

Key Points
IL-32 is a proinflammatory cytokine expressed by plasma cells in a subset of MM patients, and high expression correlates with poor survival.
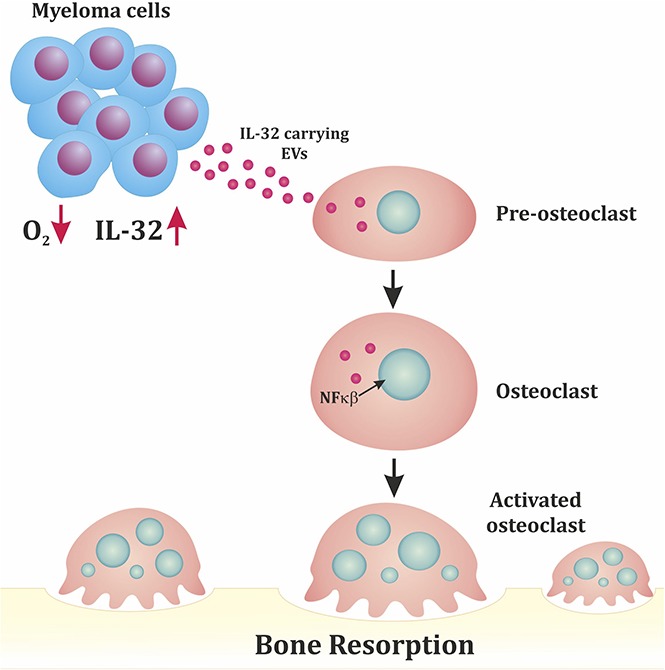
IL-32 is induced by hypoxia and secreted from MM cells in EVs that promote bone destruction.
Abstract
Multiple myeloma (MM) is a hematologic cancer characterized by expansion of malignant plasma cells in the bone marrow. Most patients develop an osteolytic bone disease, largely caused by increased osteoclastogenesis. The myeloma bone marrow is hypoxic, and hypoxia may contribute to MM disease progression, including bone loss. Here we identified interleukin-32 (IL-32) as a novel inflammatory cytokine expressed by a subset of primary MM cells and MM cell lines. We found that high IL-32 gene expression in plasma cells correlated with inferior survival in MM and that IL-32 gene expression was higher in patients with bone disease compared with those without. IL-32 was secreted from MM cells in extracellular vesicles (EVs), and those EVs, as well as recombinant human IL-32, promoted osteoclast differentiation both in vitro and in vivo. The osteoclast-promoting activity of the EVs was IL-32 dependent. Hypoxia increased plasma-cell IL-32 messenger RNA and protein levels in a hypoxia-inducible factor 1α–dependent manner, and high expression of IL-32 was associated with a hypoxic signature in patient samples, suggesting that hypoxia may promote expression of IL-32 in MM cells. Taken together, our results indicate that targeting IL-32 might be beneficial in the treatment of MM bone disease in a subset of patients.
Visual Abstract
Introduction
Multiple myeloma (MM) is a hematologic cancer caused by accumulation of malignant plasma cells in the bone marrow (BM).1 Osteolytic lesions are a hallmark of this disease, and the loss of bone is largely caused by increased osteoclast activity.2
Extracellular vesicles (EVs) are different types of vesicles released from cells, including exosomes and microvesicles.3 EVs shuttle proteins, nucleic acids, and lipids and are important in the interaction between tumor cells and the microenvironment.4,5 EVs produced by MM cells may regulate angiogenesis, inflammation, and osteoclast differentiation.6-8 Also, EVs secreted from BM stromal cells may promote MM tumor growth.9,10 The factors mediating these effects are, however, to a large extent not identified. Moreover, if or how hypoxia influences MM cell–derived EV content is not known.
Unlike most other organs, the BM microenvironment is physiologically hypoxic,11 and the myeloma BM seems to be even more hypoxic.12-14 Cells adapt to low oxygen levels by activating hypoxia-inducible factors (HIFs), which regulate the expression of several genes. HIF1α may also be activated as a result of increased oncogene signaling (eg, gain of function of Myc, Raf, or Ras).15 Hypoxia is associated with resistance to therapy and poor patient survival in several types of cancer.16 In MM mouse models, targeting HIF1α blocked tumor growth and inhibited angiogenesis and bone destruction.17 These data suggest that hypoxic stress may promote myeloma tumor progression.
We identified interleukin-32 (IL-32) as a novel cytokine produced by MM cells under hypoxic stress. We also found that this cytokine is secreted in EVs and that IL-32–containing EVs potently promote osteoclast differentiation in vitro and in vivo. This is the first study to describe a role for IL-32 in MM.
Materials and methods
Cells
MM cell lines were obtained and cultured as previously described18 (for details, see supplemental Methods). BM aspirates were obtained from Norwegian Myeloma Biobank (St. Olavs University Hospital HR, Trondheim, Norway). Myeloma cells were isolated from BM aspirates from patients using RoboSep automated cell separator and Human CD138 Positive Selection Kit (StemCell Technologies, Grenoble, France). The purity of plasma cell isolates as estimated by counting plasma cells on cytospins was >95%. The Regional Committee for Medical and Health Research Ethics (REK2011/2029) approved the study, and all patients provided informed consent.
Recombinant proteins and antibodies
Recombinant IL-32 (#4690-IL-025/CF), macrophage colony-stimulating factor (M-CSF; #216-MC-025/CF), RANKL (#390-TN-010), and transforming growth factor β1 (TGF-β1; #240-B-002) were purchased from R&D Systems. The following antibodies were used: IL-32 (#AF3040) from R&D Systems and CD63 (#HPA010088), CD81 (# HPA007234), and β-actin (A2066) from Sigma. TGN38 (#sc-33783) and tubulin (sc-5286)19 were from Santa Cruz, and HIF1α (#Ab51608), GM130 (#EP892Y), TSG101 (#Ab125011),20 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; #Ab8245) were from Abcam. Antibodies toward IkB (44D4), Alix (3A9; #2171S), and p65 (D14E12) were purchased from Cell Signaling Technology.
Nanostring analysis
For messenger RNA (mRNA) transcript counting, the nCounter Human Immunology v2 Kit (#GXA-HIM2-24) was used following the manufacturer’s protocol. Total RNA (50 ng) was hybridized overnight with capture/reporter probes. nSolver Analysis Software (Nanostring Technologies) was used for calculations of transcript numbers, applying normalization against the following housekeeping genes: ABCF1, ALAS1, EEF1G, G6PD, GAPDH, GUSB, HPRT1, OAZ1, POLR1B, POLR2A, PPIA, RPL19, SDHA, TBP, and TUBB.
Gene expression data
RNA sequencing data (IA8b_E74GTF_Sailfish_GENE_TPM) and clinical data were downloaded from the Multiple Myeloma Research Foundation CoMMpass IA8 release. RNA sequencing data from CD138+ cells were available for 591 samples from patients with MM; 548 of these were diagnostic samples taken from the BM. Data on progression-free survival were available for all these patients. For survival analyses, patient samples taken at diagnosis were divided into high and low IL-32 expression based on the upper 15th percentile (n = 82; transcripts per million [TPM] > 1.874) and lower 85th percentile (n = 466; TPM < 1.874). To identify differently expressed genes between patients with high and low IL-32 expression, we divided the group of patients into those with IL-32 expression in the upper 15th percentile and those in the lower 15th percentile (n = 82; TPM < 0.0798). Differently expressed genes between these 2 patient groups were examined using GeneSpring GX software (Agilent), and the resulting ranked list was analyzed using GSEA (Gene Set Enrichment Analysis v.2.2.1; Broad Institute). To test for a possible association between IL-32 and genes described to be linked with hypoxia in MM (ie, myeloma hypoxic signature),21 we used the CoMMpass RNA sequencing data and normalized the expression of individual genes by the median. The average of all hypoxia-regulated genes was then calculated and termed average hypoxia signature. The log2 of the average hypoxia signature was checked for correlation with IL-32 expression using Spearman tests. The microarray gene expression data set GSE75522 was used to compare IL-32 gene expression in patients with (n = 137) and without (n = 36) bone loss. This data set included patients with relapsed myeloma enrolled in phase 2 and 3 clinical trials of bortezomib. Bone disease was assessed by magnetic resonance imaging, and presence of bone disease was defined as presence of at least 1 osteolytic lesion.
Real-time quantitative PCR
mRNA was isolated from cells and reversely transcribed, and polymerase chain reaction (PCR) analysis was performed using StepOne Real-Time PCR System and TaqMan Gene Expression Assay (Applied Biosystems) as described previously.23 Probes were as follows: human IL-32 (Hs00992441_m1), HIF1A (Hs00153153_m1), and housekeeping gene TATA-binding protein (TBP; Hs00427620_m1) or GAPDH (Hs99999905_m1).
Immunoblotting
Cells were lysed, electrophoresed, and blotted as previously described.23 For details, see supplemental Methods.
EV isolation and characterization
Fetal calf serum (FCS) was depleted for EVs by ultracentrifugation at 120 000g for 16 hours followed by filtration with 0.2 μm of membrane filter (#83.1826.001; Sarstedt); 0.8 × 106 myeloma cells per mL were cultured for 48 hours in media with 10% FCS depleted for EVs before isolation of EVs by stepwise centrifugation as reported previously (supplemental Figure 5A).24 In brief, cells and culture media were harvested and spun down at 450g to remove the cells. The supernatant was then centrifuged at 16 000g for 15 minutes to remove cell debris and larger vesicles, followed by ultracentrifugation at 120 000g for 70 minutes. The pellet was dissolved in 10 mL of phosphate-buffered saline (PBS) and ultracentrifuged once more at 120 000g for 70 minutes (PBS wash). The pellet containing EVs from 50 mL of conditioned media was finally dissolved in 200 μL of PBS and used for additional experiments. The amount of protein in the vesicle fraction was determined by Bradford assay (#B6916; Sigma) according to the manufacturer’s instructions. Amount of EVs (5 μL) used in experiments corresponded to ∼100 ng of total protein. For NanoSight analyses, 40 μL of EVs was diluted up to 500 μL of PBS and run on a NanoSight LM10 (NanoSight Ltd., Amesbury, United Kingdom) for analysis using blue laser (532 nm) for 60 seconds. The average size was calculated from 5 independent replicates using NTA 3.0. (NanoSight Ltd.). The NanoSight was calibrated using standard 100-nm beads in PBS.
IL-32 ELISA
Amounts of IL-32 in BM plasma, cell culture supernatant, and EVs were quantified by IL-32 Duo-set enzyme-linked immunosorbent assay (ELISA; #DY3040; R&D Systems). Cell culture supernatant was harvested from 0.8 × 106 cells per mL cultured for 2 days. EVs were isolated as described in the previous paragraph and resuspended in 200 μL of reagent diluent (#DY995; R&D Systems) before analysis. To release IL-32, the EVs were treated with NP-40 (IGEPAL CA-630; #18896; Sigma) to a final concentration of 1% before analysis.
Confocal microscopy
Cells were fixed with 4% paraformaldehyde for 10 minutes, washed with PBS, and quenched with 50 mM of ammonium chloride in PBS. Cells were permeabilized with 0.25% Triton X-100 in PBS for 10 minutes at room temperature and then blocked with 10% bovine serum albumin in PBS containing 0.02% Tween-20 for 1 hour. Primary antibodies were incubated overnight at 4°C, and secondary antibodies were added for 45 minutes at room temperature. The cells were observed by confocal microscopy using an Axiovert 100-M inverted microscope, equipped with an LSM 510 laser scanning unit and a ×63 1.4-NA plan Apochromat oil-immersion objective (Zeiss, Jena, Germany). ImageJ 1.5i software was used for visualizing, merging, and analyzing the confocal images.
Generation of IL-32–depleted cells
JJN3 cells were electroporated with IL-32 CRISPR/Cas9 plasmid containing green fluorescent protein (GFP) for selection (#sc-406489; Santa Cruz), using buffer R (Amaxa Nucleofector Kit R VCA-1003; Lonza) and program T-001. After 24 hours, cells were sorted for GFP positivity by fluorescence-activated cell sorting and cloned. Clones were screened for IL-32 expression by flow cytometry. IL-32 protein knockout (KO) was thereafter confirmed by immunoblotting. Clones that had been transfected with the plasmid (GFP+) but still expressed IL-32 were used as control cells (IL-32 wild type [WT]).
HIF1α silencing
JJN3 cells were electroporated with small interfering (siRNA) HIF1A 11 and 12 (GeneSolution; #1027416; Qiagen) or control siRNA (#1027310; Qiagen) as described in the previous paragraph. The transfected cells were isolated as previously described.25
Osteoclast differentiation
Blood mononuclear cells were isolated from healthy donors using Ficoll gradients (Lymphoprep; #1114547; Alere Technologies). Monocytes were isolated via adherence or CD14 sorting (CD14 magnetic beads; #130-050-201; Miltenyi). Isolated cells were seeded at 8000 cells per well in 96-well plates, and preosteoclasts were generated as reported.26,27 Briefly, human monocytes were grown in minimum essential medium α (#4160; Thermo Fischer) supplemented with 10% heat-inactivated human serum, 20 μg/mL of gentamicin, and 30 ng/mL of M-CSF, 10 ng/mL of RANKL, and 1 ng/mL of TGF-β1. After 6 to 9 days, when binucleated cells appeared, cells were treated with recombinant IL-32 (25 ng/mL) or EVs with or without 50 ng/mL of RANKL and 1 ng/mL of M-CSF for 3 days, as indicated in the figure legends. Osteoclast differentiation was evaluated by staining for tartrate resistant acidic phosphatase (TRAP) using the Leukocyte Acid Phosphatase Kit (387A-1KIT; Sigma) according to the manufacturer’s instructions. TRAP+ multinuclear cells with ≥ 3 nuclei were counted as osteoclasts. All samples were run in triplicate, and every experiment was counted by at least 2 persons, blinded.
In vivo experiments
Regarding calvarial inection of EVs, 5 consecutive subcutaneous injections of 3 µg of recombinant human IL-32 (rhIL-32) in 30 µL of PBS, 30 µL of EVs, or 30 µL of PBS were performed over the calvariae at 24-hour intervals in 8-week-old male NOD SCID mice as previously described.28,29 Three days after the last injection, calvariae were harvested upon euthanization, fixed in 4% paraformaldehyde for 24 hours, and decalcified in 15% of EDTA in PBS for 12 days. The decalcified calvariae were embedded in paraffin, sectioned, and stained for TRAP using the Leukocyte Acid Phosphatase Kit (387A-1KIT; Sigma) according to the manufacturer’s instructions and counterstained with fast green (#F7252; Sigma). Images were obtained with a Nikon Eclipse Ci (Nikon Instruments), using a 20× objective (Plan Fluor 20×/0.5). TRAP+ bone surface and total bone surface were quantified using NIS Elements BR 4.00.00 (Nikon Instruments). The mice were obtained from Taconic Biosciences at 4 weeks of age.
JJN3 IL-32 KO (n = 5) or IL-32 WT (n = 5) cells (1 × 105) in 10 µL of PBS were injected into the tibiae of 9- to 12-week-old RAG2−/−γc−/− mice. After 20 days, the mice were euthanized. Tibiae were harvested for micro–computed tomography (µCT) analyses, and blood was collected for quantification of collagen type 1 degradation product. For details, see supplemental Methods.
The experiments were approved by the Norwegian Food Safety Authority (FOTS ID 7984 and FOTS ID 10517).
Statistics
Statistical analyses were performed using Prism 6 and SPSS version 24 for Mac OS X. Mann-Whitney U test was used to determine significance between 2 groups, and 1-way analysis of variance and Fisher’s least significant difference post hoc tests were used to determine significance when comparing multiple factors. Correlation between 2 parameters was estimated by the Spearman rank correlation analysis. The Kaplan-Meier method was used to generate survival curves, and differences between the survival curves was determined using the log-rank test. Influence of risk factors on survival was assessed by multivariate Cox regression analysis. Results were considered statistically significant when P < .05.
Results
MM cells express IL-32, and high expression of IL-32 is associated with a poor prognosis
We assessed mRNA copy numbers of immune-associated genes in MM cells. Strikingly, 1 of the most highly expressed genes in freshly isolated CD138+ MM cells from BM of patients was IL32 (Figure 1A). We further detected IL32 mRNA by quantitative PCR in BM-derived CD138+ cells in 34 of 40 patients (patient characteristics; supplemental Table 1) and in MM cell lines (Figure 1B). Importantly, a subgroup of patients and a fraction (4 of 9) of the cell lines expressed high levels of IL32 (Figure 1B). The cell lines that had high IL32 mRNA levels also expressed IL-32 protein (Figure 1C; supplemental Table 2). Moreover, 6 of 6 primary MM cells expressed IL-32 protein (Figure 1D). Bands of different molecular weights in western blots (Figure 1C-D) indicated that MM cells express several isoforms of IL-32.30-32 Moreover, we detected IL-32 in BM plasma obtained from healthy persons, patients with monoclonal gammopathy of unknown significance, and patients with MM (Figure 1E), suggesting that IL-32 is present extracellularly in the BM microenvironment.
Figure 1.

IL-32 is expressed by MM cells, and high expression is correlated with inferior survival. (A) Heat map of top 25 most highly expressed genes as determined by Nanostring analyses in BM-derived CD138+ MM cells obtained from patients (n = 8). (B) IL32 mRNA expression was quantified by quantitative PCR in BM-derived CD138+ MM cells (n = 40) and MM cell lines (n = 9). Immunoblotting of total cell lysates from MM cell lines (n = 9) (C) and CD138+ primary MM cells (n = 6) (D). (E) IL-32 was quantified by ELISA in BM plasma obtained from healthy persons (n = 5; median, 360 pg/mL), patients with monoclonal gammopathy of unknown significance (MGUS; n = 16; median, 565.6 pg/mL), and patients with MM (n = 41; median, 906.5 pg/mL). The differences between the groups are not statistically significant. (F) Survival curves generated from the CoMMpass data (IA8 release) by comparing the IL32 upper 15th percentile (IL-32 expressors; n = 82; median survival, 464 days) with the lower 85th percentile (IL-32 nonexpressors; n = 466; median survival, 914 days). Log-rank P < .0001. Bars indicate median values in panels A and C.
Analyzing the CoMMpass RNA sequence database (CoMMpass IA8 release), we found that ∼15% of all patients expressed IL32 (cutoff TPM > 2; supplemental Figure 1). At diagnosis, patients who expressed IL32 had more advanced disease compared with IL-32 nonexpressing patients (supplemental Figure 2). Moreover, progression-free survival was significantly shorter in IL32–expressing patients (n = 82) compared with patients with lower IL-32 levels (IL-32 levels in the lower 85th percentile; n = 466; log-rank P < .0001; Figure 1F). Also, when adjusting for International Staging System stage, IL32 retained prognostic information (supplemental Table 3). The association between high IL32 and inferior survival was confirmed in another data set (GSE9782; supplemental Figure 3). Thus, high IL32 expression in malignant plasma cells correlated with reduced survival for patients with MM.
IL-32 is secreted in EVs
IL-32 is secreted from MM cells (data not shown), although it lacks a classical secretory signal peptide. When we stained MM cells for IL-32, we observed that it was present on distinct intracellular vesicles. Furthermore, IL-32 was found in Golgi, because IL-32 colocalized with a Golgi marker, GM130 (Figure 2A upper panel). IL-32 also to some extent colocalized with the tetraspanins CD81 and CD63, commonly used markers for exosomes3 (Figure 2A), which may suggest that IL-32 is secreted on EVs. Importantly, we next isolated EVs from JJN3 cells cultured for 48 hours (supplemental Figure 4A). The EVs had a mean diameter of ∼150 nm and appeared to have double lipid membranes (supplemental Figure 4B-C). IL-32 was detected in EVs obtained from the IL-32–expressing cell lines JJN3 and H929 but not in EVs isolated from IL-32− CAG cells (Figure 2B). The EV-associated proteins TSG101 and Alix were also detected in the EV fraction, whereas proteins that are not enriched in EVs, like the Golgi-specific protein GM130 and α-tubulin, were not detected, suggesting that the EVs may be of endosomal origin.33-35 In contrast, all these proteins were present in cellular lysates of the same cell lines (Figure 2B).
Figure 2.

IL-32 is secreted from myeloma cells on EVs. (A) JJN3 cells were stained for IL-32 (red), GM130 (Golgi marker; green), CD63 (EV marker; green), and CD81 (EV marker; green). Scale bar, 15 μm. (B) Total cell lysates and lysates of EVs isolated from cell culture supernatants were immunoblotted as indicated. (C) EVs were isolated from media from JJN3 cells cultured for 48 hours. IL-32 in the EV fraction treated with or without NP-40 detergent was quantified by ELISA. (D) EVs were isolated from BM plasma obtained from patients with myeloma (n = 18), and amount of IL-32 in the EV fraction was determined by ELISA.
We could detect IL-32 in the EV fraction by ELISA, suggesting that IL-32 may bind to the surface of the EVs. Importantly, when we treated the EV fraction with NP-40 to lyse the vesicles, we detected more IL-32, suggesting that a significant amount of IL-32 can be released from EVs by detergent treatment (Figure 2C). We also found IL-32 in EVs isolated from BM plasma from patients (Figure 2D), supporting that IL-32 is present in EVs in the BM.
IL-32 levels increase in response to hypoxia, and the increase is dependent of HIF1α
In breast cancer cells, IL-32 was shown to increase in response to hypoxia.36 Therefore, we cultured CD138+ cells obtained from patients with MM (n = 5) in 2% oxygen (hypoxia) for 24 hours and examined IL-32 protein expression. In 3 of 5 samples, IL-32 protein levels increased more than twofold when cells were cultured in hypoxia compared with normoxia (20% oxygen; Figure 3A; supplemental Figure 5A). It was also evident that IL-32 protein and mRNA expression were upregulated during hypoxia in MM cell lines (Figure 3B and supplemental Figure 5B, respectively). Importantly, hypoxia also increased IL-32 protein in the EV fraction (Figure 3B lower panels). Culture in hypoxic conditions for 24 hours did not induce IL-32 expression in the nonexpressing cell line CAG (Figure 3B). In JJN3 cells, IL32 mRNA increased by approximately twofold after 24 hours of hypoxia and quickly declined when the cells were returned to normoxia (Figure 3C).
Figure 3.

Hypoxia increases IL-32 expression in an HIF1α-dependent manner. (A) BM-derived CD138+ MM cells were cultured for 24 hours in normoxia (20% oxygen) or hypoxia (2% oxygen) as indicated, and IL-32, HIF1α, and β-actin were detected by immunoblotting in total cellular lysates. (B) MM cell lines were cultured for 48 hours under normoxia or hypoxia before collection of cells and cell culture supernatant for EV isolation. Total cell lysates and isolated EVs were immunoblotted as indicated. (C) IL-32 mRNA expression was determined by quantitative PCR in JJN3 cells cultured in hypoxia for the indicated time before reoxygenation as indicated. (D) JJN3 cells were transfected with scrambled control siRNA (siCtr) or HIF1α siRNA (siHIF1α), and expression of IL-32 and HIF1α was examined by immunoblotting after 24 hours. (E) Density of IL-32 and HIF1α bands was quantified by Image Studio Software and normalized to levels of β-actin. Data presented are mean + standard error of the mean from 4 independent experiments. (F) RNA sequencing data were downloaded from the CoMMPass IA8 release and analyzed using GSEA software v2.2.3 to identify functionally related groups of genes with statistically significant enrichment. The figure shows the enrichment plot for the hypoxia-related gene set for patients with IL32 levels in the upper 15th percentile. (G) The same RNA sequencing data in panel F were also analyzed to determine the correlation between relative IL32 mRNA expression vs relative hypoxic gene signature expression in MM as defined previously.21 Relative IL32 gene expression significantly correlated with the hypoxic signature (Spearman ρ = 0.4; P < .0001). ***P < .001.
HIF1α expression is elevated by hypoxia but is also high in some cancer cells because of oncogene expression.15 In MM cell lines, IL-32 mRNA levels correlated with HIF1A mRNA levels (Spearman ρ = 0.9; P = .02; supplemental Figure 6A). When we knocked down HIF1α in JJN3 cells using siRNA, HIF1A mRNA was reduced by 60% to 70% (supplemental Figure 6B), whereas IL-32 protein and mRNA expression remained unchanged, under normoxic conditions (Figure 3D-E and supplemental Figure 6B, respectively). However, the increase of IL-32 in response to hypoxia was clearly blunted by HIF1α knockdown (Figure 3D-E; supplemental Figure 6B).
To examine if high IL32 expression is associated with a hypoxic signature in patients’ MM cells, we analyzed RNA sequence data from the CoMMpass IA8 release. Gene set enrichment analyses revealed that high IL32 mRNA levels (upper 15th percentile; n = 82) were significantly associated with a hypoxic signaling pathway (Figure 3F; supplemental Table 4). Moreover, in the same data set, we found that when IL32 gene expression was high, expression of genes associated with hypoxia in MM21 was also high (Spearman ρ = 0.4; P < .0001; n = 546; Figure 3G). Taken together, these data suggest that hypoxia increases IL-32 in plasma cells in the BM.
rhIL-32 and EVs obtained from myeloma cells promote osteoclast differentiation in vitro and in vivo
Several proinflammatory cytokines, including IL-32, may promote osteoclast differentiation.37,38 To address if IL-32–carrying EVs from MM cells influence osteoclast differentiation, we exposed preosteoclasts to JJN3-derived EVs or rhIL-32 for 3 days and counted numbers of multinucleated TRAP+ osteoclasts. Both rhIL-32 and EVs increased numbers of osteoclasts, independent of the presence of RANKL (Figure 4A-B). The EVs were taken up by the osteoclast precursors, as demonstrated by confocal microscopy (supplemental Figure 7). Next, we injected rhIL-32 and EVs from JJN3 cells cultured in normoxia and hypoxia on top of the calvaria of NOD-SCID mice every day for 5 days. Strikingly, both rhIL-32 and EVs from JJN3 cells increased TRAP+ bone surface. Importantly, EVs from JJN3 cells cultured in hypoxia caused a greater increase in TRAP+ bone surface than EVs from cells in normoxia (Figure 4C-D).
Figure 4.

IL-32 in EVs induces osteoclast differentiation in vitro and in vivo. (A-B) Preosteoclasts were treated with rhIL-32 (25 ng/mL), RANKL (50 ng/mL), or JJN3-derived EVs (5 μL) for 3 days before the cells were TRAP stained. (Original magnification ×200 in panel A.) Number of osteoclasts was determined by counting TRAP+ cells with >2 nuclei. Bars indicate relative number of osteoclasts (mean ± standard error of the mean [SEM]) from 5 independent experiments using peripheral blood mononuclear cells (PBMCs) from 5 different donors. (C-D) EVs (30 μL) from JJN3 cells cultured in normoxia (N) or hypoxia (H), rhIL-32 (3 μg in 30 μL of PBS), and PBS control (30 μL) were injected on top of the calvaria of NOD-SCID mice (n = 8 per group) every day for 5 days. After 3 more days, the mice were euthanized and the calvaria harvested. (Original magnification ×200 in panel C.) Amount TRAP+ bone surface (BS) was quantified using NIS-Elements BR software. (E) JJN3 cells in which IL-32 was silenced using CRISPR/Cas9 (KO) or control cells (WT) were cultured in normoxia or hypoxia for 48 hours. Cells and EVs obtained from the culture media were harvested and lysed before immunoblotting as indicated. (F) Preosteoclasts were treated with rhIL-32 (25 ng/mL), RANKL (50 ng/mL), or EVs (5 μL) obtained from JJN3 KO and JJN3 WT cells as indicated for 3 days. Number of osteoclasts was determined by counting TRAP+ cells with >2 nuclei. Bars indicate relative number of osteoclasts (mean + SEM) from 6 independent experiments using PBMCs from 6 different donors. (G-H) 100 000 JJN3 WT or JJN3 KO cells were injected intratibially in RAG2/GC KO mice. After 20 days, tibiae were harvested and examined by µCT. Representative images from the 2 groups are shown in panel G. Bars in panel H represents number of osteolytic lesions (mean + SEM). Significance was determined by Student t test. (I) NFκB p65 nuclear localization was examined in preosteoclast serum starved for 3 hours and then treated with EVs isolated from IL-32–expressing JJN3 cells (WT) or from IL-32 KO cells for 1 hour. p65 in red; Dapi nuclear stain in blue. (Original magnification ×400.) (J) Number of cells with NFκB p65 in the nucleus was counted to estimate percentage of nuclear localization. Data were obtained from 4 independent experiments using 4 different PBMC donors. For all experiments, unless otherwise stated, significance was determined by 1-way analysis of variance followed by Fisher’s least significant difference post hoc test. *P < .05, **P < .01, ***P < .001, ****P < .0001. TBS, total bone surface.
To clarify whether the pro-osteoclastogenic effect of the EVs was mediated by IL-32, we generated JJN3 cells depleted for IL-32 using the CRISPR/Cas9 system. In these cells, IL-32 was not detected in cellular lysates or EVs, even when the cells were cultured in hypoxia, confirming depletion of IL-32 (Figure 4E). When we treated preosteoclasts with EVs obtained from IL-32 KO cells, these EVs did not promote osteoclast differentiation, whereas EVs from the WT cells did (Figure 4F). We also examined the effect of a different KO clone, which similarly did not promote osteoclast differentiation (supplemental Figure 8). Moreover, adding rhIL-32 to the cultures treated with KO EVs rescued the effect on osteoclastogenesis, suggesting that the lack of effect of the EVs is a result of the loss of IL-32 (supplemental Figure 9). Furthermore, to investigate if depletion of IL-32 reduced the osteolytic capacity of myeloma cells in vivo, we injected 1 × 105 JJN3 IL-32 WT or KO cells into the tibiae of male RAG2−/−γc−/− mice. After 20 days, the mice were euthanized, and bone resorption was assessed by µCT. µCT images demonstrated extensive osteolytic lesions in the tibiae of mice injected with WT cells, compared with mice injected with KO cells (Figure 4G). The difference in number of osteolytic lesions between the 2 groups was statistically significant (Figure 4H; Student t test P = .012). Moreover, mice injected with IL-32 WT cells had significantly less trabecular bone volume, increased trabecular separation, and reduced numbers of trabeculae as compared with mice injected with KO cells (supplemental Figure 10A-D). Finally, the µCT data were supported by increased levels of circulating type 1 collagen degradation product, a serum marker for osteoclast activity, in 3 of 5 mice injected with WT JJN3 cells but in none of the mice injected with KO cells (supplemental Figure 10E). Taken together, these data strongly support that IL-32 is important for the osteolytic capacity of the cells in vivo.
NFκB is an important pro-osteoclastogenic transcription factor. To address if EVs influenced NFκB activation, we treated preosteoclasts with EVs obtained from IL-32 WT cells or IL-32 KO cells and quantified NFκB p65 nuclear localization (Figure 4I-J). Although EVs from IL-32 WT cells promoted nuclear translocation of NFκB, EVs from IL-32 KO cells did not. Activation of NFκB by WT EVs, but not by KO EVs, was also confirmed by immunoblotting (supplemental Figure 11). Hence, IL-32–containing EVs promote NFkB signaling in preosteoclasts. Together, these data suggest that the pro-osteoclastogenic effect of MM cell–derived EVs is mediated by IL-32.
We finally examined if IL-32 gene expression is associated with MM bone disease by using a gene expression data set with available bone status for 173 patients (GSE755).22 From these data, we found that patients with focal lesions assessed by magnetic resonance imaging (n = 137) had significantly higher IL-32 mRNA in their plasma cells than patients without focal lesions (n = 36; Mann-Whitney U test P < .001; Figure 5A), supporting that IL-32 might play a role in MM bone disease.
Figure 5.

High IL-32 expression is associated with osteolytic bone disease. (A) IL32 gene expression in MM cells from patients without bone disease (BD; median, 0.42; n = 36) compared with patients with BD (median, 0.53; n = 137). Data obtained from GSE755. BD was defined as having ≥1 osteolytic lesion as assessed by magnetic resonance imaging. Mann-Whitney U test P < .001. (B) Summary of main findings presented in the article. In response to hypoxia, MM cells secrete IL-32 in EVs, which promote osteoclast differentiation.
Discussion
We report here that MM cells express IL-32, IL-32 expression is increased in response to hypoxia, and IL-32 in EVs promotes osteoclast differentiation in vitro and in vivo. High ρ expression in MM cells is associated with inferior survival and bone disease.
IL-32 is a poorly characterized cytokine associated with certain inflammatory diseases and cancers.39 It does not share sequence homology with other cytokines but is classified as proinflammatory because it induces production of cytokines like TNFα, IL-6, and IL-18 from macrophages and dendritic cells.30,40 We found that a subgroup of patients expressed high ρ mRNA levels and that high ρ gene expression was associated with a hypoxic signature. Hypoxia has for decades been known to increase solid tumor aggressiveness, and it is also emerging as an important factor in MM.15,41 Our data support that HIF1α is key in promoting IL-32 expression in MM cells, because knockdown of HIF1α blunted IL-32 expression in response to hypoxia. In contrast, knockdown of HIF1α did not change IL-32 basal level under normoxic conditions, and primary myeloma cells express IL-32 even when HIF1α expression is low. Hence, hypoxia leads to an increase in IL-32 expression, but we cannot determine whether HIF1α is needed per se for IL-32 to be constitutively expressed by MM cells. Assessing HIF1α expression in patient samples is challenging, because HIF1α is degraded when the cells are transferred to normoxia (based on our own observations). Even so, staining of BM biopsies from patients with MM demonstrated stabilization of HIF1α in ∼1 in 3 patients.14,42 In another study, c-Myc was shown to stabilize HIF1α in MM cells both in normoxia and hypoxia.43 We found, however, no association between mRNA levels of c-Myc and IL32 in the large CoMMpass database (data not shown). Thus, we suggest that hypoxia-induced HIF1α expression is a driver for IL-32 in MM cells.
Osteolytic lesions often develop adjacent to MM cells, suggesting that MM cell–derived factors directly contribute to osteoclast differentiation.2 We found that EVs obtained from JJN3 cells promoted osteoclast differentiation both in vitro and in vivo dependent on IL-32. Others have also shown a pro-osteoclastogenic effect of MM cell–derived EVs,7 although which factors might mediate the effect was not investigated in that study. Our data strongly support that IL-32 is an important mediator of the pro-osteoclastogenic effect of the EVs. Importantly, the osteolytic capacity of JJN3 cells in vivo was significantly reduced when the cells were depleted of IL-32. A previous study showed that HIF1α knockdown in JJN3 cells reduced CCL3 levels and nearly completely abolished bone destruction in mice.17 It is possible, based on our findings presented here, that IL-32 is 1 of several factors that are induced by hypoxia and contribute to bone destruction in MM.
IL-32 lacks a secretory signal peptide but is still secreted from cells.30,44 Unconventional secretion, independent of the classical endoplasmic reticulum–Golgi route, has been shown for a number of different proteins, including IL-1β.45,46 Some of these proteins are transported by organelle carriers like exosomes, microvesicles, and secretory autophagosomes. In our study, IL-32 colocalized with the tetraspanins CD63 and CD81, commonly used as markers for exosomes. Moreover, the isolated EVs expressed TSG101, further supporting that they may be of endosomal origin (exosomes). However, the size of the vesicles, with a mean diameter of 150 nm, might be more similar to that of microvesicles, and it is not easy to distinguish these 2 types of vesicles with current available purification methods. We also observed that a fraction (∼50%) of IL-32 was secreted as a free, nonvesicular protein. Whether this is due to disruption of vesicles and release of IL-3247 or whether IL-32 is also released from the cells as a free protein remains to be investigated.
In conclusion, our results show that MM cells in response to hypoxia produce IL-32 and that IL-32 is needed for the pro-osteoclastogenic effect of MM cells and MM cell–derived EVs (Figure 5B). Receptors and signaling pathways downstream of IL-32 are poorly characterized but could be potential targets for treatment of MM bone disease.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Berit Størdal, Hanne Hella, Lill-Anny Gunnes Grøseth, Solveig Kvam, and Linh Hoang for technical assistance and Miriam Giambelluca for making the summary figure. The confocal imaging services provided by the Cellular and Molecular Imaging Core Facility (CMIC), Norwegian University of Science and Technology (NTNU). CMIC is funded by the Faculty of Medicine at NTNU and Central Norway Regional Health Authority. The Research Council of Norway is acknowledged for the support to the Norwegian Micro- and Nano-Fabrication Facility (NorFab; project #245963/F50).
This work was supported by funds from the Norwegian Cancer Society (#4500930), the Kristian Gerhard Jebsen Foundation (#SKGJ‐MED‐007), the Norwegian Research Council (#223255 and #193072), and the Liaison Committee for Education, Research and Innovation in Central Norway (#90061000 and #90171600).
Authorship
Contribution: T.S. and M.Z. designed experiments; M.Z., M.W., S.H.M., K.R.A., K.M., K.M.P.-A., G.B., M.G., and A.S. conducted experiments and acquired and analyzed data; A.W. provided patient samples and clinical data; A.-M.S. assisted in data interpretation and edited the paper; T.S. and M.Z. wrote the paper; and all authors revised the paper and approved final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Therese Standal, Centre of Molecular Inflammation Research, Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology (NTNU), MTFS, Olav Kyrresgt 9, N-7489 Trondheim, Norway; e-mail: therese.standal@ntnu.no.
References
- 1.Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood. 2015;125(20):3049-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roodman GD. Pathogenesis of myeloma bone disease. J Cell Biochem. 2010;109(2):283-291. [DOI] [PubMed] [Google Scholar]
- 3.Yáñez-Mó M, Siljander PR, Andreu Z, et al. . Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4(1):27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30(6):836-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyiadzis M, Whiteside TL. The emerging roles of tumor-derived exosomes in hematological malignancies. Leukemia. 2017;31(6):1259-1268. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, De Veirman K, Faict S, et al. . Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol. 2016;239(2):162-173. [DOI] [PubMed] [Google Scholar]
- 7.Raimondi L, De Luca A, Amodio N, et al. . Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget. 2015;6(15):13772-13789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124(25):3748-3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roccaro AM, Sacco A, Maiso P, et al. . BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123(4):1542-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Hendrix A, Hernot S, et al. . Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014;124(4):555-566. [DOI] [PubMed] [Google Scholar]
- 11.Spencer JA, Ferraro F, Roussakis E, et al. . Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J, Handisides DR, Van Valckenborgh E, et al. . Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. 2010;116(9):1524-1527. [DOI] [PubMed] [Google Scholar]
- 13.Azab AK, Hu J, Quang P, et al. . Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119(24):5782-5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colla S, Storti P, Donofrio G, et al. . Low bone marrow oxygen tension and hypoxia-inducible factor-1α overexpression characterize patients with multiple myeloma: role on the transcriptional and proangiogenic profiles of CD138(+) cells. Leukemia. 2010;24(11):1967-1970. [DOI] [PubMed] [Google Scholar]
- 15.Martin SK, Diamond P, Gronthos S, Peet DJ, Zannettino AC. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia. 2011;25(10):1533-1542. [DOI] [PubMed] [Google Scholar]
- 16.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352(6282):175-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storti P, Bolzoni M, Donofrio G, et al. . Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia. 2013;27(8):1697-1706. [DOI] [PubMed] [Google Scholar]
- 18.Baranowska K, Misund K, Starheim KK, et al. . Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget. 2016;7(43):70845-70856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lessnick SL, Dacwag CS, Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell. 2002;1(4):393-401. [DOI] [PubMed] [Google Scholar]
- 20.Abdeen A, Sonoda H, Oshikawa S, Hoshino Y, Kondo H, Ikeda M. Acetazolamide enhances the release of urinary exosomal aquaporin-1. Nephrol Dial Transplant. 2016;31(10):1623-1632. [DOI] [PubMed] [Google Scholar]
- 21.Maiso P, Huynh D, Moschetta M, et al. . Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015;75(10):2071-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian E, Zhan F, Walker R, et al. . The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349(26):2483-2494. [DOI] [PubMed] [Google Scholar]
- 23.Moen SH, Westhrin M, Zahoor M, et al. . Caspase-8 regulates the expression of pro- and anti-inflammatory cytokines in human bone marrow-derived mesenchymal stromal cells. Immun Inflamm Dis. 2016;4(3):327-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witwer KW, Buzás EI, Bemis LT, et al. . Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holien T, Våtsveen TK, Hella H, et al. . Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by Smad-dependent repression of MYC. Leukemia. 2012;26(5):1073-1080. [DOI] [PubMed] [Google Scholar]
- 26.Westhrin M, Moen SH, Holien T, et al. . Growth differentiation factor 15 (GDF15) promotes osteoclast differentiation and inhibits osteoblast differentiation and high serum GDF15 levels are associated with multiple myeloma bone disease. Haematologica. 2015;100(12):e511-e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harre U, Georgess D, Bang H, et al. . Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. 2012;122(5):1791-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van ’t Hof RJ. The calvarial injection assay. Methods Mol Biol. 2012;816:537-544. [DOI] [PubMed] [Google Scholar]
- 29.Johnson RW, Brennan HJ, Vrahnas C, et al. . The primary function of gp130 signaling in osteoblasts is to maintain bone formation and strength, rather than promote osteoclast formation. J Bone Miner Res. 2014;29(6):1492-1505. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22(1):131-142. [DOI] [PubMed] [Google Scholar]
- 31.Choi JD, Bae SY, Hong JW, et al. . Identification of the most active interleukin-32 isoform. Immunology. 2009;126(4):535-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinhuis B, Koenders MI, van de Loo FA, Netea MG, van den Berg WB, Joosten LA. Inflammation-dependent secretion and splicing of IL-32{gamma} in rheumatoid arthritis. Proc Natl Acad Sci USA. 2011;108(12):4962-4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowal J, Arras G, Colombo M, et al. . Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 2016;113(8):E968-E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baietti MF, Zhang Z, Mortier E, et al. . Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14(7):677-685. [DOI] [PubMed] [Google Scholar]
- 35.Lötvall J, Hill AF, Hochberg F, et al. . Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park JS, Lee S, Jeong AL, et al. . Hypoxia-induced IL-32β increases glycolysis in breast cancer cells. Cancer Lett. 2015;356(2 Pt B):800-808. [DOI] [PubMed] [Google Scholar]
- 37.Kim YG, Lee CK, Oh JS, Kim SH, Kim KA, Yoo B. Effect of interleukin-32gamma on differentiation of osteoclasts from CD14+ monocytes. Arthritis Rheum. 2010;62(2):515-523. [DOI] [PubMed] [Google Scholar]
- 38.Mabilleau G, Sabokbar A. Interleukin-32 promotes osteoclast differentiation but not osteoclast activation. PLoS One. 2009;4(1):e4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joosten LA, Heinhuis B, Netea MG, Dinarello CA. Novel insights into the biology of interleukin-32. Cell Mol Life Sci. 2013;70(20):3883-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ribeiro-Dias F, Saar Gomes R, de Lima Silva LL, Dos Santos JC, Joosten LA. Interleukin 32: a novel player in the control of infectious diseases. J Leukoc Biol. 2017;101(1):39-52. [DOI] [PubMed] [Google Scholar]
- 41.Hu J, Van Valckenborgh E, Menu E, De Bruyne E, Vanderkerken K. Understanding the hypoxic niche of multiple myeloma: therapeutic implications and contributions of mouse models. Dis Model Mech. 2012;5(6):763-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giatromanolaki A, Bai M, Margaritis D, et al. . Hypoxia and activated VEGF/receptor pathway in multiple myeloma. Anticancer Res. 2010;30(7):2831-2836. [PubMed] [Google Scholar]
- 43.Zhang J, Sattler M, Tonon G, et al. . Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 2009;69(12):5082-5090. [DOI] [PubMed] [Google Scholar]
- 44.Hasegawa H, Thomas HJ, Schooley K, Born TL. Native IL-32 is released from intestinal epithelial cells via a non-classical secretory pathway as a membrane-associated protein. Cytokine. 2011;53(1):74-83. [DOI] [PubMed] [Google Scholar]
- 45.Zhang M, Schekman R. Cell biology. Unconventional secretion, unconventional solutions. Science. 2013;340(6132):559-561. [DOI] [PubMed] [Google Scholar]
- 46.Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine. 2015;74(2):213-218. [DOI] [PubMed] [Google Scholar]
- 47.Bosch S, de Beaurepaire L, Allard M, et al. . Trehalose prevents aggregation of exosomes and cryodamage. Sci Rep. 2016;6:36162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.