
Figure 3. Genetic origins of retinoblastoma.
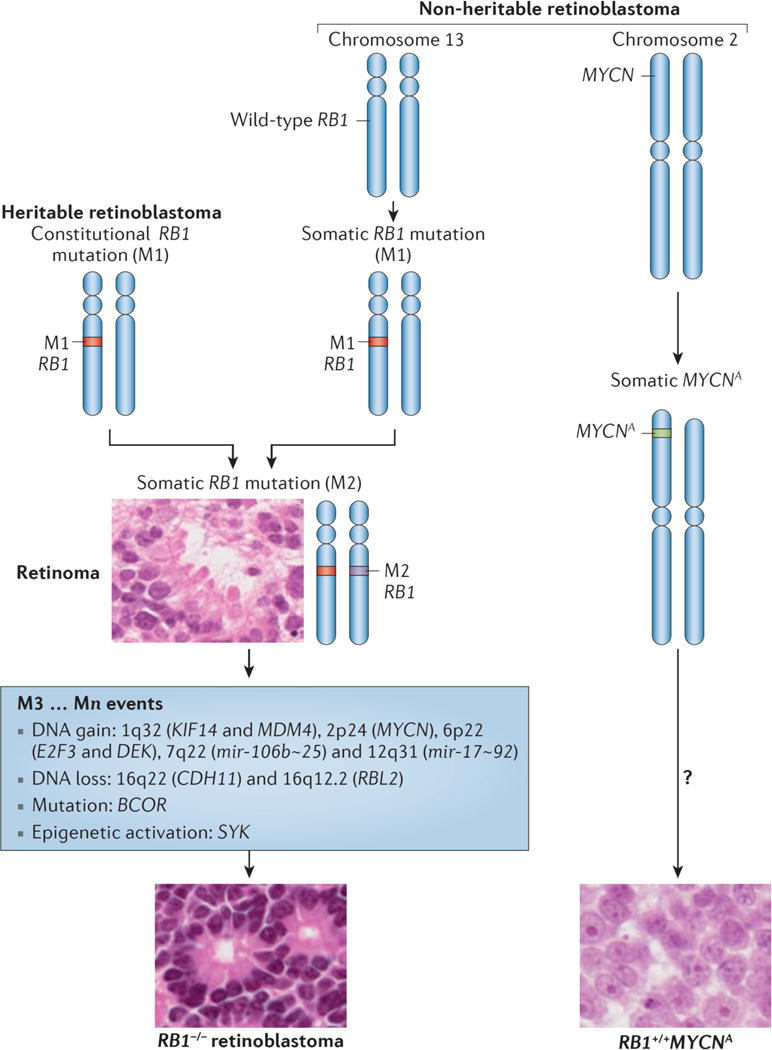
Three genetic subtypes of retinoblastoma are known. Heritable retinoblastoma patients have a constitutive inactivating mutation (M1) in the RB1tumor suppressor gene in all cells of their body. A second, somatic mutation (M2) in a susceptible retinal cell can lead to benign retinoma. Further genetic and/or epigenetic events (M3…Mn) are required to transform to retinoblastoma. Non-heritable, RB1−/− retinoblastomas progress similarly, except both M1 and M2 occur in one susceptible retinal cell. RB1+/+MYCN-amplified (RB1+/+MYCNA) retinoblastoma is a rare, non-heritable retinoblastoma subtype driven by amplification of MYCN with normal RB1; other changes in these tumours remain uncharacterized. Retinoma histology shows distinct photoreceptor-like fleurettes, whereas RB1−/− retinoblastoma can show Flexner-Wintersteiner (insert) and Homer Wright rosettes (not shown). RB1+/+MYCNA retinoblastoma have a distinct morphology with rounded nuclei and prominent nucleoli related to the high MYCN protein.