
Figure 5. Harnessing p53.
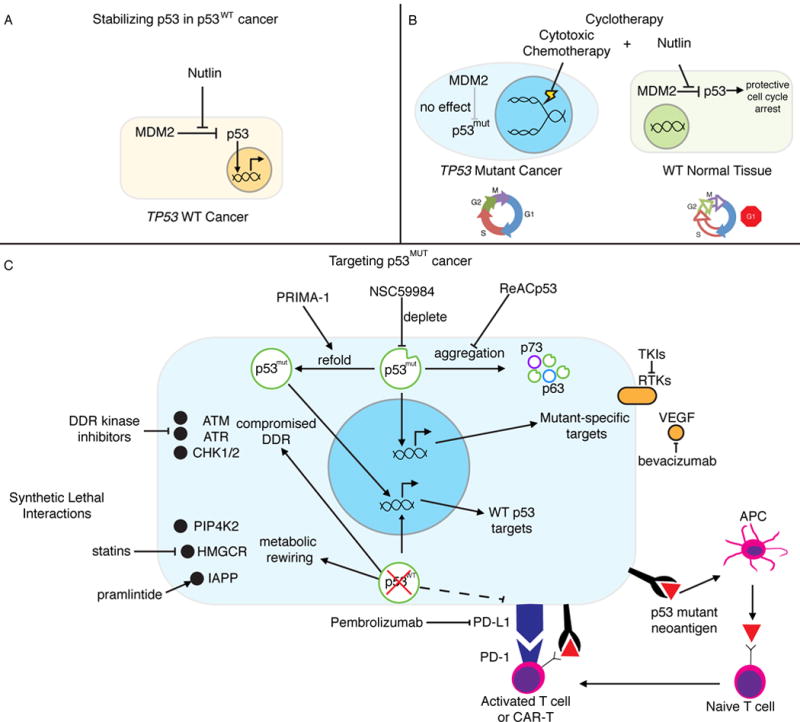
(A) Stabilizing p53 in p53WT cancer. Nutlin and other MDM2/MDMX inhibitors (RG7112, RO5503781, SAR405838, HDM201, MK4828, AMG232, and RG7388) allow for the accumulation and activity of p53 in cancer in which it is not mutated. (B) Cyclotherapy. Nutlin is used to transiently arrest p53WT normal cells, while p53MUT cancer cells continue to cycle and remain vulnerable to genotoxic chemotherapy. Sparing normal tissue allows for increased dosing and reduced toxicity. (C) Targeting p53MUT cancer. PRIMA-1 and other agents (APR-246, RITA, PK7088, p53R3, and ZMC1) are used to support proper folding of mutant p53 and restore wild type-like structure and activity. p53 mutant protein is depleted through a number of indirect mechanisms including inhibition of HSP90 (17-AAG), HDAC (SAHA), and SIRT1 (YK-3-237). The aggregation and inactivation of mutant p53 and its family members is inhibited by ReACp53. Synthetic lethal interactions are dependencies in p53 mutant cancer but not in p53WT cells. p53 deficient cells have a compromised DDR, leaving then vulnerable to even further genomic instability by inhibiting DDR-related kinases. Metabolic rewiring introduces druggable dependencies on PIP4K2, cholesterol biosynthesis/HMGCR (statins), and IAPP (pramlinitide). Some p53 mutations can result in recognizable neoantigens, which has led to the development of mutant p53-targeted immunotherapy. p53 ablation can also modify antigen presentation efficiency justifying the investigation of immune checkpoint inhibition, especially when combined with other strategies.