent universal anti-proliferative effects in all tested cancer cell lines in the NCI-60 cell line panel as well as in our in vitro assays. Cytostasis is accompanied by fast progression into cell death in only a few cell lines, notably in melanoma cell lines. Subsequent experiments demonstrated that in melanoma cells, mensacarin activates caspase-dependent apoptotic pathways. We also observed early markers of apoptosis, including DNA damage measured using a comet assay (6 h post-exposure) and nuclear alterations visualized with confocal microscopy (15 h post-exposure). Chemical coupling of mensacarcin with a rhodamine dye yielded a fluorescent probe that localized to mitochondria within 20 min of exposure. Metabolic flux analysis of mensacarcin-treated melanoma cells revealed changes in oxidative consumption rate within minutes of exposure, consistent with a mitochondria-perturbing mode of action.
In recent years, mitochondria have emerged as a potential target for anticancer therapy as cancer cell mitochondria are structurally and functionally different from their non-cancerous counterparts (48). Resistance to apoptotic stimuli, inactivation of the tumor suppressor protein p53, and overexpression of the proto-oncogene Bcl-2 are commonly seen traits in neoplasia. Mitochondria play a pivotal role in cell death signaling, and small molecules that can selectively target mitochondria and activate the apoptotic cascade without relying on intact upstream apoptotic pathways could represent a directed approach in cancer therapy. Malignant melanoma patients are in urgent need of new therapeutic options, as drug resistance to vemurafenib and other B-Raf-targeting agents is readily observed. Mitochondrial respiration is up-regulated in response to vemurafenib treatment in melanoma cells and reagents like mensacarcin that alter mitochondrial respiration may be good candidates for drug combination therapy.
In conclusion, this study shows that mensacarcin strongly inhibits cell growth universally in human cancer cell lines and potently induces apoptosis in melanoma cells. Our results indicate that mensacarcin localizes to mitochondria, affects energy metabolism in mitochondria, and activates caspase-dependent apoptotic pathways. Because mensacarcin is produced in high titers by S. bottropensis, it is possible to perform in vivo compound evaluations, and initial studies show that mensacarcin is well tolerated in mice, with a maximum tolerated dose of 30 mg/kg/day (i.p.) being most effective after 0, 3, and 7 days (Oncotest Freiburg, now Charles River Laboratories). Our results suggest that mesacarcin acts via a unique and rapid effect on cell metabolism. This is consistent with the observed cell type specificity, where it induced strong cytostasis in all cell types tested but was most effective in cancer types with low oxygen respiration ratios like melanomas. Mensacarcin is a promising new compound with potentially important applications as both an anti-melanoma drug lead and a molecular probe.
Experimental procedures
Reagents and cell lines
Unless otherwise stated, all chemicals were purchased from Amresco (Solon, OH) or TCI (Portland, OR). HCT-116 colon cancer cells were purchased from ATCC (Manassas, VA), SK-Mel-28 cells were a kind gift from Dr. Nupur Pande (Oregon Health and Science University, Portland, OR). SK-Mel-5 cells were obtained from the NCI cell line repository (Frederick, MD). Minimum essential medium, DMEM, trypsin/EDTA (0.25%/2.21 mm), and penicillin/streptomycin solution were obtained from Corning Life Sciences (Corning, NY). FBS was obtained from Atlanta Biologicals (Flowery Branch, GA). PBS, Halt® protease inhibitor mixture, HRP-labeled goat anti-rabbit IgG (catalog no. 65-6120), doxorubicin, PI, TMRCA, and ER-Tracker Green (BODIPY® FL glibenclamide) fluorescence stain were acquired from Thermo Fisher Scientific (Waltham, MA). CytoPainter MitoGreen fluorescence stain was obtained from Abcam (Cambridge, UK). Hoechst 33342 was purchased from BioVision Inc. (Milpitas, CA). Anti-PARP-1 (catalog no. 9542), anti-caspase-3 (catalog no. 9662), and cytochrome c (catalog no. 11940S) antibodies were obtained from Cell Signaling Technologies (Danvers, MA). The PARP antibody detects full-length PARP-1 (116 kDa) as well as the large fragment (89 kDa) of PARP-1 resulting from caspase cleavage. The antibody does not cross-react with related proteins or other PARP isoforms. The caspase-3 antibody detects full-length caspase-3 (35 kDa) and the large fragment of caspase-3 resulting from cleavage (17 kDa). Anti-actin antibody (catalog no. PA1872) was obtained from Boster Biological Technology (Pleasanton, CA). Luminol sodium salt was purchased from Biosynth (Itasca, IL), and p-coumaric acid was purchased from MP Biomedicals (Solon, OH). Antimycin A was obtained from Enzo Life Sciences (Lörrach, Germany), FCCP and rotenone were from Cayman Chemical Co. (Ann Arbor, MI), and oligomycin was from Merck Millipore (Billerica, MA). Staurosporine and Z-VAD-FMK were purchased from Adooq Bioscience. Mensacarcin was received as a gift from Bioviotica (Göttingen, Germany), and its purity and stability of stock solutions were tested via LC-MS analysis before usage.
Cell culture
HCT-116, SK-Mel-5, and SK-Mel-28 cells were grown in minimum essential medium supplemented with 10% (v/v) FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). The cell lines were maintained in a humidified chamber at 37 °C with 5% CO2. The passage number for cells used in experiments never exceeded 15 passages. All cell lines were tested mycoplasma-negative by real-time PCR (MycoSolutions mycoplasma detection kit, Akron Biotech, Boca Raton, FL).
Cell proliferation assay
Cell viability was determined by measuring the reduction of the tetrazolium salt MTT by metabolically active cells (49). Cells were plated into 96-well plates and maintained overnight before treatment was started with the addition of mensacarcin to each well. After the designated time, MTT (5 mg/ml in PBS) was added to each well at a final concentration of 0.5 mg/ml. The plates were incubated for 2 h at 37 °C. The medium was removed, cells were lysed, and the purple formazan product was solubilized by the addition of 50 μl of DMSO. Absorbance was measured at 550 nm. Metabolic activity of vehicle-treated cells (0.5% DMSO unless otherwise stated) was defined as 100% cell growth.
Cell cytotoxicity assay
Cell cytotoxicity was determined by measuring the extent of lactate dehydrogenase (LDH) release of cells (50). Cell were plated into 96-well plates and maintained overnight before treatment was started with the addition of mensacarcin. After the designated time, LDH activity of the cell supernatant was measured. Briefly, 50 μl of supernatant of each well was transferred to a new well plate, and 50 μl of iodonitrotetrazolium-lactate mix was added (100 μl of iodonitrotetrazolium solution (25 mg/ml in DMSO), 100 μl of phenazine methosulfate solution (7.75 mg/ml in PBS), 2.3 ml of NAD+ (2.8 mg/ml in PBS), and 2.5 ml of lithium lactate solution (150 mm lithium lactate in 50 mm Tris, pH 8.5). Well plates were incubated for 15 min at room temperature in the dark, and the reaction was stopped by the addition of 50 μl of 1 m acetic acid. Absorbance was measured at 490 nm. LDH activity of lysed vehicle-treated cells (0.5% DMSO unless otherwise stated) was defined as 100% cell cytotoxicity. To lyse vehicle-treated cells, lysis solution (0.9% (v/v) Triton X-100) was added to the cell culture medium, and cells were incubated for 15 min at 37 °C before the assay was performed.
Western blot analysis
For extraction of total cellular protein, cells were detached, and culture medium and cells were collected by centrifugation. Pellets were washed with PBS and resuspended in ice-cold lysis buffer (1% (v/v) Nonidet P-40, 150 mm NaCl, 50 mm Tris, pH 8.0, protease inhibitor mixture) and kept for 20 min on ice. After a 20-s sonication, cell lysates were cleared by centrifugation. The supernatant contained the total cellular protein extract. Protein concentrations were determined using the BCA protein assay kit from Pierce according to the manufacturer's instructions. Lysates were mixed with Laemmli loading buffer and incubated for 6 min at 95 °C. The same amount of protein for each cell lysate was loaded and separated by SDS-PAGE. After electrophoresis, the proteins were transferred onto Hybond-P PVDF membranes (GE Healthcare, Chalfont St Giles, UK). Membranes were blocked in TBST (20 mm Tris, 140 mm NaCl, 0.1% (v/v) Tween 20, pH 7.6) containing 5% (w/v) milk powder and incubated with primary antibodies at 4 °C overnight and then washed with TBST. The membranes were incubated with HRP-labeled goat anti-rabbit IgG. After washing with TBST, membranes were developed with ECL reagent (2 ml of luminol solution (1.25 mm luminol, 100 mm Tris, pH 8.6), 1 μl of 30% H2O2, 200 μl of p-coumaric acid (1.1 mg/ml in DMSO)) and imaged with a ChemiDocTMMP imaging system (Bio-Rad). For application of multiple antibodies, the same blot was probed repeatedly or simultaneously with multiple antibodies once the possibility of false interpretation of nonspecific binding was eliminated through validation. Protein levels were quantified by densitometric measurements using ImageJ software (51). The intensity of each band was normalized to the loading control and quantified relative to the vehicle control.
Assessment of nuclear morphology with fluorescence staining
Cells were plated in glass bottom 24-well plates (MatTek Corp., Ashland, MA) and maintained overnight before treatment was started. Hoechst staining was used to detect changes in chromatin morphology as a typical characteristic of apoptosis (22). A stock solution of Hoechst 33342 was prepared in water. ER-Tracker Green fluorescence stain was used to visualize the endoplasmic reticulum. After 14 h of treatment, aliquots of the stains were directly added to the culture medium (0.001% (w/v) Hoechst and 1 μm ER-Tracker). After incubating for 15 min at 37 °C, cells were fixed with 4% formaldehyde for 2 min at 37 °C. Cells were then carefully washed twice with PBS. Nuclear chromatin was afterward evaluated in PBS by inverted confocal fluorescence microscopy (Zeiss LSM 780, Oberkochen, Germany) using Zen 2 Software.
Single-cell gel electrophoresis
The alkaline comet assay was performed to detect DNA strand breaks in single cells based on the method described by Singh et al. (52). SK-Mel-5 cells were plated in 6-well plates, maintained overnight, and treated with mensacarcin for 6 or 15 h. Cells were detached, and cell suspensions were centrifuged for 10 min at 1000 × g and washed once with PBS. 75,000 cells were resuspended in 70 μl of molten 0.5% low-melting point agarose and pipetted onto a glass microscope slide (precoated in 1% normal melting point agarose), spread with a coverglass, and allowed to solidify on ice. After removal of the coverglass, another layer of low-melting point agarose was added on top, spread with a coverglass, and allowed to solidify on ice. Then the coverglass was removed, and the slide was immersed into cold lysis solution (10 mm Tris, 100 mm EDTA, 2.5 m NaCl, pH 10.0, 1% Triton X-100, 10% DMSO) for 1 h at 4 °C. Slides were carefully rinsed with H2O, transferred to the gel electrophoresis tank, and incubated in cold electrophoresis buffer (300 mm NaOH, 1 mm EDTA) for 30 min. The electrophoresis was then run in the same buffer at 25 V (0.8 V/cm) for 30 min at 4 °C. After electrophoresis, slides were neutralized with Tris buffer (0.4 m Tris, pH 7.5) and then carefully washed with H2O. Slides were stained with 60 μl of ethidium bromide (10 μg/ml) and covered with a coverglass. For each sample, two slides were prepared, and images were captured with a Nikon E400 fluorescence microscope. For each slide, 50 cells were randomly analyzed, and comets were scored using Comet Assay IV analysis software (Perceptive Instruments, Bury St. Edmunds, UK). DNA damage was quantified as percentage of DNA in the comet tail.
Cell cycle analysis and Nicoletti apoptosis assay
Flow cytometric cell cycle analysis was performed using PI DNA staining. Briefly, cells were plated in 12-well plates, maintained overnight, and directly treated with mensacarcin or synchronized before treatment. For synchronization in G0/G1 phase, SK-Mel-5 cells were serum-starved for 26 h following release into serum-containing culture medium with or without mensacarcin. For synchronization in S-phase, a double thymidine block modified from the method described by Ma and Poon was carried out (53). SK-Mel-5 cells were treated with 2 mm thymidine for 24 h and released from the block for 9 h, followed by a second thymidine block for 23 h. This arrested cells at the G1/S-phase border. A release into complete culture medium for 3 h led to a synchronized population in S phase. After treatment with mensacarcin, the culture medium was collected. For detachment, cells were incubated for 2 min with trypsin/EDTA at 37 °C, carefully singularized by pipetting, and added to the collected culture medium. Cell suspensions were centrifuged for 10 min at 1000 × g, washed once, and resuspended in 600 μl of cold PBS. Cells were fixed by slowly adding 1400 μl of ice-cold 100% ethanol and incubation at −20 °C for 30 min. Cells were pelleted, washed once with cold PBS, and resuspended in 500 μl of PBS containing 0.1% Triton X-100 and RNase A (10 μg/ml), following staining with PI (20 μg/ml) for 15 min at 37 °C in the dark. Cell suspensions were then analyzed by flow cytometry using a Cytomics FC500 flow cytometer (Beckman Coulter, Brea, CA). For each measurement, at least 10,000 events were counted. Apoptosis was analyzed according to Nicoletti et al. (2, 54) by determining the percentage of cells with hypodiploid DNA content. Analysis was performed using WinList 3D version 7.1 software (Verity Software House, Topsham, ME).
Cytochrome c release
Approximately 2 × 106 SK-Mel-5 cells were seeded in 96-well clear-bottom plates (Greiner Bio-One, Monroe, NC). Cells were treated for 6 h with mensacarcin (50 μm). Staurosporine (10 μm) served as positive control, and DMSO (10 μl) served as vehicle control.
Subcellular fractionation
For cell fractionation, cells were washed twice with PBS and collected. The cell pellet was resuspended in 170 μl of hypo-osmotic buffer (10 mm NaCl, 1.5 mm MgCl2, 10 mm Tris, pH 7.5) and left on ice for 10 min. The cell suspension was transferred into a prechilled 2-ml Dounce homogenizer, and cells were disrupted using 50 strokes of a tight B pestle followed by the addition of 100 μl of 2.5× iso-osmotic buffer (0.625 m sucrose, 2.5 mm EDTA, 2.5 mm DTT, 10 mm Tris, pH 7.5, protease inhibitor mixture). The homogenate was centrifuged at 700 × g for 10 min to pellet intact cells, nuclei, and cell debris. The supernatant was further subjected to a 17,000 × g (for cytochrome c release assay) or 7000 × g (for the co-localization study) centrifugation step for 15 min to obtain the mitochondrial pellet. The pellet was resuspended in 1× iso-osmotic buffer (0.25 m sucrose, 1 mm EDTA, 1 mm DTT, 10 mm Tris, pH 7.5, protease inhibitor mixture) and centrifuged a second time at 17,000 × g or 7000 × g. The mitochondrial pellet was then resuspended in ice-cold lysis buffer, resulting in the mitochondrial fraction. For the cytochrome c assay, the post-mitochondrial supernatant was obtained as the cytosolic fraction. For the co-localization study, the post-mitochondrial supernatant was centrifuged at 100,000 × g for 20 min to obtain the light membrane pellet and the cytosolic fraction. The pellet was resuspended in iso-osmotic buffer, the centrifugation step was repeated, and the pellet was resuspended in ice-cold lysis buffer, resulting in the light membrane fraction. All fractionation steps were carried out at 4 °C.
Caspase activity assay
Cells were seeded as mentioned in 96-well white-walled, clear bottom plates. Treatment with staurosporine (10 μm) served as positive control, and DMSO (10 μl) served as vehicle control. Cells were treated with mensacarcin (0.5, 2, 10, or 50 μm), broad-spectrum caspase inhibitor Z-VAD-FMK (10 μm), or a combination of mensacarcin (0.5, 2, 10, or 50 μm) and Z-VAD-FMK (10 μm) for 12 and 24 h. Caspase-3 and -7 activity was measured using a luminescence-based Caspase- GloH 3/7 assay (Promega). The Caspase-GloH reagent, which also serves to lyse the cells, was added directly to each well (50 μl) and incubated for 1 h. The resulting luminescence was measured using a Synergy HT microplate reader.
Fluorescence labeling of mensacarcin
For synthesis of the rhodamine-mensacarcin conjugate, 3 mg of TMRCA and 4.5 mg of mensacarcin were dissolved in 800 μl of dried DMSO and heated at 80 °C for 1 h under nitrogen. The reaction mixture was purified by preparative HPLC using an Agilent 1100 HPLC equipped with a photodiode array detector using a Phenomenex Kinetex C18, 5 μm × 150 mm × 4.6-mm column. The mobile phase consisted of ultrapure water (solution A) and acetonitrile (solution B) using a gradient method (0–2 min 5% B; 2–10 min 5–35% B; 10–28 min 35% B; 28–32 min 35–50% B; 32–45 min 50% B) with a flow rate of 0.8 ml/min at room temperature. Elution was monitored by UV-visible detection at λ = 330 nm (Amax mensacarcin) and λ = 545 nm (Amax TMR). Chromatograms revealed three major rhodamine-mensacarcin conjugates (TMR-mensa) with retention times between 25 and 30 min, respectively. All three TMR-mensa conjugates were collected and subjected to mass spectrometric analysis. Low-resolution ESI-MS showed m/z values of 848.3 for all three peaks indicate singly labeled mensacarcin conjugates. HR-TOF-MS (ESI+) (C46H46N3O13) found m/z = 848.2986 [M + H]+, calculated m/z = 848.3025 [M + H]+, Δ ppm = 4.6. LC-LRMS spectra were recorded on Agilent 1100 series LC with MSD 1946 and LC-HRMS spectra on Agilent 1200 series LC with 6230 TOF MS, respectively.
Subcellular co-localization studies
SK-Mel-5 cells were plated in glass bottom 24-well plates (MatTek Corp.; Ashland, MA) and maintained overnight. Cells were then incubated for 20 min at 37 °C with the fluorescence stains directly added to the culture medium (0.001% (w/v) Hoechst, 1 μm TMR-mensa or TMRCA, and 1 μm ER-Tracker or MitoGreen according to the manufacturer). Cells were carefully washed twice with PBS. Images were captured, and co-localization was analyzed by inverted confocal fluorescence microscopy (Zeiss LSM 780, Oberkochen, Germany) using Zen 2 software (Hoechst: λex = 405, detection 410–477 nm; TMR-mensa/TMRCA: λex = 561, detection 575–638 nm; ER-Tracker: λex = 488, detection 495–589 nm; MitoGreen: λex = 488, detection 491–562 nm). Cell fractionation was used to verify accumulation of TMR-mensa in cell organelles. Approximately 2 × 106 SK-Mel-5 cells were plated and maintained overnight before treatment with 50 μm mensa was started. After 6 h, cells were washed twice with PBS and collected and fractionated following the protocol described previously.
Bioenergetic analysis and assessment of mitochondrial function
OCR of living cells and ECAR measurements were performed using the Seahorse XF24 extracellular flux analyzer (Seahorse Bioscience, Billerica, MA) as described (28). The Seahorse XF24 extracellular flux analyzer makes continuous measurements of oxygen concentration and proton flux over time. These measurements of OCR and ECAR enable a direct quantification of mitochondrial respiration and glycolysis. To assess basal mitochondrial function and mitochondrial stress in response to mensacarcin, cells were plated in XF24 cell culture plates (HCT-116, 35,000 cells/well; SK-Mel-28, 15,000 cells/well; SK-Mel-5, 20,000 cells/well) and maintained for 24 h at 37 °C at 5% CO2. Before analysis, the culture medium was replaced with unbuffered DMEM, pH 7.4. Cells were allowed to equilibrate for 1 h at 37 °C in a non-CO2 incubator immediately before the XF assay. OCR and ECAR were measured under basal conditions and after injection of various concentration of mensacarcin. To assess key parameters of mitochondrial function, the drug injection ports of the XF24 assay cartridge were loaded with mitochondrial toxins (final concentration in wells: 1 μm oligomycin, 0.5 μm FCCP, or 0.5 μm rotenone/antimycin A), and the mensacarcin injection was followed by injection of the mitochondria-perturbing reagents.
Data analysis
Statistical analyses were carried out using GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA). The significance of observed differences was evaluated by one-way analysis of variance followed by Bonferroni multiple-comparison post hoc test. The non-parametric Kruskal–Wallis test followed by Dunn's multiple-comparison post hoc test was used for analysis of the comet assay results and the basal bioenergetics analysis of cells. In all cases, p < 0.05 was considered to be significant. Unless otherwise stated, all experimental values are reported as mean ± S.D.
Author contributions
B. P., E. N. K., and S. L. designed the study and wrote the paper. B. P. performed most experiments and prepared figures, and E. N. K. tested the caspase-dependent activity, subcellular fractionation, and cytochrome c release (Figs. 4 and 5). All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Napur Pande for providing SK-Mel-28 cells, Bioviotica (Prof. Dr. Axel Zeeck and Hans-Peter Kroll) for providing mensacarcin, and NCI, National Institutes of Health, for NCI-60 cell screening. We thank Kerry McPhail, Jane Ishmael, Paul Blakemore, B. J. Philmus, James Strother, April Risinger, and Susan Mooberry for fruitful discussions.
This work was primarily supported by Oregon State University startup funds. The authors declare that they have no conflicts of interest with the contents of this article.
A. Zeeck and M. Arnold, unpublished results.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- GI50
- 50% growth inhibition
- LC50
- 50% lethal concentration
- PARP-1
- poly(ADP-ribose)polymerase-1
- Z
- benzyloxycarbonyl
- FMK
- fluoromethyl ketone
- TMRCA
- tetramethylrhodamine-5-carbonyl azide
- TMR
- tetramethylrhodamine
- mensa
- mensacarcin
- OCR
- oxygen consumption rate(s)
- ECAR
- extracellular acidification rate(s)
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- PI
- propidium iodide
- MTT
- 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide
- LDH
- lactate dehydrogenase
- ESI
- electrospray ionization.
References
- 1. Newman D. J., and Cragg G. M. (2016) Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 [DOI] [PubMed] [Google Scholar]
- 2. Riccardi C., and Nicoletti I. (2006) Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 1, 1458–1461 [DOI] [PubMed] [Google Scholar]
- 3. Nagatsu J., and Suzuki S. (1963) Studies on an antitumor antibiotic, cervicarcin. J. Antibiot. 16, 203–206 [PubMed] [Google Scholar]
- 4. Tietze L. F., Stewart S. G., Polomska M. E., Modi A., and Zeeck A. (2004) Towards a total synthesis of the new anticancer agent mensacarcin: synthesis of the carbocyclic core. Chemistry 10, 5233–5242 [DOI] [PubMed] [Google Scholar]
- 5. Singal P. K., and Iliskovic N. (1998) Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 339, 900–905 [DOI] [PubMed] [Google Scholar]
- 6. Tietze L. F., Güntner C., Gericke K. M., Schuberth I., and Bunkoczi G. (2005) A Diels-Alder reaction for the total synthesis of the novel antibiotic antitumor agent mensacarcin. Eur. J. Org. Chem. 2005, 2459–2467 [Google Scholar]
- 7. Tietze L. F., Stewart S. G., and Polomska M. E. (2005) Intramolecular Heck reactions for the synthesis of the novel antibiotic mensacarcin: investigation of catalytic, electronic and conjugative effects in the preparation of the hexahydroanthracene core. Eur. J. Org. Chem. 2005, 1752–1759 [Google Scholar]
- 8. Tietze L. F., Gericke K. M., and Schuberth I. (2007) Synthesis of highly functionalized anthraquinones and evaluation of their antitumor activity. Eur. J. Org. Chem. 2007, 4563–4577 [Google Scholar]
- 9. Maier S., Pflüger T., Loesgen S., Asmus K., Brötz E., Paululat T., Zeeck A., Andrade S., and Bechthold A. (2014) Insights into the bioactivity of mensacarcin and epoxide formation by MsnO8. Chembiochem 15, 749–756 [DOI] [PubMed] [Google Scholar]
- 10. Yan X., Probst K., Linnenbrink A., Arnold M., Paululat T., Zeeck A., and Bechthold A. (2012) Cloning and heterologous expression of three type II PKS gene clusters from Streptomyces bottropensis. Chembiochem 13, 224–230 [DOI] [PubMed] [Google Scholar]
- 11. Maier S., Heitzler T., Asmus K., Brötz E., Hardter U., Hesselbach K., Paululat T., and Bechthold A. (2015) Functional characterization of different ORFs including luciferase-like monooxygenase genes from the mensacarcin gene cluster. Chembiochem 16, 1175–1182 [DOI] [PubMed] [Google Scholar]
- 12. Paull K. D., Shoemaker R. H., Hodes L., Monks A., Scudiero D. A., Rubinstein L., Plowman J., and Boyd M. R. (1989) Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 81, 1088–1092 [DOI] [PubMed] [Google Scholar]
- 13. American Cancer Society (2017) Cancer Facts and Figures 2017, pp. 24–25, American Cancer Society, Atlanta, GA [Google Scholar]
- 14. Beck D., Niessner H., Smalley K. S. M., Flaherty K., Paraiso K. H. T., Busch C., Sinnberg T., Vasseur S., Iovanna J. L., Driessen S., Stork B., Wesselborg S., Schaller M., Biedermann T., Bauer J., et al. (2013) Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci. Signal. 6, ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balch C. M., Gershenwald J. E., Soong S.-J., Thompson J. F., Atkins M. B., Byrd D. R., Buzaid A. C., Cochran A. J., Coit D. G., Ding S., Eggermont A. M., Flaherty K. T., Gimotty P. A., Kirkwood J. M., McMasters K. M., et al. (2009) Final version of 2009 AJCC melanoma staging and classification, J. Clin. Oncol. 27, 6199–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., et al. (2002) Mutations of the BRAF gene in human cancer, Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 17. Moreau S., Saiag P., Aegerter P., Bosset D., Longvert C., Hélias-Rodzewicz Z., Marin C., Peschaud F., Chagnon S., Zimmermann U., Clerici T., and Emile J.-F. (2012) Prognostic value of BRAFV600 mutations in melanoma patients after resection of metastatic lymph nodes. Ann. Surg. Oncol. 19, 4314–4321 [DOI] [PubMed] [Google Scholar]
- 18. Poulikakos P. I., and Rosen N. (2011) Mutant BRAF melanomas–dependence and resistance. Cancer Cell 19, 11–15 [DOI] [PubMed] [Google Scholar]
- 19. Gray-Schopfer V., Wellbrock C., and Marais R. (2007) Melanoma biology and new targeted therapy. Nature 445, 851–857 [DOI] [PubMed] [Google Scholar]
- 20. Paull K. D., Lin C. M., Malspeis L., and Hamel E. (1992) Identification of novel antimitotic agents acting at the tubulin level by computer-assisted evaluation of differential cytotoxicity data. Cancer Res. 52, 3892–3900 [PubMed] [Google Scholar]
- 21. Boyd M. R., and Paull K. D. (1995) Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 34, 91–109 [Google Scholar]
- 22. Toné S., Sugimoto K., Tanda K., Suda T., Uehira K., Kanouchi H., Samejima K., Minatogawa Y., and Earnshaw W. C. (2007) Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp. Cell Res. 313, 3635–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Los M., Mozoluk M., Ferrari D., Stepczynska A., Stroh C., Renz A., Herceg Z., Wang Z.-Q., and Schulze-Osthoff K. (2002) Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 13, 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soldani C., and Scovassi A. I. (2002) Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis 7, 321–328 [DOI] [PubMed] [Google Scholar]
- 25. Bröker L. E., Kruyt F. A. E., and Giaccone G. (2005) Cell death independent of caspases: a review. Clin. Cancer Res. 11, 3155–3162 [DOI] [PubMed] [Google Scholar]
- 26. Olive P. L., and Banáth J. P. (2006) The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 1, 23–29 [DOI] [PubMed] [Google Scholar]
- 27. Rundell M. S., Wagner E. D., and Plewa M. J. (2003) The comet assay: genotoxic damage or nuclear fragmentation? Environ. Mol. Mutagen. 42, 61–67 [DOI] [PubMed] [Google Scholar]
- 28. Van Noorden C. J. F. (2001) The history of Z-VAD-FMK, a tool for understanding the significance of caspase inhibition. Acta Histochem. 103, 241–251 [DOI] [PubMed] [Google Scholar]
- 29. Jiang X., and Wang X. (2004) Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 [DOI] [PubMed] [Google Scholar]
- 30. Tse W. C., and Boger D. L. (2004) Sequence-selective DNA recognition: natural products and nature's lessons. Chem. Biol. 11, 1607–1617 [DOI] [PubMed] [Google Scholar]
- 31. Piehler J., Brecht A., Gauglitz G., Zerlin M., Maul C., Thiericke R., and Grabley S. (1997) Label-free monitoring of DNA-ligand interactions. Anal. Biochem. 249, 94–102 [DOI] [PubMed] [Google Scholar]
- 32. Lüpertz R., Wätjen W., Kahl R., and Chovolou Y. (2010) Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 271, 115–121 [DOI] [PubMed] [Google Scholar]
- 33. Cliby W. A., Lewis K. A., Lilly K. K., and Kaufmann S. H. (2002) S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 277, 1599–1606 [DOI] [PubMed] [Google Scholar]
- 34. Zhao J., Li W., Ma R., Chen S., Ren S., and Jiang T. (2013) Design, synthesis and DNA interaction study of new potential DNA bis-intercalators based on glucuronic acid, Int. J. Mol. Sci. 14, 16851–16865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang S.-N., Phelps M. A., and Swaan P. W. (2003) Involvement of endocytic organelles in the subcellular trafficking and localization of riboflavin. J. Pharmacol. Exp. Ther. 306, 681–687 [DOI] [PubMed] [Google Scholar]
- 36. Curtius T. (1894) 20. Hydrazide und Azide organischer Säuren I. Abhandlung, J. Prakt. Chem. 50, 275–294 [Google Scholar]
- 37. Smith P. A. S. (2004) The Curtius reaction. In Organic Reactions, Vol. 3, pp. 337–449, John Wiley & Sons, Inc., New York [Google Scholar]
- 38. Vult von Steyern F., Josefsson J. O., and Tågerud S. (1996) Rhodamine B, a fluorescent probe for acidic organelles in denervated skeletal muscle, J. Histochem. Cytochem. 44, 267–274 [DOI] [PubMed] [Google Scholar]
- 39. Hall A., Meyle K. D., Lange M. K., Klima M., Sanderhoff M., Dahl C., Abildgaard C., Thorup K., Moghimi S. M., Jensen P. B., Bartek J., Guldberg P., and Christensen C. (2013) Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 4, 584–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schöckel L., Glasauer A., Basit F., Bitschar K., Truong H., Erdmann G., Algire C., Hägebarth A., Willems P. H., Kopitz C., Koopman W. J., and Héroult M. (2015) Targeting mitochondrial complex I using BAY 87–2243 reduces melanoma tumor growth, Cancer Metab. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrill J. D., Tan M., Fotso S., Sikorska J., Kasanah N., Hau A. M., McPhail K. L., Santosa D. A., Zabriskie T. M., Mahmud T., Viollet B., Proteau P. J., and Ishmael J. E. (2015) Apoptolodins A and C activate AMPK in metabolically sensitive cell types and are mechanistically distinct from oligomycin A. Biochem. Pharmacol. 93, 251–265 [DOI] [PubMed] [Google Scholar]
- 42. Giordano S., Lee J., Darley-Usmar V. M., and Zhang J. (2012) Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One 7, e44610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu M., Neilson A., Swift A. L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., Chomicz S., and Ferrick D. A. (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 292, C125–C136 [DOI] [PubMed] [Google Scholar]
- 44. Dykens J. A., and Will Y. (2007) The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 12, 777–785 [DOI] [PubMed] [Google Scholar]
- 45. Koppenol W. H., Bounds P. L., and Dang C. V. (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 [DOI] [PubMed] [Google Scholar]
- 46. Zheng J. (2012) Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 4, 1151–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parmenter T. J., Kleinschmidt M., Kinross K. M., Bond S. T., Li J., Kaadige M. R., Rao A., Sheppard K. E., Hugo W., Pupo G. M., Pearson R. B., McGee S. L., Long G. V., Scolyer R. A., Rizos H., et al. (2014) Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 4, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. D'Souza G. G. M., Wagle M. A., Saxena V., and Shah A. (2011) Approaches for targeting mitochondria in cancer therapy. Biochim. Biophys. Acta 1807, 689–696 [DOI] [PubMed] [Google Scholar]
- 49. Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 [DOI] [PubMed] [Google Scholar]
- 50. Korzeniewski C., and Callewaert D. M. (1983) An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 64, 313–320 [DOI] [PubMed] [Google Scholar]
- 51. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singh N. P., McCoy M. T., Tice R. R., and Schneider E. L. (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 175, 184–191 [DOI] [PubMed] [Google Scholar]
- 53. Ma H. T., and Poon R. Y. C. (2011) Synchronization of HeLa cells. Methods Mol. Biol. 761, 151–161 [DOI] [PubMed] [Google Scholar]
- 54. Nicoletti I., Migliorati G., Pagliacci M. C., Grignani F., and Riccardi C. (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139, 271–279 [DOI] [PubMed] [Google Scholar]