

Abstract
Key points
Omecamtiv mecarbil and blebbistatin perturb the regulatory state of the thick filament in heart muscle.
Omecamtiv mecarbil increases contractility at low levels of activation by stabilizing the ON state of the thick filament.
Omecamtiv mecarbil decreases contractility at high levels of activation by disrupting the acto‐myosin ATPase cycle.
Blebbistatin reduces contractility by stabilizing the thick filament OFF state and inhibiting acto‐myosin ATPase.
Thick filament regulation is a promising target for novel therapeutics in heart disease.
Abstract
Contraction of heart muscle is triggered by a transient rise in intracellular free calcium concentration linked to a change in the structure of the actin‐containing thin filaments that allows the head or motor domains of myosin from the thick filaments to bind to them and induce filament sliding. It is becoming increasingly clear that cardiac contractility is also regulated through structural changes in the thick filaments, although the molecular mechanisms underlying thick filament regulation are still relatively poorly understood. Here we investigated those mechanisms using small molecules – omecamtiv mecarbil (OM) and blebbistatin (BS) – that bind specifically to myosin and respectively activate or inhibit contractility in demembranated cardiac muscle cells. We measured isometric force and ATP utilization at different calcium and small‐molecule concentrations in parallel with in situ structural changes determined using fluorescent probes on the myosin regulatory light chain in the thick filaments and on troponin C in the thin filaments. The results show that BS inhibits contractility and actin‐myosin ATPase by stabilizing the OFF state of the thick filament in which myosin head domains are more parallel to the filament axis. In contrast, OM stabilizes the ON state of the thick filament, but inhibits contractility at high intracellular calcium concentration by disrupting the actin‐myosin ATPase pathway. The effects of BS and OM on the calcium sensitivity of isometric force and filament structural changes suggest that the co‐operativity of calcium activation in physiological conditions is due to positive coupling between the regulatory states of the thin and thick filaments.
Keywords: cardiac myosin, muscle contraction, cardiac muscle regulation, fluorescence polarization, Omecamtiv Mecarbil
Key points
Omecamtiv mecarbil and blebbistatin perturb the regulatory state of the thick filament in heart muscle.
Omecamtiv mecarbil increases contractility at low levels of activation by stabilizing the ON state of the thick filament.
Omecamtiv mecarbil decreases contractility at high levels of activation by disrupting the acto‐myosin ATPase cycle.
Blebbistatin reduces contractility by stabilizing the thick filament OFF state and inhibiting acto‐myosin ATPase.
Thick filament regulation is a promising target for novel therapeutics in heart disease.
Introduction
Contraction of heart muscle is driven by transient interactions between the myosin‐containing thick filaments and actin‐containing thin filaments, coupled to the hydrolysis of ATP. Each heartbeat is triggered by a transient rise in intracellular calcium concentration followed by calcium binding to the troponin complex in the thin filaments, which in turn induces a structural change in the thin filaments that allows the head or motor domain of myosin to interact with actin (Gordon et al. 2000). Recently, however, the canonical calcium–thin filament paradigm of contractile regulation has been extended in both cardiac and skeletal muscle by the emerging concept of regulatory states of the thick filament. That concept originates from structural studies of myosin and thick filaments from myosin‐regulated muscles, including vertebrate smooth muscle (Wendt et al. 2001) and invertebrate skeletal muscle (Woodhead et al. 2005; Alamo, 2008; Pinto et al. 2012), which are not regulated via their thin filaments, but by a change in the structure of the thick filaments controlled by phosphorylation of the myosin regulatory light chain (RLC) (Brito, 2011) or by calcium binding to its essential light chain (ELC) (Woodhead et al. 2013). In the OFF states of those thick filaments, the myosin head domains are prevented from either binding actin or hydrolysing ATP by a network of intra‐ and inter‐molecular interactions that stabilize a conformation in which they are folded back onto the myosin tails and the thick filament surface. Activation of these myosin‐regulated muscles releases the myosin heads from this OFF state, making them available for actin binding and ATP hydrolysis.
Multiple lines of evidence now indicate that this regulatory transition in the thick filament has been retained in muscle types and species that are regulated by the calcium–thin filament regulatory pathway to work in parallel with it (Linari et al. 2015; Fusi et al. 2016; Kampourakis et al. 2016). In resting skeletal and cardiac muscles of vertebrates, the myosin head domains lie in helical tracks similar to those seen in the OFF state of thick filaments from myosin‐regulated muscles (Huxley & Brown, 1967; Linari et al. 2015; Ait‐Mou et al. 2016). Many of the intra‐ and inter‐molecular interactions that stabilize that OFF structure are conserved in isolated thick filaments from mammalian heart muscle (Zoghbi et al. 2008; Al‐Khayat et al. 2013). Biochemical studies of vertebrate skeletal and cardiac muscle suggested that this OFF structure corresponds to a myosin state with very low ATP turnover called the ‘super‐relaxed’ state (Hooijman et al. 2011; Nogara et al. 2016b). Orientation‐sensitive probes on the myosin RLC in vertebrate skeletal and cardiac muscle cells show that this OFF state is destabilized by calcium activation, RLC phosphorylation, and by increased sarcomere length (Kampourakis et al. 2015, 2016; Fusi et al. 2016). Finally, a recent X‐ray diffraction study on intact trabeculae suggested a stress‐dependent activation of the thick filament structure (Reconditi et al. 2017).
Together, the results from these diverse approaches suggest that control of the structure of both the thin and thick filaments is required for the physiological regulation of cardiac contractility, including the strength and kinetics of the heartbeat. Thick filament regulation is likely to mediate the functional effects of phosphorylation of the myosin regulatory light chain (RLC) and myosin binding protein‐C, and probably length‐dependent activation and the Frank‐Starling relation (Colson et al. 2012; Ait‐Mou et al. 2016; Kampourakis et al. 2016). However, the molecular mechanisms underlying these effects remain poorly understood. Here we investigated those mechanisms using two small molecules that bind specifically to cardiac myosin: omecamtiv mecarbil (OM), a myosin activator in clinical trials for the treatment of heart disease (Malik et al. 2011; Teerlink et al. 2011; Liu et al. 2015), and (−)‐blebbistatin (BS), a widely used and well‐characterized myosin inhibitor (Kovacs et al. 2004; Dou et al. 2007; Farman et al. 2008). Both OM and BS bind to identified sites on the myosin head domain with micromolar affinity (Fig. 1) but have no known effects on other sarcomeric proteins. We determined the effects of OM and BS on the regulatory state of the thick filaments in cardiac muscle cells, in which the native conformation and organization of the myofilament sub‐proteome is preserved, using polarized fluorescence from bifunctional rhodamine probes on the myosin RLC (Kampourakis et al. 2014, 2015) (Fig. 1). To check the effects of OM and BS on the canonical calcium–thin filament regulatory pathway, we carried out a similar set of studies using a probe attached to troponin C. In both cases we made measurements over a range of calcium concentrations bracketing the physiological range, and correlated the changes induced by OM and BS in thick and thin filament structure with those on myocardial force in the same preparation and on myofibrillar ATPase activity.
Figure 1. Labelling site of bifunctional sulforhodamine and binding sites of blebbistatin and omecamtiv mecarbil in the myosin head domain and labelling site of bifunctional rhodamine on troponin.
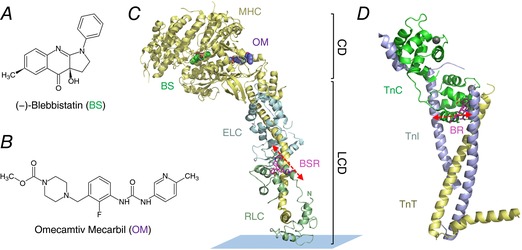
Chemical structures of (−)‐blebbistatin (BS) and omecamtiv mecarbil (OM) are shown in A and B, respectively. C, atomic model of squid myosin S1 in the detached conformation (PDB 1KK8). Bifunctional sulforhodamine (BSR) attached to the RLC (green) E‐helix is shown in pink, and BS and OM are shown in green and purple, respectively. The myosin heavy chain (MHC; CD‐catalytic domain; LCD‐light chain domain) and essential light chain (ELC) are shown in yellow and blue, respectively. The orientation of the BSR fluorescence dipole with respect to the motor domain is indicated by red dashed double arrow. The model was created by superimposition of the catalytic domains of myosin II bound to OM (PDB 4PA0) and BS (PDB 1YV3), with the catalytic domain of the squid myosin S1. D, labelling site of bifunctional rhodamine (BR, pink) in the troponin complex (PDB 1J1D). Troponin C, troponin I, troponin T and Ca2+ are shown in green, blue, yellow and grey, respectively. The BR probe is attached to the troponin C E‐helix. The orientation of the BR fluorescence dipole with respect to IT arm is indicated by red dashed double arrow. [Color figure can be viewed at wileyonlinelibrary.com]
The results show that the functional effects of OM and BS on cardiac contractility can be understood in terms of their effects on two regulatory pathways: the previously established effects on the acto‐myosin ATPase pathway and a novel effect on thick filament‐based regulation controlling the availability of myosin heads for contraction. OM stabilizes the ON state of the thick filament and myosin states with bound ADP (Malik et al. 2011; Liu et al. 2015; Rohde et al. 2017; Swenson et al. 2017), whereas BS stabilizes myosin states with bound ADP.Pi (Kovacs et al. 2004; Dou et al. 2007; Farman et al. 2008) and the OFF state of the thick filament. Moreover, our results give new insights into the physiological coupling between the regulatory states of the thick and thin filaments and establish mechanistic principles that may guide the development of new myosin modulators with improved therapeutic properties.
Methods
Animals and ethical approval
All animals were treated in accordance with the guidelines approved by the UK Animal Scientific procedures Act (1986) and European Union Directive 2010/63/EU. Wistar rats (male, 200–250 g) were killed by cervical dislocation (Schedule 1 procedure in accordance with UK Animal Scientific Procedure Act, 1986) and demembranated right ventricular trabeculae were prepared as described previously (Sun et al. 2009). All procedures were carried out in accordance with the guidelines of the Animal Welfare and Ethical Review Body (AWERB, King's College London). All animals have been kept with free access to food and water prior to use.
Reagents
Omecamtiv mecarbil and (−)‐blebbistatin were purchased from Selleck (S2623) and Sigma (B0560), respectively. Stock solutions were prepared in DMSO (molecular biology grade, Sigma, D8418) and compound purities were estimated to be >95% by RP‐HPLC and ESI mass spectrometry.
Preparation of BSR labelled cRLC and BR labelled cTnC
Bifunctional rhodamine labelled cTnC (BR‐cTnC‐E) and bifunctional sulforhodamine labelled cRLC (BSR‐cRLC‐E) were prepared as previously described (Sun et al. 2009; Kampourakis et al. 2014; Kampourakis & Irving, 2015).
Reconstitution of labelled cRLCs and cTnCs into ventricular trabeculae
BR‐cTnC‐E was reconstituted into demembranated trabeculae by overnight soak in relaxing buffer (composition in mmol L−1: 25 imidazole, 15 disodium creatine phosphate (Na2CrP), 78.4 potassium propionate (KPr), 5.65 Na2ATP, 6.8 MgCl2, 10 K2EGTA, 1 DTT, pH 7.1) containing 0.5 mg ml−1 of BR‐cTnC‐E at 4°C, replacing about 80% of the endogenous cTnC (Sevrieva et al. 2014). The maximal calcium‐activated force after reconstitution with BR‐cTnC‐E was 40.7 ± 2.5 mN mm−2 (mean ± SEM, n = 11) at ∼2.0 μm sarcomere length.
BSR‐cRLC‐E was exchanged into demembranated trabeculae by extraction in CDTA‐rigor solution (composition in mmol L−1: 5 CDTA, 50 KCl, 40 Tris‐HCl pH 8.4, 0.1% (v/v) Triton X‐100) for 30 min followed by reconstitution with 40 μmol L−1 BSR‐cRLC‐E in relaxing solution for 1 h, replacing ∼50% of the endogenous cRLC (Kampourakis et al. 2014). The average maximal calcium‐activated force after cRLC exchange (37.4 ± 3.2 mN mm−2, mean ± SEM, n = 9) was 85 ± 7% of that before cRLC exchange.
Fluorescence polarization experiments
Composition of experimental solutions and activation protocols were identical to those described previously for fluorescence polarization experiments (Sun et al. 2009; Kampourakis et al. 2014; Kampourakis & Irving, 2015). Polarized fluorescence intensities were measured as described previously for skeletal and cardiac muscle fibres (Corrie et al. 1999; Brack et al. 2004; Sun et al. 2009; Kampourakis et al. 2014). Fluorescence emission from BSR‐cRLC‐E and BR‐cTnC‐E in trabeculae was collected by a 0.25 NA objective using an excitation light beam in line with the emission path. The polarization of the excitation and emitted beams was set either parallel or perpendicular to the trabecular axis, allowing determination of the order parameter <P 2> that describes the dipole orientations in the trabeculae (Dale et al. 1999).
The sarcomere length of trabeculae was measured by laser diffraction in relaxing solution prior to each activation. Activating solution contained (in mmol L−1): 25 imidazole, 15 Na2CrP, 58.7 KPr, 5.65 Na2ATP, 6.3 MgCl2, 10 CaCl2, 10 K2EGTA, 1 DTT, pH 7.1. Each activation was preceded by a 2‐min incubation in pre‐activating solution (composition in mmol L−1: 25 imidazole, 15 Na2CrP, 108.2 KPr, 5.65 Na2ATP, 6.3 MgCl2, 0.2 K2EGTA, 1 DTT, pH 7.1). Solutions with varying concentrations of free [Ca2+] were prepared by mixing relaxing and activating solutions using MAXCHELATOR software (maxchelator.stanford.edu). Isometric force and steady‐state fluorescence polarization values were measured once steady force had been established. The dependence on force and order parameters on free calcium concentration was fitted to data from individual trabeculae using non‐linear least‐squares regression to the modified Hill equation:
where pCa50 is the negative logarithm of [Ca2+] corresponding to half‐maximal change in F, n H is the Hill coefficient, Y 0 is the baseline, and A is the amplitude (for normalized force data: Y 0 = 0 and A = 1). Trabeculae which showed a decline in maximal calcium‐activated force of more than 15% after the pCa titrations were discarded.
Compound stocks were directly diluted in physiological buffers used for trabeculae experiments. The DMSO concentration was typically not more than 0.2% (v/v) and control experiments showed that addition of 0.2% (v/v) DMSO had no effect on calcium sensitivity or cooperativity of force and probe orientation. Prior to the experiments, demembranated rat ventricular trabeculae were incubated in relaxing solution containing the appropriate concentration of either omecamtiv mecarbil or blebbistatin for 25–30 min at 22°C.
Preparation of cardiomyofibrils and ATPase activity measurements
Cardiomyofibrils (CMFs) were prepared by homogenizing freshly frozen ventricular tissue samples in myofibril buffer (composition in mmol L−1: 20 imidazole pH 7.4, 75 KCl, 2 MgCl2, 2 EDTA, 1 DTT, 1% (v/v) Triton X‐100, protease inhibitor cocktail (Roche), PhosStop cocktail (Roche)) followed by centrifugation at 5000 g for 5 min at 4°C. CMFs were washed and homogenized three more times in the same buffer without Triton X‐100.
CMFs were washed three times in ATPase assay buffer (composition in mmol L−1: 20 MOPS pH 7.0, 35 NaCl, 5 MgCl2, 1 EGTA, 1 DTT) with varying concentrations of CaCl2 (pCa 9 to pCa 4.3) and the CMF concentration adjusted to 0.5 mg ml−1 (Utter et al. 2015). For ATPase measurements at activating Ca2+ concentrations, CMFs were partially crosslinked with 5 mmol L−1 N‐hydroxysuccinimide (NHS) and 2 mmol L−1 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride (EDC) in myofibril buffer on ice for 90 min (Herrmann et al. 1993). The crosslinking reaction was stopped by the addition of 25 mmol L−1 glycine pH 8.0 and 10 mmol L−1 DTT for 30 min on ice and CMFs processed for experiments as mentioned above. Chemical crosslinking prevents CMFs from shortening during calcium activation. Reactions were started by the addition of 2.5 mmol L−1 ATP and samples taken at the indicated time points were quenched with 0.5 volumes ice cold 25% (w/v) TCA solution. Samples were kept on ice at all times, diluted with double‐deionized water and inorganic phosphate content measured using the malachite green assay according to manufacturer's instructions (Sigma, MAK030).
Statistical analysis
All data sets were normally distributed as assessed by Shapiro‐Wilk test (P > 0.05). Statistical significance of difference between groups was assessed with a one‐way ANOVA followed by Turkey's post hoc test. Paired data sets were analysed by a two‐tailed paired Student's t test. Details of significance levels are shown in the figure captions.
Results
Effects of omecamtiv mecarbil and blebbistatin on isometric force production in rat ventricular trabeculae
Although omecamtiv mecarbil (OM) is generally regarded as an activator of cardiac myosin, and OM increases cardiac output under therapeutic conditions (Malik et al. 2011), its effects depend strongly on calcium concentration [Ca2+] and are inhibitory at high [Ca2+] (Fig. 2 A). OM does activate isometric force in isolated trabeculae both at very low [Ca2+] (pCa 9, Fig. 2, black circles), and also around 1 μmol L−1 [Ca2+], close to the physiological value at systole under basal conditions. The largest effects were observed at micromolar concentrations of OM, and the increases in isometric force were statistically significant (P < 0.05) for [OM] = 1 μmol L−1 and higher. Isometric force was lower at higher OM concentrations, as reported previously for isolated rat cardiomyocytes (Nagy et al. 2015). In contrast, OM strongly inhibited force production at full calcium activation (pCa 4.3, Fig. 2 A, open circles) with an EC50 of 2.12 ± 0.17 μmol L−1 (mean ± SEM, n = 3), in agreement with its reported affinity for isolated β‐cardiac myosin S1 (Malik et al. 2011). Interestingly, the inhibition of maximal active isometric force has not been observed in human myocardium for [OM] ≤ 10 μmol L−1 (Swenson et al. 2017). OM slowed the rate of isometric force development at all [Ca2+] (data not shown), consistent with previous studies on isolated rodent cardiomyocytes (Nagy et al. 2015).
Figure 2. Concentration dependence of the effects of omecamtiv mecarbil (OM) and blebbistatin (BS) on isometric force development of native demembranated rat right ventricular trabeculae.
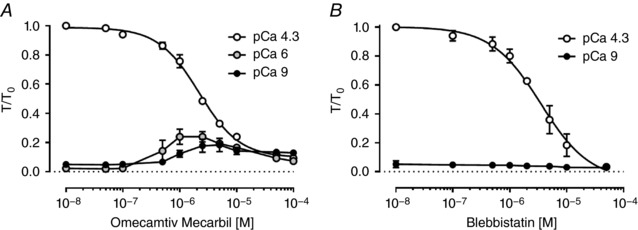
A and B, dose–response curves for OM and BS, respectively, on passive force at pCa 9 (filled circles), and active force at pCa 6 (grey circles) and pCa 4.3 (open circles). Isometric forces were normalized to that at pCa 4.3 in the absence of drugs (T 0). Values indicate means ± SEM (n = 3). BS reduced passive force at pCa 9 (filled circles) by ∼60% with an EC50 of 2.3 ± 0.9 μmol L−1.
In contrast, blebbistatin (BS) inhibited isometric force in ventricular trabeculae at all [Ca2+] (Fig. 2 B). At full calcium activation (pCa 4.3) force inhibition by BS had an EC50 of 3.17 ± 0.43 μmol L−1, consistent with the binding affinity of BS for isolated skeletal myosin S1 in the ADP.Pi state (Kovacs et al. 2004) and with the EC50 for force inhibition by BS in mouse papillary muscle (∼2.8 μmol L−1) (Dou et al. 2007). BS also reduced passive force slightly at pCa 9, with a similar EC50 (Fig. 2 B). BS did not significantly affect the apparent rate of isometric force development (data not shown).
Omecamtiv mecarbil and blebbistatin induce changes in thick filament structure
We determined the effect of OM and BS on thick filament structure in trabeculae using a bifunctional sulforhodamine (BSR) probe attached to the myosin regulatory light chain (cRLC, Fig. 1). The probe was cross‐linked to the cRLC E‐helix (BSR‐cRLC‐E), which is almost parallel to the long axis of the myosin light chain domain (LCD) (Kampourakis et al. 2015). The orientation of the RLC region of the myosin heads with respect to the thick filament axis can be determined from the polarization of the fluorescence from this probe. The results are expressed in terms of the order parameter <P2> (Dale et al. 1999), which would be +1 if all the probe dipoles (red arrow in Fig. 1) are parallel to the trabecular or thick filament axis, and −0.5 if they are perpendicular. <P2> decreases when trabeculae are activated, indicating a more perpendicular orientation of the cRLC E‐helix and LCD with respect to the thick filament axis, and a more ON state of the thick filament.
Incubating relaxed trabeculae in OM produced a decrease in <P2> for the cRLC E‐helix probe (Fig. 3 C, pCa 9, filled circles), with an EC50 of 1.12 ± 0.09 μmol L−1 (mean ± SEM, n = 4), and a Hill coefficient of 1.75 ± 0.14 (mean ± SEM, n = 4), indicating positive cooperativity. The maximum effect of OM on <P2> was ∼70% larger than that of maximal calcium activation of ventricular trabeculae in the absence of drug. OM also reduced <P2> during maximal calcium activation (pCa 4.3, open circles), with a similar EC50 (1.03 ± 0.14 μmol L−1). These EC50 values are similar to those for active isometric force of native and BSR‐cRLC‐E exchanged trabeculae (Figs 2 A and 3 A), and to that of the ATPase rate of isolated β‐cardiac myosin (Malik et al. 2011). However, whilst addition of OM was associated with a more ON thick filament (a lower <P2> indicating that the LCD of the myosin heads is more perpendicular to the filament axis) in both relaxing and activating conditions (Fig. 3 C), it had opposite effects on isometric force in these two conditions (pCa 9 and pCa 4.3; Fig. 3 A). Moreover, there was no sign in the <P2>–[OM] relation (Fig. 3 C) of the bell‐shaped dose–response relation of force at low [Ca2+] (pCa 9, Figs 2 A and 3 A). Thus, although OM binds stoichiometrically to a single site on the myosin catalytic domain (CD) (Malik et al. 2011; Winkelmann et al. 2015), its effect on contractility, in contrast with that on cRLC E‐helix orientation, depends strongly on [Ca2+].
Figure 3. Concentration dependence of the effect of omecamtiv mecarbil and blebbistatin on force and myosin head orientation measured from BSR‐cRLC‐E exchanged trabeculae.
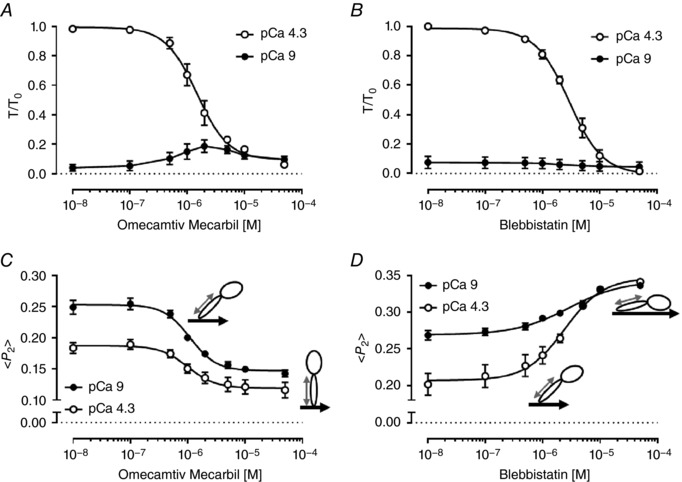
A and C, effect of OM on isometric force (A) and order parameter <P2> (C) for BSR‐cRLC‐E at pCa 9 (closed circles) and pCa 4.3 (open circles). B and D, concentration‐dependent effect of BS on isometric force (B) and order parameter <P2> (D) at pCa 9 (closed circles) and pCa 4.3 (open circles). Note that the effects of OM and BS on isometric force are identical to those for native, untreated trabeculae (Fig. 1). Values indicate means ± SEM (n = 4). The pictograms indicate the orientation of BSR probe (grey double arrow) and myosin heads with respect to the thick filament axis (black thick arrow). The angular changes are exaggerated for clarity.
In contrast to OM, incubation of trabeculae in BS increased <P2> for the BSR‐cRLC‐E probe, indicating that BS stabilizes the OFF conformation of the thick filament in which the LCD of the myosin heads is more parallel to the filament axis (Kampourakis et al. 2014, 2015). BS increased <P2> at both low and high [Ca2+], with EC50 values of 2.91 ± 0.78 μmol L−1 and 2.33 ± 0.38 μmol L−1, respectively (Fig. 3 D), similar to those for passive and active force in the same trabeculae (Fig. 3 B) and in trabeculae containing the native cRLC (Fig. 2 B). The change in cRLC E‐helix orientation associated with calcium activation was attenuated by BS (Fig. 3 D), and became insignificant at [BS] = 5 μmol L−1, despite the fact that active isometric force (T/T 0) at this BS concentration is still 31 ± 7% (mean ± SEM, n = 4) of its control value (Fig. 3 B).
Effect of omecamtiv mecarbil and blebbistatin on cardiomyofibrillar ATPase activity
To better understand the mechanisms underlying the ability of OM to either activate or inhibit isometric force depending on the conditions, we measured the ATPase activity of freshly prepared rat ventricular cardiomyofibrils at 50 μmol L−1 OM, a concentration at which the thick filament structure is fully ON, but calcium‐activated force is largely inhibited (Fig. 3). For comparison, we also measured the effects on myofibrillar ATPase of 50 μmol L−1 BS, a concentration that induces the OFF state of the thick filament and also inhibits calcium‐activated force.
Addition of 50 μmol L−1 OM inhibited the cardiomyofibrillar ATPase rate at both low (pCa 9) and high [Ca2+] (pCa 4.3) (Fig. 4, grey bars). OM reduced the ATPase of relaxed myofibrils by about 24%, and the ATPase rate of isometrically contracting myofibrils at pCa 4.3 by about 37% with an EC50 of ∼1.3 μmol L−1 (Fig. 4 D), consistent with its effects on thin filament‐activated ATPase activity of isolated porcine heavy meromyosin (HMM) (Liu et al. 2015) and the actin‐activated ATPase activity of recombinant human β‐cardiac myosin S1 (EC50 ∼0.5 μmol L−1) (Swenson et al. 2017). Thus, although addition of 50 μmol L−1 OM at low and high [Ca2+] induces a more ON thick filament structure (Fig. 2 C) and activates isometric force at low [Ca2+] (Figs 2 A and 3 A), it inhibits the ATPase under the same conditions. Interestingly, the fractional inhibition of myofibrillar ATPase at maximal calcium activation (∼37%) is much less than that of isometric force (∼90%). However, OM increases the ATPase activity of cardiac myofibrils at intermediate levels of activation (pCa 6) (Fig. 4 B), similar to its effect on isometric force under the same conditions (Fig. 2 A). Moreover, the similar EC50 for OM's effect on myosin head orientation (Fig. 3 C) and ATPase activity suggests coupling between biochemical and structural states of myosin.
Figure 4. Effects of omecamtiv mecarbil and blebbistatin on the ATPase activity of cardiomyofibrils.
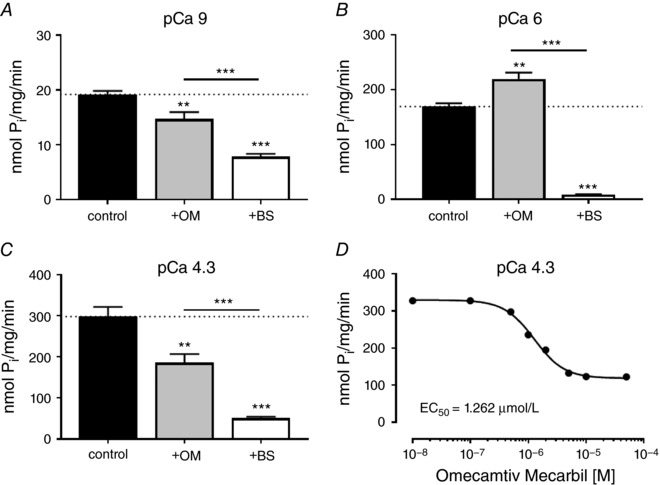
A–C, steady‐state ATPase rates for control, OM treated and BS treated CMFs at pCa 9 (A), pCa 6 (B) and pCa 4.3 (C). Values indicate means ± SEM (n = 4–8). D, dose–response curve for the effect of omecamtiv mecarbil on steady‐state cardiomyofibriliar ATPase activity at pCa 4.3. Statistical significance of differences between groups were assessed by a one‐way ANOVA with Turkey's post hoc test: ** P < 0.01; *** P < 0.001.
Addition of 50 μmol L−1 BS reduced the ATPase rate of cardiac myofibrils at all [Ca2+] tested by 60–90%, as expected from the inhibitory effects of BS on isometric force and thick filament structure (Fig. 3 B and D).
Omecamtiv mecarbil and blebbistatin have contrasting effects on the calcium sensitivity of isometric force and thick filament structure
The activating effect of OM on isometric force at low and intermediate [Ca2+] implies that it sensitizes the myofilaments to calcium, an effect that might be associated with the more ON thick filament structure induced by the drug, although in apparent contradiction with its inhibition of myofibrillar ATPase at high [Ca2+]. To understand better the action of OM on the calcium sensitivity of contractile activation, we determined the effect of a clinically relevant concentration of OM of 1 μmol L−1, which is within the range of plasma concentrations measured during clinical trials (0.25–2.5 μmol L−1) (Cleland et al. 2011; Teerlink et al. 2011, 2016; Greenberg et al. 2015), on the [Ca2+] dependence of myocardial force and thick filament structure in the physiological range. As before we contrasted the effects of OM with those of BS, at a concentration (5 μmol L−1) that induced a largely OFF state of the thick filament, but left enough isometric force to allow us to determine its [Ca2+] dependence.
At 1 μmol L−1 OM had a large effect on the [Ca2+] dependence of isometric force (Fig. 5 A, open circles), particularly in the physiologically relevant range around pCa 6. The pCa required for half‐maximal force (pCa50, Fig. 6 A, Table 1) increased from 5.56 ± 0.01 (mean ± SEM, n = 9) in the absence of OM to 5.90 ± 0.03 in its presence, but the Hill coefficient (n H, Fig. 6 B), a measure of the cooperativity of calcium activation, decreased from 6.85 ± 0.65 to 2.78 ± 0.12. The effects of 1 μmol L−1 OM on the calcium sensitivity of thick filament structure as reported by the cRLC E‐helix probe were even larger (Fig. 6 B). pCa50 for probe orientation increased from 5.56 ± 0.01 (the same as the value for force in the absence of OM) to 6.11 ± 0.06, and n H decreased from 8.41 ± 0.66 to 2.61 ± 0.17. The [Ca2+] dependence of the orientation of the cRLC E‐helix probe in the absence of OM is similar to that reported previously for a BSR probe on the N‐lobe of the cRLC (Kampourakis et al. 2014).
Figure 5. Calcium dependence of force, and cRLC E‐helix and cTnC E‐helix orientation in the absence (filled circles), and presence of 1 μmol L−1 omecamtiv mecarbil (+OM, open circles) and 5 μmol L−1 blebbistatin (+BS, grey triangles).
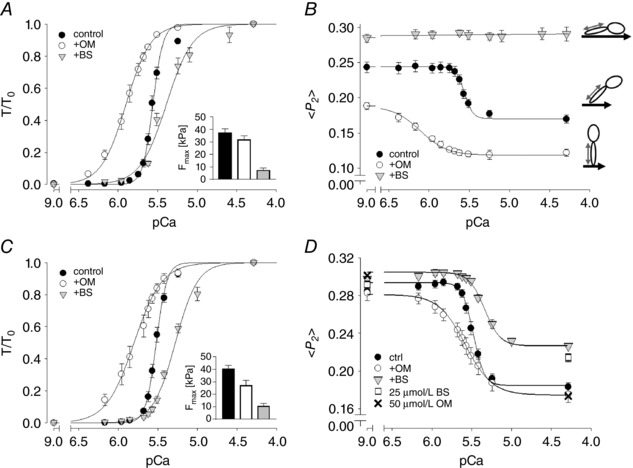
A and C, normalized force–pCa relation for BSR‐cRLC‐E and BR‐cTnC‐E exchanged trabeculae, respectively. The maximal forces at pCa 4.3 are shown in the inset. B and D, <P2>–pCa relation measured in parallel with force for BSR‐cRLC‐E and BR‐cTnC‐E exchanged trabeculae, respectively. Means ± SEM (n = 4–9). Pictograms indicate the orientation of BSR probe (grey double arrow) and myosin heads with respect to the thick filament axis (black thick arrow)
Figure 6. Summary of force–pCa (circles) and <P2>–pCa (squares) parameters.
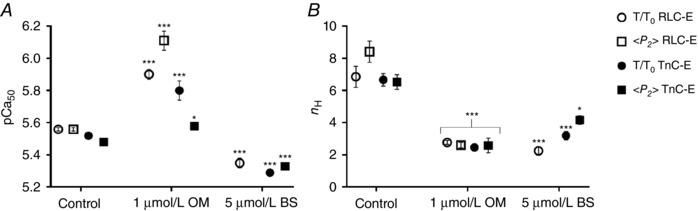
A and B, pCa50 (A) and n H values (B) for BSR‐cRLC‐E (open symbols) and BR‐cTnC‐E (closed symbols) exchanged trabeculae. Means ± SEM (n = 4–9). Statistical significance of differences from the control group was assessed by a one‐way ANOVA with Turkey's post hoc test: * P < 0.05; *** P < 0.001.
Table 1.
Summary of force–pCa and <P2>–pCa parameters for BSR‐cRLC‐E and BR‐cTnC‐E exchanged trabeulae
| BSR‐cRLC‐E | BR‐cTnC‐E | ||||||
|---|---|---|---|---|---|---|---|
| Control | + 1 μmol L−1 OM | + 5 μmol L−1 BS | Control | + 1 μmol L−1 OM | + 5 μmol L−1 BS | ||
| n | 9 | 5 | 4 | 11 | 5 | 4 | |
| Force | F max (kPa) | 37.4 ± 3.2 | 32.0 ± 3.0 | 7.4 ± 3.5$$$ | 40.7 ± 2.5 | 27.2 ± 4.3$ | 11.1 ± 2.6$$$ |
| pCa50 | 5.56 ± 0.01 | 5.90 ± 0.03$$$ | 5.35 ± 0.03$$$ | 5.52 ± 0.01 | 5.80 ± 0.06$$$ | 5.29 ± 0.02$$$ | |
| n H | 6.85 ± 0.65 | 2.78 ± 0.12$$$ | 2.25 ± 0.26$$$ | 6.66 ± 0.39 | 2.47 ± 0.22$$$ | 3.20 ± 0.26$$$ | |
| <P2> | pCa50 | 5.56 ± 0.01 | 6.11 ± 0.06$$$ , ** | — | 5.48 ± 0.01 | 5.58 ± 0.02$ , * | 5.33 ± 0.02$$$ , * |
| n H | 8.41 ± 0.66 | 2.61± 0.17$$$ | — | 6.53 ± 0.45 | 2.59 ± 0.46$$$ | 4.17 ± 0.26$ , * | |
| <P2> ‐ amplitude (A) | 0.074 ± 0.007 | 0.070 ± 0.002 | — | 0.109 ± 0.003 | 0.107 ± 0.007 | 0.081 ± 0.002$$ | |
| <P2> ‐ baseline pCa 4.3 (Y 0) | 0.171 ± 0.006 | 0.119 ± 0.004$$$ | — | 0.185 ± 0.005 | 0.175 ± 0.008 | 0.226 ± 0.002$$$ | |
Values indicate means ± SEM. Statistical significance of differences between groups was assessed by a one‐way ANOVA with Turkey's post hoc test: $ P < 0.05; $$ P < 0.01, $$$ P < 0.001. Statistical significance of differences within groups was assessed with a paired two‐tailed Student's t test: * P < 0.05; ** P < 0.01
In marked contrast with the effects of OM, 5 μmol L−1 BS decreased pCa50 for normalized force by ∼0.25 pCa units (Fig. 5 A, inverted triangles; Fig. 6 A). BS, like OM, reduced n H to ∼2.5 (Fig. 6 B; Table 1). Thick filament structure as reported by the RLC probe was independent of [Ca2+] at 5 μmol L−1 BS (Figs 5 B and 3 D). Strikingly, the [Ca2+] dependences of force and thick filament structure in the presence of OM and BS (Fig. 5 A and B; open circles, inverted triangles) bracket the control relationship (filled circles). OM produces a more ON thick filament structure and sensitizes force to calcium; BS does the opposite. However both OM and BS greatly reduce the co‐operativity of calcium activation (Fig. 6 B), suggesting that the steep co‐operativity in control conditions requires a thick filament structure that is finely poised between the OFF and ON states.
Effects of omecamtiv mecarbil and blebbistatin on thin filament activation
The correlation described above, between the changes in thick filament structure induced by OM and BS and the calcium dependence of active force mediated by Ca2+ binding to troponin in the thin filaments, suggests that the activation state of the thin filament is sensitive to that of the thick filament (Kampourakis et al. 2016). To further investigate that possibility we used a bifunctional rhodamine (BR) probe on the E‐helix of cardiac troponin C (cTnC) in the so‐called ‘IT arm’ of troponin to determine directly the effects of OM and BS on the activation state of the thin filament (Sun et al. 2009; Kampourakis et al. 2014).
The effects of OM (1 μmol L−1) and BS (5 μmol L−1) on isometric force in trabeculae containing BR‐cTnC‐E (Fig. 5 C) were similar to those reported above. However, in contrast to their effects on the activation state of the thick filament, neither OM nor BS changed the activation state of the thin filament as reported by the cTnC E‐helix probe in relaxing conditions (pCa 9) (Fig. 5 D). Since 1 μmol L−1 OM activates isometric force in these conditions (Figs 2 A and 3 A), this is a surprising result. To check it, we measured the orientation of the cTnC E‐helix probe at a series of OM concentrations up to 50 μmol L−1 and found no effect of OM on probe orientation in either relaxing conditions (pCa 9) or during maximal calcium activation (pCa 4.3) (crosses in Fig. 5 D), in contrast with its effect on thick filament structure in the same conditions (Fig. 3 C). Moreover, lattice compression by 1–3% (w/v) dextran had no significant effect on probe orientation in the presence of 1 μmol L−1 OM at either pCa 9 or pCa 4.3 (data not shown). Neither activation of isometric force by OM at pCa 9 nor inhibition of force at pCa 4.3 are mediated by changes in thin filament structure as reported by the TnC E‐helix probe.
At 5 μmol L−1 BS did reduce the level of thin filament activation at pCa 4.3 reported by the cTnC E‐helix probe by ∼30% (Fig. 5 D and Table 1), in agreement with previous results during almost complete force inhibition at 25 μmol L−1 BS (Sun et al. 2009). However, increasing [BS] to 25 μmol L−1 in the present experiments had no further effect on cTnC E‐helix probe orientation at pCa 4.3 (squares in Fig. 5 D), despite the further inhibition of isometric force from ∼25% to ∼1%. Thus, as in the case of OM, the effects of BS on isometric force are not simply related to thin filament structure as monitored by the TnC E‐helix probe.
The calcium sensitivity (pCa50) of the change in thin filament activation reported by the cTnC E‐helix probe was increased by 1 μmol L−1 OM and decreased by 5 μmol L−1 BS (Figs 5 D and 6 A), consistent with the hypothesis that the activation state of the thin filament is sensitive to that of the thick filament at physiological [Ca2+] (Kampourakis et al. 2016). OM (1 μmol L−1) reduced the steepness (n H) of the calcium dependence of TnC E‐helix probe orientation to 2.5 (Fig. 6 B), similar to the effect on force and thick filament structure reported by the cRLC probe (Figs 5 B and 6 B), but BS had a smaller effect on n H for TnC E‐helix probe orientation, which was still 4.2 in the presence of 5 μmol L−1 BS (Fig. 6 D, Table 1). The effect of 1 μmol L−1 OM on pCa50 for TnC probe orientation was significantly smaller than that for force, in contrast with its effect on pCa50 for the cRLC probe in the thick filament, which was larger than that for force (Fig. 6 A).
Discussion
OM and BS stabilize ON and OFF states of the thick filament respectively in the absence of Ca2+
In this study we have for the first time established detailed structure–function relationships for OM and BS that integrate functional, biochemical and structural measurements in the native environment of the intact muscle lattice. Our results establish a novel mechanistic basis for the modulation of myosin function via thick filament‐based regulation and coupling between the regulatory states of the thick and thin filaments, and suggest how those mechanisms contribute to the normal physiological regulation of contractility in the heart.
We have interpreted the antagonistic effects of OM and BS on the orientation of the myosin cRLC in terms of structural OFF and ON states of the thick filament. Those states were monitored using a probe that has its fluorescence dipole parallel to the cRLC E‐helix (Kampourakis et al. 2014, 2015, 2016) (Fig. 1). This probe is more parallel to the thick filament axis in the myosin OFF state or interacting heads motif (IHM) observed in EM reconstructions of isolated thick filaments (Al‐Khayat et al. 2013) and this orientation is measured as a higher value of the order parameter <P 2>. The results from this cRLC E‐helix probe showed that, at very low [Ca2+] (pCa 9), OM induces a more ON state of the thick filament, whereas BS induces a more OFF state (Fig. 3). This conclusion is consistent with the contrasting effects of OM and BS on the ATPase of isolated myosin head domains (Kovacs et al. 2004; Malik et al. 2011; Liu et al. 2015; Swenson et al. 2017); OM accelerates the rate of release of inorganic phosphate (Pi) from the myosin.ADP.Pi intermediate, whereas BS inhibits it. Thus OM increases the fraction of myosins in the ADP state that can bind strongly to actin and generate force, whereas BS increases the fraction in the weak‐binding or low‐force ADP.Pi state. These two states are associated with different conformations of the myosin head domain, the switch‐2 open and closed states, respectively. Moreover, the ADP.Pi or switch‐2 closed conformation is required for the OFF structure of the thick filament (Xu et al. 1999; Zoghbi et al. 2004; Zhao et al. 2008). Furthermore, the quantitative effect of both compounds (OM and BS) is most likely modulated by post‐translational modifications of proteins associated with regulatory structural transitions in the thick filament, e.g. phosphorylation of cRLC and cMyBP‐C.
The biochemical correlate of the myosin OFF state has been described for both skeletal and cardiac muscle, and termed the super‐relaxed state (SRX), characterized by a population of myosin heads with ultra‐low ATPase activity (Stewart et al. 2010; Hooijman et al. 2011). Structural and functional studies in isolated skeletal muscle fibres (Wilson et al. 2014) and human myocardium (Tang et al. 2016) suggested that BS and its derivatives stabilize the SRX.
Consistent with these structural and biochemical correlations, OM and BS have opposite effects on isometric force at pCa 9; OM activates force and BS inhibits it (Fig. 2). The ability of OM to activate force in the absence of calcium, when the thin filament would normally be switched OFF, implies that myosin heads with bound OM can bind to thin filaments and displace tropomyosin to its ON position in the absence of calcium binding to troponin (Fig. 7), consistent with the decreased rate of relaxation observed in isolated cardiomyocytes in the presence of OM (Nagy et al. 2015).
Figure 7. Proposed model for the effects of OM and BS on myofilament Ca2+ activation.
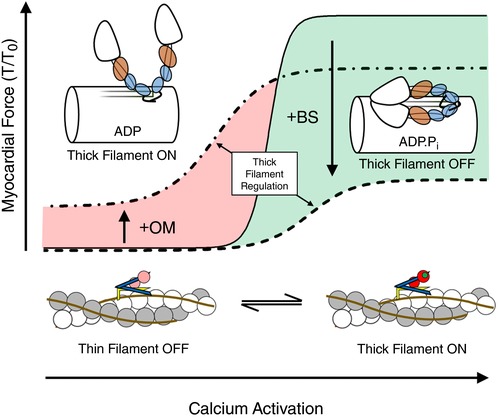
Under control conditions in the absence of any drug, normalized myocardial force shows a steep dependence on [Ca2+] (continuous line). OM induces a perpendicular myosin head orientation, activates the thick filament in the absence of Ca2+ and stabilizes the myosin.ADP state (left). Thick filament activation by OM increases myocardial calcium sensitivity of force, but decreases cooperativity of activation (dashed‐dotted line). In contrast, BS stabilizes the thick filament OFF state characterized by a parallel myosin head orientation in the ADP.Pi state (right), and decreases calcium sensitivity and cooperativity of force (dashed line). [Color figure can be viewed at wileyonlinelibrary.com]
However, the orientation of the cTnC‐E helix probe did not change when the thick filament and isometric force were activated by OM in these conditions (Fig. 5), in marked contrast to the large change in cTnC E‐helix probe orientation associated with calcium activation and the even larger change associated with activation by rigor myosin heads (Sun et al. 2009). Although all these thin filament activation pathways presumably involve a change in the azimuthal position of tropomyosin, activation by OM appears to work through a distinct inter‐filament signalling pathway which is not directly coupled to a change in the cTnC E‐helix orientation. Activation by OM at pCa 9 is also associated with a modest decrease in the myofibrillar ATPase activity, in contrast with the ∼15‐fold increase associated with calcium activation (Fig. 4), and this is presumably associated with direct inhibition of ATPase in myosin heads with bound OM.
Effects of OM and BS in the presence of calcium reveal regulatory coupling between thick and thin filaments
The competing activating and inhibitory effects of OM are modified in the presence of calcium. At full calcium activation (pCa 4.3) OM strongly inhibits isometric force (Figs 2 A and 3 A) and cross‐bridge kinetics (Mamidi et al. 2015), and modestly reduces myofibrillar ATPase (Fig. 4), despite inducing a more ON state of the thick filament (Fig. 3 C). These inhibitory and activating effects have the same EC50, suggesting that both are the consequence of OM binding to a single site on the myosin head to stabilize a myosin.ADP state (Rohde et al. 2017; Swenson et al. 2017). Similar to results in the absence of calcium discussed above, OM binding to myosin does not alter the regulatory state of the thin filament as reported by the TnC E‐helix probe at pCa 4.3 (Fig. 5). As in that case, we conclude that the competing activating and inhibitory effects of OM at pCa 4.3 are mediated by changes in thick filament structure and actin‐activated myosin ATPase, respectively. The activating effect dominates contractility at low [OM] and [Ca2+], probably as a result of the co‐operativity of thick filament activation, with a Hill co‐efficient for [OM] dependence of 1.75, whereas the inhibitory effect dominates at higher [OM] and [Ca2+], when both thick and thin filaments are largely ON. These competing effects may be responsible for the bell‐shaped dependence of isometric force on [OM] at low and intermediate [Ca2+] (Figs 2 A and 3 A).
BS, in contrast, stabilizes the OFF structure of the thick filament and inhibits force production (Fig. 3) and myofibrillar ATPase in all conditions studied. The effects of BS on active force and thick filament structure have the same EC50, consistent with both being associated with stabilization of the ADP.Pi state of the myosin head presumably in the pre‐power‐stroke conformation (Alamo et al. 2017), reducing ATP turnover and promoting the OFF structure of the thick filament. BS had no effect on the level of activation of the thin filament as reported by the cTnC E‐helix probe at pCa 9, but reduced its activation level at pCa 4.3 (Fig. 5), indicating positive coupling between the activation states of the thick and thin filaments in the presence of calcium.
Although OM does not affect the regulatory state of the thin filament reported by the cTnC E‐helix probe at either pCa 9 or 4.3, at intermediate [Ca2+] in the physiological range it switches ON the thin filament as well as the thick filament, and BS switches OFF both filaments (Figs 5 and 6). Both small molecules can perturb the normal coupling between the regulatory states of the thick and thin filaments, as shown by the altered Ca2+ dependence of structural changes in the myofilaments and force development. In the absence of either OM or BS, activation of the thick and thin filaments and isometric force have similar but not identical [Ca2+] dependence; calcium sensitivity (pCa50) is close to 5.5, and Hill coefficients (n H) are greater than 5.5 (Fig. 6; Table 1). However, n H for thick filament structure is significantly higher than that for thin filament structure or force, as reported previously for a different probe on the N‐lobe of the cRLC (Kampourakis et al. 2016); calcium activation of the thick filament is more co‐operative than that of the thin filament. Co‐operativity of the thin filament is likely to be associated with end‐to‐end joining between tropomyosin regulatory units (Gordon et al. 2000); that of the thick filament to the multiple inter‐ and intra‐molecular interactions that stabilize the thick filament OFF state (Al‐Khayat et al. 2013).
The effects of BS and OM reported above have implications for the roles of thin and thick filament co‐operativity during normal contractile activation. When the thick filament is locked in its OFF state by 5 μmol L−1 BS, so that its structure becomes independent of [Ca2+] (Fig. 5 B), calcium activation becomes less co‐operative – n H for force is reduced from more than 6 to less than 3, and calcium sensitivity pCa50 is reduced by about 0.25 pCa units (Table 1). Pushing the thick filament towards the ON state with 1 μmol L−1 OM also reduces n H to less than 3, but increases calcium sensitivity of force by about 0.3 pCa50 units (Figs 5 and 6).
These effects suggest that the intrinsic co‐operativity of the thin filament activation is low, and that its calcium sensitivity depends on the regulatory state of the thick filament (Fig. 7). When the thick filament is OFF, pCa50 is about 5.3 (as in the presence of BS; Fig. 7, dashed line); when the thick filament is ON, pCa50 is about 5.5 or higher (as in the presence of OM; Fig. 7, dashed‐dotted line). In physiological conditions, the thick filament is OFF at low [Ca2+], and no active force is produced at pCa > 5.9. At slightly higher [Ca2+], around pCa 5.7, the thin filament starts to be activated and this starts to activate the thick filament, which in turn increases the calcium sensitivity of the thin filament (Fig. 7, red shaded area). The net result of this inter‐filament coupling is to produce a very steep dependence of the activation state of both filaments and isometric force on [Ca2+] (continuous line).
Taken together, our results suggest that the force–calcium relation of cardiac muscle is controlled by three regulatory pathways working in parallel: calcium activation of the thin filament, the regulatory state of the thick filament and intermediates in the acto‐myosin ATPase pathway.
This scheme also provides a qualitative explanation for the effects of other interventions that affect the regulatory state of the thick filament, like RLC phosphorylation and increased sarcomere length (Kampourakis et al. 2016), on the calcium sensitivity of the thin filament and isometric force. Moreover, hypertrophic and dilated cardiomyopathy mutations have been shown to alter the calcium dependence of myofilament activation in a similar manner to OM and BS (Spudich, 2014), implicating disturbed inter‐filament coupling in the aetiology of cardiomyopathy‐associated heart failure. An uncoupling between the regulatory structural changes in the thick filament and force generation similar to that produced by OM has been recently suggested as a mechanism underlying the hyper‐contractility associated with hypertrophic cardiomyopathy mutations in the myosin head domain (Alamo et al. 2017; Nag et al. 2017; Trivedi et al. 2017).
The molecular mechanisms responsible for coupling between the regulatory states of the thin and thick filaments remain to be elucidated, but candidate mechanisms would include binding of myosin heads to thin filaments (Gordon et al. 2000) and binding of the N‐terminus of cMyBP‐C to the thin filaments (Pfuhl & Gautel, 2012; Kampourakis et al. 2014). Binding of myosin heads to thin filaments stabilizes their ON state by preventing tropomyosin moving back to its OFF position, so an ON thick filament can promote an ON thin filament. Similarly, binding of the N‐terminus of cMyBP‐C to the thin filaments also stabilizes their ON state, although it interacts with myosin head domains with similar affinity, and also has an inhibitory effect on the thick filament structure (Pfuhl & Gautel, 2012; Kampourakis et al. 2014; Mun et al. 2014). The major rearrangement of the myosin heads associated with activation of the thick filament might disrupt an interaction between cMyBP‐C and myosin heads, promoting a competing activating interaction with the thin filament (Kampourakis et al. 2014). Reciprocal (thin to thick) effects are also possible and additional mechanisms may contribute to inter‐filament signalling. Thick filament mechano‐sensing (Linari et al. 2015), for example, postulates that the regulatory state of the thick filament is controlled by mechanical stress in the filament, which is itself a consequence of force generation and therefore of thin filament activation. This mechanism would also generate positive co‐operativity, but requires a population of myosin heads outside thick filament control to initiate the positive feedback loop.
Further consideration of the relative significance of these and other potential mechanisms of interfilament signalling is likely to require a more detailed understanding of the structural basis of the regulatory states of the thin and thick filaments, at a level beyond the simple concept of OFF and ON states. For the thin filaments, the distinct responses of probes on the cTnC C and E‐helices to BS (Sun et al. 2009; Kampourakis et al. 2014) provide further support for a concept of thin filament regulation in the intact sarcomere as a signalling pathway in which different structural components may be activated to different extents by the different inputs to the pathway, including calcium, post‐translational modifications, binding of myosin heads and the actions of cMyBP‐C. Thick filament regulation is also likely to constitute an analogous multi‐component pathway with multiple inputs for which simple OFF and ON descriptions are an oversimplification. Moreover, different regions of the thick filament, for example the C‐zone containing cMyBP‐C, are likely to have distinct regulatory states. Finally, it will be important to extend the present consideration of steady‐state regulatory states to the dynamic switching that underlies the cardiac cycle.
Therapeutic mechanism of OM and implications for drug design
Inherited cardiomyopathies including hypertrophic, dilated and restricted cardiomyopathy are commonly linked to mutations in the protein components of the thick and thin filaments, and filament dysfunction is a major cause of heart failure (Morita et al. 2010; McNally et al. 2013). Available treatments for all these types of heart disease are clearly inadequate, and much effort continues to be directed towards establishing more effective therapies with reduced off‐target effects. Drugs that enhance contractility by boosting the calcium transient suffer from the consequences of the multiple intracellular targets for calcium, and altered calcium handling can induce arrhythmias. This limitation led to an increased emphasis on targeting the thick and thin filaments themselves, initially focused on troponin in the thin filament, which was perceived to be the primary site of regulation of contraction (Malik et al. 2011; Radke et al. 2014; Spudich, 2014; Hwang & Sykes, 2015; Green et al. 2016). More recently attention has been directed towards myosin itself (Malik et al. 2011; Radke et al. 2014; Spudich, 2014; Green et al. 2016), and the search for myosin‐targeted therapeutics has been directed by biochemical and structural models of the interaction between the myosin head domain, actin and ATP, which have been used to screen for and characterize the function of small molecules that bind specifically to cardiac myosin, and activate (Malik et al. 2011), inhibit (Green et al. 2016), or restore (Radke et al. 2014) its function. Omecamtiv mecarbil (OM), the activator used in the present study, has completed phase II clinical trials for the treatment of systolic heart failure (Teerlink et al. 2016).
The results presented above showed that, at clinically relevant concentrations, OM activates cardiac muscle cells by disrupting the OFF state of the thick filament, which in turn increases the calcium sensitivity of the thin filament, producing a large increase in contractility at physiological calcium concentrations. OM also inhibits the ATPase activity of myosin and this inhibitory effect becomes dominant at higher [OM] and [Ca2+]. Both the activating and inhibitory effects of OM are mediated by stabilization of a myosin.ADP state, and the therapeutic window of [OM] and [Ca2+] is created by the intrinsic co‐operativity of the thick and thin filament regulatory mechanisms.
Our results also suggests a novel approach to the development of myosin‐targeted small molecules that modulate the contractility of cardiac muscle, either positively, to enhance the performance of the failing heart, or negatively, as might be useful in the treatment of diastolic failure associated with hypertrophic cardiomyopathy. To supplement the approach of looking for small molecules like OM and BS that perturb the actin‐myosin ATPase cycle as determined with soluble myosin fragments that do not form filaments, it might be possible to design or screen for molecules that perturb the regulatory state of the thick filament without large effects on the actin‐myosin ATPase (Nogara et al. 2016a). In principle, and in contrast with OM, such molecules could be simple activators without competing inhibitory effects, which might facilitate the selection of a therapeutic dose.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
Conception and design: T.K., Y.B.S., M.I. Acquisition, analysis and interpretation of data: T.K., X.Z., M.I. Manuscript preparation: T.K., M.I. All authors have approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the British Heart Foundation (PG/12/52/29713, FS/16/3/31887, FS/09/001/26329).
Acknowledgements
We wish to thank Mathias Gautel, Ziqian Yan and David Trentham for help and advice.
Linked articles This article is highlighted by a Perspective by Tang & Yengo. To read this Perspective, visit https://doi.org/10.1113/JP275414.
Edited by: Harold Schultz & Bruce Smaill
References
- Ait‐Mou Y, Hsu K, Farman GP, Kumar M, Greaser ML, Irving TC & de Tombe PP (2016). Titin strain contributes to the Frank‐Starling law of the heart by structural rearrangements of both thin‐ and thick‐filament proteins. Proc Natl Acad Sci USA 113, 2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamo L (2008). Three‐dimensional reconstruction of tarantula myosin filaments suggests how phosphorylation may regulate myosin activity. J Mol Biol 384, 780–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE & Padron R (2017). Effects of myosin variants on interacting‐heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 6, e24634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Khayat HA, Kensler RW, Squire JM, Marston SB & Morris EP (2013). Atomic model of the human cardiac muscle myosin filament. Proc Natl Acad Sci USA 110, 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Brandmeier BD, Ferguson RE, Criddle S, Dale RE & Irving M (2004). Bifunctional rhodamine probes of Myosin regulatory light chain orientation in relaxed skeletal muscle fibers. Biophys J 86, 2329–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito R (2011). A molecular model of phosphorylation‐based activation and potentiation of tarantula muscle thick filaments. J Mol Biol 414, 44–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland JG, Teerlink JR, Senior R, Nifontov EM, Mc Murray JJ, Lang CC, Tsyrlin VA, Greenberg BH, Mayet J, Francis DP, Shaburishvili T, Monaghan M, Saltzberg M, Neyses L, Wasserman SM, Lee JH, Saikali KG, Clarke CP, Goldman JH, Wolff AA & Malik FI (2011). The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double‐blind, placebo‐controlled, crossover, dose‐ranging phase 2 trial. Lancet 378, 676–683. [DOI] [PubMed] [Google Scholar]
- Colson BA, Patel JR, Chen PP, Bekyarova T, Abdalla MI, Tong CW, Fitzsimons DP, Irving TC & Moss RL (2012). Myosin binding protein‐C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J Mol Cell Cardiol 53, 609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrie JE, Brandmeier BD, Ferguson RE, Trentham DR, Kendrick‐Jones J, Hopkins SC, van der Heide UA, Goldman YE, Sabido‐David C, Dale RE, Criddle S & Irving M (1999). Dynamic measurement of myosin light‐chain‐domain tilt and twist in muscle contraction. Nature 400, 425–430. [DOI] [PubMed] [Google Scholar]
- Dale RE, Hopkins SC, an der Heide UA, Marszalek T, Irving M & Goldman YE (1999). Model‐independent analysis of the orientation of fluorescent probes with restricted mobility in muscle fibers. Biophys J 76, 1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Arlock P & Arner A (2007). Blebbistatin specifically inhibits actin‐myosin interaction in mouse cardiac muscle. Am J Physiol Cell Physiol 293, C1148–C1153. [DOI] [PubMed] [Google Scholar]
- Farman GP, Tachampa K, Mateja R, Cazorla O, Lacampagne A & de Tombe PP (2008). Blebbistatin: use as inhibitor of muscle contraction. Pflugers Arch 455, 995–1005. [DOI] [PubMed] [Google Scholar]
- Fusi L, Brunello E, Yan Z & Irving M (2016). Thick filament mechano‐sensing is a calcium‐independent regulatory mechanism in skeletal muscle. Nat Commun 7, 13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon AM, Homsher E & Regnier M (2000). Regulation of contraction in striated muscle. Physiol Rev 80, 853–924. [DOI] [PubMed] [Google Scholar]
- Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG & Seidman CE (2016). A small‐molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg BH, Chou W, Saikali KG, Escandon R, Lee JH, Chen MM, Treshkur T, Megreladze I, Wasserman SM, Eisenberg P, Malik FI, Wolff AA & Shaburishvili T (2015). Safety and tolerability of omecamtiv mecarbil during exercise in patients with ischemic cardiomyopathy and angina. JACC Heart Fail 3, 22–29. [DOI] [PubMed] [Google Scholar]
- Herrmann C, Sleep J, Chaussepied P, Travers F & Barman T (1993). A structural and kinetic study on myofibrils prevented from shortening by chemical cross‐linking. Biochemistry 32, 7255–7263. [DOI] [PubMed] [Google Scholar]
- Hooijman P, Stewart MA & Cooke R (2011). A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J 100, 1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley HE & Brown W (1967). The low‐angle x‐ray diagram of vertebrate striated muscle and its behaviour during contraction and rigor. J Mol Biol 30, 383–434. [DOI] [PubMed] [Google Scholar]
- Hwang PM & Sykes BD (2015). Targeting the sarcomere to correct muscle function. Nat Rev Drug Discov 14, 313–328. [DOI] [PubMed] [Google Scholar]
- Kampourakis T & Irving M (2015). Phosphorylation of myosin regulatory light chain controls myosin head conformation in cardiac muscle. J Mol Cell Cardiol 85, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampourakis T, Sun YB & Irving M (2015). Orientation of the N‐ and C‐terminal lobes of the Myosin regulatory light chain in cardiac muscle. Biophys J 108, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampourakis T, Sun YB & Irving M (2016). Myosin light chain phosphorylation enhances contraction of heart muscle via structural changes in both thick and thin filaments. Proc Natl Acad Sci USA 113, E3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampourakis T, Yan Z, Gautel M, Sun YB & Irving M (2014). Myosin binding protein‐C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc Natl Acad Sci USA 111, 18763–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs M, Toth J, Hetenyi C, Malnasi‐Csizmadia A & Sellers JR (2004). Mechanism of blebbistatin inhibition of myosin II. J Biol Chem 279, 35557–35563. [DOI] [PubMed] [Google Scholar]
- Linari M, Brunello E, Reconditi M, Fusi L, Caremani M, Narayanan T, Piazzesi G, Lombardi V & Irving M (2015). Force generation by skeletal muscle is controlled by mechanosensing in myosin filaments. Nature 528, 276–279. [DOI] [PubMed] [Google Scholar]
- Liu Y, White HD, Belknap B, Winkelmann DA & Forgacs E (2015). Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine beta‐cardiac myosin. Biochemistry 54, 1963–1975. [DOI] [PubMed] [Google Scholar]
- McNally EM, Golbus JR & Puckelwartz MJ (2013). Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest 123, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen YT, Vatner SF & Morgans DJ (2011). Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331, 1439–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidi R, Gresham KS, Li A, dos Remedios CG & Stelzer JE (2015). Molecular effects of the myosin activator omecamtiv mecarbil on contractile properties of skinned myocardium lacking cardiac myosin binding protein‐C. J Mol Cell Cardiol 85, 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, Nagai R, Seidman JG & Seidman CE (2010). Sarcomere gene mutations in hypertrophy and heart failure. J Cardiovasc Transl Res 3, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, Robbins J, Warshaw DM & Craig R (2014). Myosin‐binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci USA 111, 2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Trivedi DV, Sarkar SS, Adhikari AS, Sunitha MS, Sutton S, Ruppel KM & Spudich JA (2017). The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat Struct Mol Biol 24, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Kovacs A, Bodi B, Pasztor ET, Fulop GA, Toth A, Edes I & Papp Z (2015). The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br J Pharmacol 172, 4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogara L, Naber N, Pate E, Canton M, Reggiani C & Cooke R (2016a). Piperine's mitigation of obesity and diabetes can be explained by its up‐regulation of the metabolic rate of resting muscle. Proc Natl Acad Sci USA 113, 13009–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogara L, Naber N, Pate E, Canton M, Reggiani C & Cooke R (2016b). Spectroscopic studies of the super relaxed state of skeletal muscle. PLoS One 11, e0160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfuhl M & Gautel M (2012). Structure, interactions and function of the N‐terminus of cardiac myosin binding protein C (MyBP‐C): who does what, with what, and to whom? J Muscle Res Cell Motil 33, 83–94. [DOI] [PubMed] [Google Scholar]
- Pinto A, Sanchez F, Alamo L & Padron R (2012). The myosin interacting‐heads motif is present in the relaxed thick filament of the striated muscle of scorpion. J Struct Biol 180, 469–478. [DOI] [PubMed] [Google Scholar]
- Radke MB, Taft MH, Stapel B, Hilfiker‐Kleiner D, Preller M & Manstein DJ (2014). Small molecule‐mediated refolding and activation of myosin motor function. Elife 3, e01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reconditi M, Caremani M, Pinzauti F, Powers JD, Narayanan T, Stienen GJ, Linari M, Lombardi V & Piazzesi G (2017). Myosin filament activation in the heart is tuned to the mechanical task. Proc Natl Acad Sci USA 114, 3240–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde JA, Thomas DD & Muretta JM (2017). Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proc Natl Acad Sci USA 114, E1796–E1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrieva I, Knowles AC, Kampourakis T & Sun YB (2014). Regulatory domain of troponin moves dynamically during activation of cardiac muscle. J Mol Cell Cardiol 75, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich JA (2014). Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J 106, 1236–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MA, Franks‐Skiba K, Chen S & Cooke R (2010). Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci USA 107, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YB, Lou F & Irving M (2009). Calcium‐ and myosin‐dependent changes in troponin structure during activation of heart muscle. J Physiol 587, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson AM, Tang W, Blair CA, Fetrow CM, Unrath WC, Previs MJ, Campbell KS & Yengo CM (2017). Omecamtiv Mecarbil enhances the duty ratio of human beta‐cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J Biol Chem 292, 3768–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Blair CA, Walton SD, Malnasi‐Csizmadia A, Campbell KS & Yengo CM (2016). Modulating Beta‐cardiac myosin function at the molecular and tissue levels. Front Physiol 7, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD, Elliott L, Bee R, Habibzadeh MR, Goldman JH, Schiller NB, Malik FI & Wolff AA (2011). Dose‐dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, Omecamtiv Mecarbil: a first‐in‐man study. Lancet 378, 667–675. [DOI] [PubMed] [Google Scholar]
- Teerlink JR, Felker GM, McMurray JJ, Ponikowski P, Metra M, Filippatos GS, Ezekowitz JA, Dickstein K, Cleland JG, Kim JB, Lei L, Knusel B, Wolff AA, Malik FI, Wasserman SM & ATOMIC‐AHF Investigators (2016). Acute treatment with Omecamtiv Mecarbil to increase contractility in acute heart failure: The ATOMIC‐AHF Study. J Am Coll Cardiol 67, 1444–1455. [DOI] [PubMed] [Google Scholar]
- Trivedi DV, Adhikari AS, Sarkar SS, Ruppel KM & Spudich JA (2017). Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev (in press; https://doi.org/10.1007/s12551-017-0274-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utter MS, Ryba DM, Li BH, Wolska BM & Solaro RJ (2015). Omecamtiv Mecarbil, a cardiac myosin activator, increases Ca2+ sensitivity in myofilaments with a dilated cardiomyopathy mutant tropomyosin E54K. J Cardiovasc Pharmacol 66, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt T, Taylor D, Trybus KM & Taylor K (2001). Three‐dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc Natl Acad Sci USA 98, 4361–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Naber N, Pate E & Cooke R (2014). The myosin inhibitor blebbistatin stabilizes the super‐relaxed state in skeletal muscle. Biophys J 107, 1637–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann DA, Forgacs E, Miller MT & Stock AM (2015). Structural basis for drug‐induced allosteric changes to human beta‐cardiac myosin motor activity. Nat Commun 6, 7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead JL, Zhao FQ & Craig R (2013). Structural basis of the relaxed state of a Ca2+‐regulated myosin filament and its evolutionary implications. Proc Natl Acad Sci USA 110, 8561–8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead JL, Zhao FQ, Craig R, Egelman EH, Alamo L & Padron R (2005). Atomic model of a myosin filament in the relaxed state. Nature 436, 1195–1199. [DOI] [PubMed] [Google Scholar]
- Xu S, Gu J, Rhodes T, Belknap B, Rosenbaum G, Offer G, White H & Yu LC (1999). The M.ADP.Pi state is required for helical order in the thick filaments of skeletal muscle. Biophys J 77, 2665–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao FQ, Padron R & Craig R (2008). Blebbistatin stabilizes the helical order of myosin filaments by promoting the switch 2 closed state. Biophys J 95, 3322–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi ME, Woodhead JL, Craig R & Padron R (2004). Helical order in tarantula thick filaments requires the “closed” conformation of the myosin head. J Mol Biol 342, 1223–1236. [DOI] [PubMed] [Google Scholar]
- Zoghbi ME, Woodhead JL, Moss RL & Craig R (2008). Three‐dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci USA 105, 2386–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]