

Abstract
Background
The role of nicotinic acetylcholine receptor alpha7 subunit (a7nAchR) in the treatment of acute cerebral ischemia by VNS has not been thoroughly clarified to date. Therefore, this study aimed to investigate the specific role of a7nAchR and explore whether this process is involved in the mechanisms of VNS-induced neuroprotection in rats undergoing permanent middle cerebral artery occlusion (PMCAO) surgery.
Material/Methods
Rats received a7nAChR antagonist (A) or antagonist placebo injection for control (AC), followed by PMCAO and VNS treatment, whereas the a7nAChR agonist (P) was utilized singly without VNS treatment but only with PMCAO pretreatment. The rats were randomly divided into 6 groups: sham PMCAO, PMCAO, PMCAO+VNS, PMCAO+VNS+A, PMCAO+VNS+AC, and PMCAO+P. Neurological function and cerebral infarct volume were measured to evaluate the level of brain injury at 24 h after PMCAO or PMCAO-sham. Moreover, the related proteins levels of a7nAChR, p-JAK2, and p-STAT3 in the ischemic penumbra were assessed by Western blot analysis.
Results
Rats pretreated with VNS had significantly improved neurological function and reduced cerebral infarct volume after PMCAO injury (p<0.05). In addition, VNS enhanced the levels of a7nAchR, p-JAK2, and p-STAT3 in the ischemic penumbra (p<0.05). However, inhibition of a7nAchR not only attenuated the beneficial neuroprotective effects induced by VNS, but also decreased levels of p-JAK2 and p-STAT3. Strikingly, pharmacological activation of a7nAchR can partially substitute for VNS-induced beneficial neurological protection.
Conclusions
These results suggest that a7nAchR is a pivotal mediator of VNS-induced neuroprotective effects on PMCAO injury, which may be related to suppressed inflammation via activation of the a7nAchR/JAK2 anti-inflammatory pathway.
MeSH Keywords: alpha7 Nicotinic Acetylcholine Receptor, Brain Ischemia, Janus Kinase 2, Neuroprotective Agents, Vagus Nerve Stimulation
Background
Acute cerebral infarction is a leading cause of disability, with high mortality and morbidity, which essentially results from disturbance of the bloodstream supplying the brain [1–3]. It has been reported that intervention requiring reestablishment of the blood supply is considered as one of the most effective treatments for acute cerebral ischemia, but this inevitably causes reperfusion injury [3,4]. Multiple pathological processes, including mitochondrial dysfunction, excessive release of glutamate, and overproduction of pro-inflammatory mediators and reactive oxygen species (ROS), take part in the process of acute cerebral ischemic/reperfusion (I/R) injury, in which inflammatory response plays a vital role during this period [3,5]. Some studies have proved that pharmacological treatment has beneficial neuroprotective effects on anti-oxidation and anti-apoptotic in rat neurons, but the anti-inflammation efficacy is still uncertain [6–8]. Therefore, it is urgent to develop novel and effective therapeutic approaches to control excessive inflammation responses during acute ischemic stroke.
The original application of vagus nerve stimulation (VNS) was dedicated to the treatment of intractable epilepsy and treatment-resistant depression [9–11]. It was approved by the Food and Drug Administration (FDA) in 1997, and it has become an adjunctive therapy for other clinical diseases. Previous studies have demonstrated that VNS can effectively reduce the volume of cerebral infarction in acute cerebral infarction rats by as much as 50% [12–14]. In addition, some studies reported that VNS can exert anti-inflammatory and antioxidant effects against ischemic stroke via the cholinergic anti-inflammatory pathway of a7nAChR/Akt [15,16].
The a7nAChR pathway is a well-characterized member of the neurotransmitter-gated ion channel superfamily [17]. In neurons, a7 proteins assemble as a homopolymer composed of 5 individual a7-subunits, which is negatively regulated by phosphorylation at Tyr-386 and Tyr-442 by Src-family kinases [18,19]. Previous studies have found that the anti-inflammatory potential of the a7nAChR is mediated by the inhibition of the transcription factor and NF-κB in neural cells [20]. In VNS inhibition of peripheral inflammation, the a7nAChR triggers activation of its catalytic intracellular domain, leading to recruitment and phosphorylation of tyrosine kinase JAK2 and subsequent activation of the transcription factor STAT3 [21,22]. Overall, the process of anti-inflammatory activity caused by VNS appears to involve a7nAChR/JAK2 signaling. However, whether the a7nAChR/JAK2 pathway participates in the mechanisms of VNS-induced neuroprotective effects in protecting the brain from cerebral infarct damage remains unknown.
Therefore, we investigated the neuroprotection effect of VNS by evaluating neurological function and cerebral infarct volume, and sought to determine whether a7nAChR plays a key role in the neuroprotection process mediated by VNS after acute cerebral ischemia and to elucidate its potential molecular mechanisms.
Material and Methods
Animals
Adult male Sprague-Dawley (SD) rats weighing 250–300 g were obtained from the Center for Animal Experimentation of the Traditional Chinese Medicine University of Guangzhou, China (Certificate No: 44005800003146) and were used for these experiments. Rats were housed at 22–24°C and 50–60% humidity with a 12–12 h light/dark cycle (lights on from 07: 00 to 19: 00), 5 rats per cage, and ad libitum access to food and water. At the Laboratory Animal Center, Zhongshan University, China, we performed the establishment of the rat model of acute cerebral infarction, electrical stimulation of the vagus nerve, injection of the lateral ventricle, intraperitoneal injection treatment, behavioral testing, and TTC staining of brain tissue of rats. Western blot analysis was conducted at the Lin-Bai-Xin Medical Research Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, China. The animal experimental procedures were approved by Animal Ethics Committee of Sun Yat-sen University.
a7nAChR antagonist and agonist administration
To further investigate the function of a7nAChR in the VNS-induced neuroprotective response, the specific a7nAChR antagonist α-bungarotoxin was used to inhibit a7nAChR expression level. The rats were deeply anesthetized with 10% hydration chlorine aldehyde (0.35 ml/kg; intraperitoneal injection) and were placed in a stereotaxic frame with a head holder. The α-bungarotoxin (0.5 ug/kg; Abcam, USA) and equivalent volume of saline (antagonist placebo control) were injected into the left lateral ventricle of rats in each group. We repeated the lateral cerebral ventricle injection test alone in advance to finally determine the injection stereotaxic coordinates: 1 mm before the anterior fontanel, 1.5 mm to the left, and a depth of 4.5 mm, and marked the location on the skull surface with ink. We used a dental drill to drill the point to the dura mater, then punctured the dura mater with a needle tip, using sterile cotton swabs to stop bleeding and dry the surgical field. A catheter was inserted 4.5 mm vertically, and then we slowly injected the drugs. The catheter was removed 1 min after administration of drugs, then we sutured the scalp incision. After 30 min, rats were subjected to PMCAO surgery [26]. However, the specific agonist of a7nAChR (PHA543613; 1.0 mg/kg; R&D System, USA) was injected intraperitoneally as the reagent can pass through the blood-brain barrier.
Acute PMCAO rat model establishment
To establish a permanent acute cerebral infarction model, rats undergo left middle cerebral artery occlusion (MCAO) surgery according to the intraluminal occlusion technique previously described by Longa et al. [23]. Rats were anesthetized with 10% hydration chlorine aldehyde (0.35 ml/kg intraperitoneal injection). After a midline neck incision with the animal in supine position, the left common carotid artery (CCA), external carotid artery (ECA), internal carotid artery (ICA), and vagus nerve were isolated. Then, we ligatured the CCA and ECA, and clipped the ICA. A small cut was made in the common carotid artery with an eye scissors, the monofilament nylon suture was inserted, and the artery clamp was opened simultaneously. A monofilament nylon suture (Sunbio Biotech Limited Company, Beijing, China) with a radius determined by the weight of each rat was advanced from the CCA into the lumen of the ICA until it blocked the origin of the MCA. For rats weighing >280 g, a monofilament nylon suture (diameter=0.28 mm) was inserted into the left common carotid artery about 18.5 mm; for rats weighing 260–280 g, a nylon suture (diameter=0.26 mm) was inserted about 18.0 mm; and for rats weighting <260 g, the length was 17.5 mm. Subsequently, a silk suture was then tightened around the left common carotid artery stumps and nylon filament. Finally, we sequentially sutured the skin incision, sterilized the skin with iodine, and injected 40 000 U/day penicillin into the abdominal cavity. Behavioral evaluations of the rats were performed 3 h after surgery, using the Bederson 4-point rating scale scored as: 0, no deficit; 1, failing to stretch right forepaw during tail suspension test; 2, decreasing ability of forelimb resistance to contralateral thrust; and 3, circling to the right after holding the tail [24]. The standard PMCAO model was defined as a Bederson scale score >1 point, and animals that did not meet this criterion were excluded from the study. Throughout the duration of the experiment, animals kept breathing smoothly and body temperature was maintained at 36–37°C with a heating pad.
Vagus nerve stimulation
According to a previous study, rats received VNS by use of the Taimeng biological experimental system BL-420S (Taimeng Technology Co., Ltd., Chengdu, China) initiated 30 min after PMCAO surgery [13]. The left vagus nerve was then carefully separated from the surrounding connective tissues until the exposed length of the nerve was long enough for electrode placement. The stimulating electrodes, which we made of circular double-wire wires, were connected to the VNS stimulator machine following the process described by Smith [25]; the inner metal wires of the electrodes were exposed and the outer packs were insulated to prevent electrical stimulation of the non-vagus nerves. The electrodes were sutured to the sternocleidomastoid muscle and wrapped around the left nerve using a microscope, as described in a previous study [15]. The stimulation pulses consisted of repeated square pulses of 0.5 mA in 30-s trains (0.5 ms width at 20 Hz), which were repeated every 5 min and lasted for 60 min [13]. Rats from the sham PMCAO group were treated with CCA and ECA surgical exposure but did not undergo filament insertion.
Experimental design
This study included 2 separate protocols (Figure 1).
Figure 1.
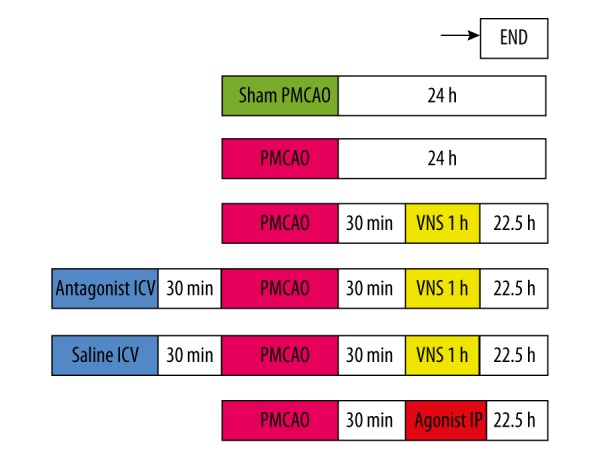
The Experimental Protocol. Before establishing the PMCAO model, a-bungarotoxin (a7nAChR antagonist) and saline (antagonist control) were administered intracerebroventricularly for 30 min in PMCAO+VNS+A and PMCAO+VNS+AC groups, respectively. The PMCAO+P group received intraperitoneal injection with a7nAChR agonist 30 min after PMCAO. Neurological function and infarct volume assessment were performed in half of the rats of each group, and the other half were subjected to Western blot analysis at 24 h after PMCAO.
Protocol 1: To further identify whether VNS treatment reduces infarct volume and improves neurological function after cerebral infarction in rats, and to determine whether the nicotinic acetylcholine receptor alpha7 subunit (a7nAChR) plays a key role in the VNS-mediated neuroprotection.
Protocol 2: To investigate whether a7nAChR/JAK2 pathway is involved in VNS-mediated neuroprotective effect against cerebral ischemia injury in rats of permanent middle cerebral artery occlusion (PMCAO) model.
The experimental animals were randomly assigned to 6 groups: sham PMCAO, PMCAO, PMCAO+VNS, PMCAO+VNS+A (α-bungarotoxin-a7nAChR antagonist), PMCAO+VNS+AC (saline-sham antagonist), and PMCAO+P (PHA543613-a7nAChR agonist). A total of 108 rats were used for this experiment: 17 rats for the PMCAO group and 1 died; 16 rats for the sham PMCAO group and no deaths; 18 rats for the PMCAO+VNS group and 2 died; 20 rats for the PMCAO+VNS+A group and 4 died; 19 rats for the PMCAO+VNS+AC group and 3 died; and 18 rats for the PMCAO+P group and 2 died. Therefore, each group has 16 rats for the experiment. Additionally, rats in each group were randomly divided into 2 subgroups, and each subgroup had 8 rats: in one subgroup we used the Garcia scale to evaluate the neurological function, and then used TTC staining to calculate the infarct volume 24 h after PMCAO or sham PMCAO; in the other subgroup we used Western blot to analyze the expression levels of a7nAChR, p-JAK2, and p-STAT3 in the ischemic penumbra 24 h after PMCAO or sham PMCAO.
Evaluation of neurological deficits scores
Neurological function of rats was evaluated 24 h after PMCAO surgery using the Garcia scale [12–14,27]. Briefly, neurological deficits were scored according to 6 aspects: autonomous movement, symmetry in the movement of 4 limbs, forepaw outstretching, climbing, body proprioception, and response to vibrissae touch. The minimum neurological score is 3 and the maximum is 18, with higher scores indicating less neurological deficits. Table 1 shows criteria of the Garcia scale [27].
Table 1.
Neurological evaluation after permanent middle cerebral artery occlusion in rats.
| Test | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Spontaneous activity (in cage for 5 min) | No movement | Barely moves | Moves but does not approach at least three sides of cage | Moves and approaches at least three sides of cage |
| Symmetry of movements (four limbs) | Right side: no movement | Right side: slight movement | Right side: moves slowly | Both sides: move symmetrically |
| Symmetry of forelimbs (outstretching while held by tail) | Right side: no movement, no outreaching | Right side: slight movement to outreach | Right side: moves and outreaches less than right side | Symmetrical outreach |
| Climbing wall of wire cage | … | Fails to climb | Right side is weak | Normal climbing |
| Reaction to touch on either side of trunk | … | No response on right side | Weak response on right side | Symmetrical response |
| Response to vibrissae touch | … | No response on right side | Weak response on right side | Symmetrical response |
Rats were housed in stainless plastic cages with wire-mesh bottoms.
Cerebral infarct volume measurement
Cerebral infarct volume was examined using 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St Louis, MO) staining, as previously described [28]. Briefly, when finishing the evaluation of neurological deficit scores, rats were deeply anesthetized and sacrificed by 10% chloral hydrate (0.5 ml/100 mg) intraperitoneal injection, and the fresh brain tissue samples were placed in a freezer at −20°C for 20 min and then removed. Brains were immediately sliced into 5 serial 2-mm-thick sections using a rodent brain matrix. The slices were stained with 1.5% TTC at 37°C for 30 min, resulting in normal tissue dyed red and the cerebral ischemic tissue white. The infarct volume of each brain slice was calculated by multiplying the infarct area by slice thickness (2.0 mm) using Image J software [29,30]. The infarct area was calculated by adding all the infarct volumes of each slice (i.e., the contralateral brain hemispheric volume minus ipsilateral non-infarct volume). Finally, the infarct volume of the brain was expressed as a percentage of the contralateral hemispheric volume.
Western blot analysis
The rats were anesthetized and decapitated at 24 h after PMCAO surgery. Protein samples of brain tissues were homogenized on ice in RIPA lysis buffers supplemented with phosphatase inhibitors. Western blot analysis was conducted according to protocols previously reported [15, 31]. Proteins were acquired by centrifugation at 12 000 rpm at 4°C for 30 min and quantified using the BCA (bicinchoninic acid) protein assay (96-well microtiter plate method; Shenneng, Bocai, Shanghai, China). The molecular weight of the detected protein determined the appropriate constitution of 10% separation gel and 5% concentrated gel. Each 20-ul sample mixed with 5 ul loading buffer was subjected to electrophoresis in a sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) at 60 V. The proteins were transferred to PVDF membranes (Millipore) in semi-dry conditions at 200 mA. The membranes were blocked with bovine serum albumin (BSA) for 60 min at room temperature, then washed in Tris-buffered saline containing 0.1% Tween-20, and subsequently incubated overnight at 4°C with the following primary antibodies: rabbit anti-a7nAChR (1: 500, Abcam, USA), GAPDH (1: 2000), p-JAK2 (1: 1000), JAK2 (1: 1000), p-STAT3 (1: 2000), and STAT3 (1: 1000) (rabbit anti-rat antibody, Cell Signaling Technology, Beverley, MA). After washing, the membranes were incubated with horseradish peroxidase (HRP) secondary antibody for 60 min at room temperature. The Western blot signals were quantified using an enhanced chemiluminescence kit and analyzed using Image J software (Bethesda, USA).
Statistical analysis
Data values are presented as mean ±SD. For normally distributed homogeneous data, statistical analysis between groups was performed using one-way analysis of variance (one-way ANOVA) followed by Fisher’s least significant differences (LSD) post hoc analysis tests. If the data did not conform to normal distribution, the Kruskal-Wallis H tests and t tests were performed to analyze the significance among the groups. The significance criterion was set at p<0.05. Data were analyzed with SPSS 17.0 for Windows (IBM, USA).
Results
Evaluation of neurological function
To determine the effects of VNS treatment on neurological behavior impairment under the acute cerebral infarction condition, neurological scores of rats were evaluated at 24 h after PMCAO surgery. As shown in Figure 2, rats in the PMCAO group exhibited more severe neurological deficits relative to the sham PMCAO group (p<0.05), showing that PMCAO surgery caused neurological deficits and brain damage. Compared with the PMCAO group, rats treated with VNS (PMCAO+VNS group) showed significant improvement in neurological function (p<0.05), which was consistent with previous studies reporting that VNS improved neurological function in rats with acute cerebral infarction. To investigate whether a7nAChR is a key protein involved in neuroprotection induced by VNS, inhibition of a7nAChR using α-bungarotoxin intracerebroventricular injection was performed. More severe neurological impairments were observed in the PMCAO+VNS+A group as compared to either the PMCAO+VNS+AC group (p<0.05) or the PMCAO+VNS group (p<0.05). However, no significant difference was observed between the PMCAO+VNS+AC and PMCAO+VNS groups (p>0.05). Additionally, rats receiving a7nAChR-specific agonist intraperitoneal injection (PMCAO+P group) had greater improvements of neurological scores as compared to the PMCAO group (p<0.05), whereas there was no statistically significant difference between the PMCAO+P group and the PMCAO+VNS group (p>0.05). These data suggest that VNS protects the brain against acute cerebral infarction damage and that a7nAChR is related to the neuroprotective effects induced by VNS in rats subjected to acute cerebral infarction.
Figure 2.
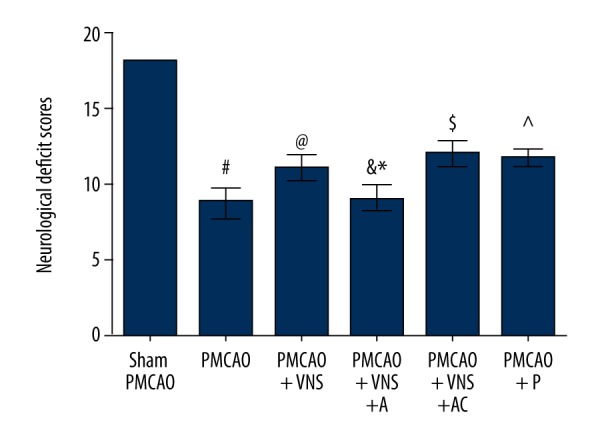
Neurological deficit scores were evaluated after PMCAO with and without VNS. The graphs indicate the neurological deficit scores at 24 h following cerebral ischemia in the 6 groups. All data are expressed as mean±SD. # p<0.05 versus the sham PMCAO group; & p<0.05 versus the sham PMCAO+VNS group; * p<0.05 versus the sham PMCAO+VNS+AC group; @, ^, $ p<0.05 versus the PMCAO group.
Measurement of cerebral infarct volume
To estimate the extent of brain damage 24 h after acute cerebral infarction, TTC staining was performed. Brain infarct volume percentage (BIVP) was expressed as cerebral infarction volume divided by contralateral hemisphere volume. As shown in Figure 3, BIVP was significantly smaller in PMCAO+VNS as compared to PMCAO groups (p<0.05), and rats in the PMCAO group had an increased BVIP relative to the sham PMCAO group (p<0.05). No significant difference was observed between PMCAO+P and PMCAO+VNS groups (p>0.05). These data reveal that VNS treatment reduced the infarct volume after PMCAO surgery, and these effects were also observed in the PMCAO+VNS+AC group. Moreover, the BIVP of rats in the PMCAO+VNS+A group was 0.52±0.037 versus 0.25±0.020 for the PMCAO+VNS group (p<0.05), and versus 0.27±0.018 for PMCAO+VNS+AC group (p<0.05), but there was no significant difference between PMCAO+VNS+A and PMCAO groups (p>0.05).
Figure 3.

Cerebral infarct volume was calculated by TTC staining at 24 h following PMCAO. (A) TTC staining in the 6 groups where red area is the healthy tissue and the white area is the infarct tissue. (B) The relative percentage of infarct volume is shown in histograms. All data are expressed as mean ±SD. # p<0.05 versus the sham PMCAO group; & p<0.05 versus the sham PMCAO+VNS group; * p<0.05 versus the sham PMCAO+VNS+AC group; @, ^, $ p<0.05 versus the PMCAO group.
Western blotting analysis
Expression of protein a7nAchR in the ischemic penumbra 24 h after MCAO
To identify the stimulation effectiveness in acute cerebral ischemic rats, Western blotting was used to detect the a7nAchR expression level induced by VNS. As shown in Figure 4, ischemic damage (PMCAO group) produced a significant reduction in a7nAchR expression level as compared with the sham PMCAO group (p<0.05); however, this phenomenon was mitigated after receiving VNS treatment, as shown in the PMCAO+VNS group, suggesting a significant improvement in a7nAchR expression level relative to the PMCAO group (p<0.05). These data indicate that VNS treatment exerts neuroprotective effects through activation of a7nAchR, the expression of which may play an important role in the activation of intracellular signaling pathways via a7nAchR expression. Strikingly, the specific antagonist and agonist of a7nAchR were used to further identify the real effectiveness of a7nAchR expression induced by VNS. Compared with the PMCAO+VNS group, the expression level of a7nAchR was significantly decreased in the PMCAO+VNS+A group (p<0.05), whereas no significant difference was observed between PMCAO+VNS+A and PMCAO+VNS+AC groups (p>0.05). These results demonstrate that the expression level of a7nAchR was inhibited due to the specific a7nAchR antagonist rather than by intracerebroventricular injection. Moreover, the expression levels of a7nAchR were remarkably increased in the PMCAO+P group compared to the PMCAO group (p<0.05), whereas we found no significant difference between the PMCAO+P and PMCAO+VNS groups (p>0.05), suggesting that a7nAchR specific agonist enhances the expression levels of a7nAchR.
Figure 4.

Expression levels of a7nAChR protein in the ischemic penumbra. (A) Western blot analysis of a7nAChR expression in each group. GAPDH was used as an internal control. (B) Quantitative results from Western blot analysis. All data are expressed as mean ±SD. # p<0.05 versus the sham PMCAO group; & p<0.05 versus the sham PMCAO+VNS group; * p<0.05 versus the sham PMCAO+VNS+AC group; @, ^, $ p<0.05 versus the PMCAO group.
Expression of p-JAK2 and p-STAT3 in the ischemic penumbra 24 h after MCAO
To better investigate the role of a7nAchR in beneficial neuroprotective effects induced by VNS following acute ischemia stroke, the expression of p-JAK2 and p-STAT3 were examined via Western blotting at 24 h after PMCAO, and data are presented in Figure 5. Consistent with the a7nAchR expression level results, PMCAO+VNS group and PMCAO+VNS+AC group had significantly increased p-JAK2 and p-STAT3 expressions levels on the ischemic penumbra compared with the PMCAO group (p<0.05); however, their levels were significantly decreased after inhibition of a7nAchR in the PMCAO+VNS+A group (p<0.05). There was no significant difference between PMCAO+VNS and PMCAO+VNS+AC groups (p>0.05). With respect to PMCAO+P group, expression levels of p-JAK2 were significantly increased compared with the PMCAO group (p<0.05), but no significant difference was found relative to the PMCAO+VNS group (p>0.05). Conversely, the expression levels of p-STAT3 in the PMCAO+P group were remarkably decreased compared with the PMCAO+VNS group or PMCAO group (p<0.05). Although a7nAChR specific agonist was partially substituting for the role of VNS in the treatment of rats subjected to acute cerebral ischemia, there may be other potential mechanisms causing VNS to exert much more resistant anti-inflammatory effects than the a7nAChR-specific agonist itself. Combined with the results of neurological scores and cerebral infarct volume, the a7nAChR/JAK signaling pathway seems to be a beneficial neuroprotection pathway against ischemic injury during acute cerebral ischemia. The neuroprotective effects induced by VNS were diminished following inhibition of a7nAchR. Hence, a7nAchR exerts neuroprotective effects induced by VNS after acute cerebral ischemia.
Figure 5.

Expression levels of p-JAK2 and p-STAT3 in the ischemic penumbra. (A) Western blot analysis of p-JAK2 and p-STAT3 expression levels in each group. GAPDH was used as internal controls. (B) Quantitative results of p-JAK2 expression levels. (C) Quantitative results of p-STAT3 expression levels. All data are expressed as mean ±SD. # p<0.05 versus the sham PMCAO group; Δ, & p<0.05 versus the sham PMCAO+VNS group; * p<0.05 versus the sham PMCAO+VNS+AC group; @, ^, $ p<0.05 versus the PMCAO group.
Discussion
This research demonstrates that VNS has beneficial neuroprotective effects in rats subjected to permanent cerebral infarction on improving in neurological function and reducing cerebral infarct volume. Inhibiting a7nAchR expression levels with specific blocker s resulted in significantly more severe aggravation of neurological dysfunction, increased cerebral infarct volume, and reduced expression levels of p-JAK2 and p-STAT3 after PMCAO. These findings suggest that a7nAchR is involved in VNS-mediated protection via activation of the a7nAChR/JAK2 anti-inflammatory signaling pathway in the ischemic penumbra area against permanent ischemia injury in vivo.
It has been shown that VNS not only successfully treats drug-resistant depression and intractable epilepsy, but also can be used to treat other clinical diseases, such as myocardial I/R injury, cerebral ischemia, migraine, cognition, and traumatic brain injury [32–34]. Previous evidence demonstrated that VNS improves neurological scores and reduces infarct volume by nearly 50% at 24 h after transient MCAO in a rat model of I/R injury, revealing that VNS has a potential neuroprotective effect in the clinical treatment of acute cerebral ischemia. Moreover, Sun et al. found that VNS provides neuroprotection against stroke in both transient MCAO and PMCAO [35]. Similarly, the results of the present study show that PMCAO model rats treated with VNS had significantly improved neurological function and decreased cerebral infarct volume relative to those without VNS. It is remarkable that lateral ventricular injection was used to better identify the effectiveness of a7nAchR, and this invasive technology might influence the observational results; however, there was no significant difference between Garcia scale data and BIVP between the PMCAO+VNS and PMCAO+VNS+AC groups, suggesting that intracerebroventricular injection itself did not change cerebral infarct volume, and did not affect the VNS-induced effects on the neurological function of rats with acute cerebral infarction. In addition, the comparison between a7nAchR antagonist and antagonist-sham showed that intracerebroventricular injection alone did not counteract the effect of VNS on acute cerebral infarction in rats.
In the central nervous system, the vagus nerve has numerous afferent nerve fibers that play a vital and complex role in information transmission and endocrine function, which can regulate serotonin and norepinephrine delivery through projecting to brainstem nuclei [36]. In recent years, the therapeutic mechanisms by which VNS acts have been shown to be involved in acute cerebral infarction and include anti-oxidation and anti-inflammation effects [14–16,37]. Previous studies showed that VNS decreases the level of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) 24 h after reperfusion in the ischemic penumbra, indicating that VNS participated in the process of anti-inflammation caused by acute cerebral ischemia injury, and exerted beneficial effects on neuroprotection [15,31]. Moreover, some studies have found that the VNS-induced neuroprotective effect on alleviating cerebral infarct volume was associated with the cholinergic anti-inflammatory pathway (CAP) [15,38]. CAP represents a physiological mechanism whereby the central nervous system regulates or inhibits local or systemic inflammatory response with cholinergic nerves and their neurotransmitters [39,40]. It has been reported that a7nAchR is a key protein of signals triggered by VNS to induce the endogenous CAP. The a7nAchR is mainly expressed in the brain, including neurons, glia, and endothelial cells [40,41]. Substantial evidence shows that activation of a7nAchR expressed on microglia exerts neuroprotective effects and acts as an integral part in the fight against various brain injuries. In vitro, the levels of a7nAchR were increased in microglia and protected against ischemic injury [42,43]. Additionally, a7nAchR-knockout mice failed to inhibit TNF synthesis in comparison to wild-type mice after utilizing VNS [41], suggesting that a7nAchR plays an important role in the prevention of cerebral ischemia. We found that the protein levels of a7nAchR in the ischemic penumbra were enhanced in the PMCAO+VNS group as compared to the PMCAO group. This may be because cerebral ischemia and hypoxia induced by acute cerebral infarction elicited ATP deficiency and protein synthesis disorder, whereas VNS treatment enhanced the expression of a7nAchR. Interestingly, activation of a7nAchR using the specific agonist preconditioning was positively associated with higher neurological function scores and a smaller infarct volume, which potentially exerted similar therapeutic effects to those that VNS induced. However, VNS therapy significantly aggravated neurological dysfunction and cerebral infarct volume by inhibiting a7nAchR with α-bungarotoxin preconditioning. These results reveal that a7nAchR may serve as an important mediator involved in the process of VNS-induced neuroprotection against acute ischemic damage.
The inflammatory response is a crucial process of brain tissue injury during the acute phases of ischemic stroke, which produces pro-inflammatory cytokines and up-regulates the production of ROS by NADPH oxidase [44,45]. The cellular and molecular mechanisms for anti-inflammation are partly attributable to acetylcholine (Ach), a neurotransmitter mainly released from vagus nerve endings. Previous studies have reported that the activation and up-regulation of Akt phosphorylation was involved in VNS-mediated neuroprotection, suppressing inflammation and apoptosis via cholinergic and a7nAchR/Akt pathways [15]. However, the mechanism of the a7nAchR/JAK2 pathway remains unreported. We observed that the protein levels of a7nAchR, p-JAK2, and p-STAT3 were significantly higher in the PMCAO+VNS group than in the PMCAO groups. In addition, the expressions of p-JAK2 and p-STAT3 were related to a7nAchR expression levels. By inhibiting the a7nAchR expression levels, expression levels of these proteins were significantly decreased, but the expressions of these proteins were significantly increased by activating a7nAchR via VNS. Therefore, these findings indicate that the protective effects provided by VNS in ischemic brain tissue may be partly due to increased expression of a7nAchR, p-JAK2, and p-STAT3.
The JAK2/STAT3 signal transduction pathway plays a critical role in regulating ischemia-induced neuronal inflammation and apoptosis [46]. De Jonge et al. revealed that a7nAChR-mediated JAK2/STAT3 pathway activation contributed to anti-inflammatory effects produced by VNS in rat intestinal inflammation. During this process, STAT3 was phosphorylated by the tyrosine kinase JAK2 that was recruited to the a7 subunit of the nicotinic acetylcholine [22]. Xiong et al. demonstrated that VNS elicits a7nAChR activation to make JAK2 phosphorylated, and then p-JAK2 further activates STAT3 phosphorylation. After that, the p-STAT3 enters the nucleus and competes with NF-κB, thereby reducing the production of pro-inflammatory cytokines in a mouse model of myocardial I/R injury [47]. Taken together, these data suggest that activation of the endogenous cholinergic pathway has an anti-inflammation effect and prevents cell injury, partly due to activation of a7nAchR expressed in the ischemic penumbra and the JAK2/STAT3 pathway in brain, respectively. p-STAT3 was not detected, whereas p-JAK2 was found under the PAMCO-sham condition from this study. This phenomenon may explain why p-JAK2 or p-STAT3 was inactive or absent unless it was triggered by acute cerebral ischemia injury. Therefore, we hypothesized that the potential mechanism by which a7nAchR mediates VNS-induced beneficial neuroprotection against acute ischemic stroke may related to the suppression of the inflammation response via the a7nAChR/JAK2 cholinergic anti-inflammatory pathway.
The strength of our study is the establishment of the PMCAO rat model, which distinguishes it from previous reports. Notably, we show that a7nAChR activated pharmacologically may be an alternative to VNS-mediated neuroprotective effects. Surprisingly, evidence is provided that a7nAChR agonist enhances neural plasticity in the hippocampus, and has advanced into clinical trials for treatment of cognitive impairment [48]. However, there are several limitations of our study. First, a model of acute permanent cerebral ischemia was presented and effects induced by VNS were observed only focusing on 24 h after PMCAO, thus later and continuous effectiveness of VNS was not demonstrated in this study. It is worth mentioning, however, that previous evidence has shown that acute VNS increased the expression of brain-derived neurotrophic factor and fibroblast growth factor in the hippocampus and cortex of rats, which were both beneficial for improving neurogenesis and functional recovery [49]. In a clinical study, Dawson et al. found that VNS paired with upper-limb rehabilitation is feasible for 6 months after cerebral infarction and did not increase safety concerns [50]. Second, our study only carried out with the determination of key proteins, but failed to further detect NF-κB and pro-inflammatory cytokines.
Conclusions
In conclusion, VNS decreased the infarct volume and improved the neurological function in a rat model of acute permanent cerebral ischemia injury by activation of a7nAchR expression in the ischemic penumbra. Inhibition of a7nAchR attenuated the neuroprotection induced by VNS in acute cerebral infarction. In addition, the a7nAchR/JAK2 pathway may be involved in the anti-inflammation processes induced by VNS. These data also provide a striking evidence that activation of a7nAchR, either with VNS or pharmacology, has potential as a novel therapeutic therapy for early intervention to prevent inflammatory injury in acute ischemic stroke.
Footnotes
Source of support: This research was supported by grants from the National Natural Science Foundation of China (grant No. 81301676) and the Natural Science Foundation of Guangdong Province (grant No. 2017A030313663)
Conflict of interest
None.
References
- 1.Czlonkowska A, Lesniak M. Pharmacotherapy in stroke rehabilitation. Expert Opin Pharmacother. 2009;10:1249–5. doi: 10.1517/14656560902941972. [DOI] [PubMed] [Google Scholar]
- 2.Norrving B, Kissela B. The global burden of stroke and need for a continuum of care. Neurology. 2013;80:S5–12. doi: 10.1212/WNL.0b013e3182762397. [DOI] [PubMed] [Google Scholar]
- 3.Fu SH, Zhang HF, Yang ZB, et al. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(1):87–94. doi: 10.1007/s00210-013-0922-8. [DOI] [PubMed] [Google Scholar]
- 4.Wang JY, Shen J, Gao Q, et al. Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke. 2008;39:983–90. doi: 10.1161/STROKEAHA.107.499079. [DOI] [PubMed] [Google Scholar]
- 5.He G, Xu W, Tong L, et al. Gadd45b prevents autophagy and apoptosis against rat cerebral neuron oxygen-glucose deprivation/reperfusion injury. Apoptosis. 2016;21(4):390–403. doi: 10.1007/s10495-016-1213-x. [DOI] [PubMed] [Google Scholar]
- 6.Cheng Y, Leng W, Zhang J. Protective effect of puerarin against oxidative stress injury of neural cells and related mechanisms. Med Sci Monit. 2016;22:1244–49. doi: 10.12659/MSM.896058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erfani S, Khaksari M, Oryan S, et al. Nampt/PBEF/visfatin exerts neuroprotective effects against ischemia/reperfusion injury via modulation of Bax/Bcl-2 ratio and prevention of caspase-3 activation. J Mol Neurosci. 2015;56(1):237–43. doi: 10.1007/s12031-014-0486-1. [DOI] [PubMed] [Google Scholar]
- 8.Sun T, Liu B, Li P. Nerve protective effect of asiaticoside against ischemia-hypoxia in cultured rat cortex neurons. Med Sci Monit. 21:3036–41. doi: 10.12659/MSM.894024. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arle JE, Carlson KW, Mei L. Investigation of mechanisms of vagus nerve stimulation for seizure using inite element modeling. Epilepsy Res. 2016;126:109–18. doi: 10.1016/j.eplepsyres.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Bauer S, Baier H, Baumgartner C, et al. Transcutaneous vagus nerve stimulation (tVNS) for treatment of drug-resistant epilepsy: a randomized, double-blind clinical trial (cMPsE02) Brain Stimul. 2016;9(3):356–63. doi: 10.1016/j.brs.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Conway CR, Sheline YI, Chibnall JT, et al. Brain blood-low change with acute vagus nerve stimulation in treatment-refractory major depressive disorder. Brain Stimuli. 2012;5(2):163–71. doi: 10.1016/j.brs.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ay I, Lu J, Ay H, et al. Vagus nerve stimulation reduces infarct size in rat focal cerebral ischemia. Neurosci Lett. 2009;459(3):147–51. doi: 10.1016/j.neulet.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 13.Ay I, Sorensen AG, Ay H. Vagus nerve stimulation reduces infarct size in rat focal cerebral ischemia: an unlikely role for cerebral blood flow. Brain Res. 2011;1392:110–15. doi: 10.1016/j.brainres.2011.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y, Li L, Liu B, et al. PPARgamma upregulation induced by vagus nerve stimulation exerts anti-inflammatory effect in cerebral ischemia/reperfusion rats. Med Sci Monit. 2015;21:268–75. doi: 10.12659/MSM.891407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Y, Li L, Liu B, et al. Vagus nerve stimulation attenuates cerebral ischemia and reperfusion injury via endogenous cholinergic pathway in rat. PLoS One. 2014;9(7):e102342. doi: 10.1371/journal.pone.0102342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y, Li L, Tan X, et al. miR-210 mediates vagus nerve stimulation-induced antioxidant stress and anti-apoptosis reactions following cerebral ischemia/reperfusion injury in rats. J Neurochem. 2015;134(1):173–81. doi: 10.1111/jnc.13097. [DOI] [PubMed] [Google Scholar]
- 17.Lukas RJ, Changeux JP, Le Novère N, et al. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51(2):397–401. [PubMed] [Google Scholar]
- 18.Drisdel RC, Green WN. Neuronal alpha-bungarotoxin receptors are alpha7 subunit homomers. J Neurosci. 2000;20(1):133–39. doi: 10.1523/JNEUROSCI.20-01-00133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charpantier E, Wiesner A, Huh KH, et al. Alpha7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci. 2005;25(43):9836–49. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saeed RW, Varma S, Peng-Nemeroff T, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201(7):1113–23. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arredondo J, Chernyavsky AI, Jolkovsky DL, et al. Receptor-mediated tobacco toxicity: Cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006;20(12):2093–101. doi: 10.1096/fj.06-6191com. [DOI] [PubMed] [Google Scholar]
- 22.de Jonge WJ, van der Zanden EP, The FO, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–51. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 23.Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 24.Bederson JB, Pitts LH, Tsuji M, et al. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–76. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 25.Smith DC, Modglin AA, Roosevelt RW, et al. Electrical stimulation of the vagus nerve enhances cognitive and motor recovery following moderate fluid percussion injury in the rat. J Neurotrauma. 2005;22(12):1485–502. doi: 10.1089/neu.2005.22.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaz GC, Bahia AP, de Figueiredo MF, et al. Cardiovascular and behavioral effects produced by administration of liposome-entrapped GABA into the rat central nervous system. J Neuroscience. 2015;285:60–69. doi: 10.1016/j.neuroscience.2014.10.067. [DOI] [PubMed] [Google Scholar]
- 27.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26(4):627–34. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 28.Burnett MG, Shimazu T, Szabados T, et al. Electrical forepaw stimulation during reversible forebrain ischemia decreases infarct volume. Stroke. 2006;37:1327–31. doi: 10.1161/01.STR.0000217305.82123.d8. [DOI] [PubMed] [Google Scholar]
- 29.Won S, Lee JH, Wali B, et al. Progesterone attenuates hemorrhagic transformation after delayed tPA treatment in an experimental model of stroke in rats: Involvement of the VEGF-MMP pathway. J Cereb Blood Flow Metab. 2014;34(1):72–80. doi: 10.1038/jcbfm.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong Y, Rogers MR, Qin X. Effective neuroprotection by ischemic postconditioning is associated with a decreased expression of RGMa and inflammation mediators in ischemic rats. Neurochem Res. 2013;38:815–25. doi: 10.1007/s11064-013-0984-5. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Ma J, Jin X, et al. L-PGDS mediates vagus nerve stimulation-induced neuroprotection in a rat model of ischemic stroke by suppressing the apoptotic response. Neurochem Res. 2017;42(2):644–55. doi: 10.1007/s11064-016-2121-8. [DOI] [PubMed] [Google Scholar]
- 32.Cai PY, Bodhit A, Derequito R, et al. Vagus nerve stimulation in ischemic stroke: Old wine in a new bottle. Front Neurol. 2014;5:107. doi: 10.3389/fneur.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neren D, Johnson MD, Legon W, et al. Vagus nerve stimulation and other neuromodulation methods for treatment of traumatic brain injury. Neurocrit Care. 2016;24(2):308–19. doi: 10.1007/s12028-015-0203-0. [DOI] [PubMed] [Google Scholar]
- 34.Chen M, Zhou X, Yu L, et al. Low-level vagus nerve stimulation attenuates myocardial ischemic reperfusion injury by antioxidative stress and antiapoptosis reactions in canines. J Cardiovasc Electrophysiol. 2016;27(2):224–31. doi: 10.1111/jce.12850. [DOI] [PubMed] [Google Scholar]
- 35.Sun Z, Baker W, Hiraki T, Greenberg JH. The effect of right vagus nerve stimulation on focal cerebral ischemia: An experimental study in the rat. Brain Stimul. 2012;5(1):1–10. doi: 10.1016/j.brs.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu AF, Zhao FB, Wang J, et al. Effects of vagus nerve stimulation on cognitive functioning in rats with cerebral ischemia reperfusion. J Transl Med. 2016;14:101. doi: 10.1186/s12967-016-0858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiang YX, Wang WX, Xue Z, et al. Electrical stimulation of the vagus nerve protects against cerebral ischemic injury through an anti-inflammatory mechanism. Neural Regen Res. 2015;10(4):576–82. doi: 10.4103/1673-5374.155430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J, Kang JW, Lee SM. Activation of the cholinergic anti-inflammatory pathway by nicotine attenuates hepatic ischemia/reperfusion injury via hemeoxygenase-1 induction. Eur J Pharmacol. 2013;707:61–70. doi: 10.1016/j.ejphar.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 39.Bonaz B, Sinniger V, Pellissier S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J Physiol. 2016;594(20):5781–90. doi: 10.1113/JP271539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tracey KJ. Physiology and immunology of the cholinergic anti-inflammatory pathway. J Clin Invest. 2017;117:289–96. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–88. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 42.Parada E, Egea J, Buendia I, et al. The Microglial a7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by Inducing Heme Oxygenase-1 via nuclear factor erythroid-2-related factor. Antioxid Redox Signal. 2013;19:1135–48. doi: 10.1089/ars.2012.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shytle RD, Mori T, Townsend K, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89(2):337–43. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 44.Cieslak M, Wojtczak A, Cieslak M. Relationship between the induction of inflammatory processes and infectious diseases in patients with ischemic stroke. Acta Biochim Pol. 2013;60(3):345–49. [PubMed] [Google Scholar]
- 45.Lin C, Wang L, Wang H, et al. Tanshinone IIA inhibits breast cancer stem cells growth in vitro and in vivo through attenuation of IL-6/STAT3/NF-κB signaling pathways. J Cell Biochem. 2013;114(9):2061–70. doi: 10.1002/jcb.24553. [DOI] [PubMed] [Google Scholar]
- 46.Hu GQ, Du X, Li YJ, et al. Inhibition of cerebral ischemia/reperfusion injury-induced apoptosis: Nicotiflorin and JAK2/STAT3 pathway. Neural Regen Res. 2017;12(1):96–102. doi: 10.4103/1673-5374.198992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiong J, Xue FS, Yuan YJ, et al. Cholinergic anti-inflammatory pathway: A possible approach to protect against myocardial ischemia reperfusion injury. Chin Med J (Engl) 2010;123(19):2720–26. [PubMed] [Google Scholar]
- 48.Townsend M, Whyment A, Walczak JS, et al. a7nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J Neurophysiol. 2016;116(6):2663–75. doi: 10.1152/jn.00243.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy TH, Corbett D. Plasticity during stroke recovery: From synapse to behaviour. Nat Rev Neurosci. 2009;10:861–72. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- 50.Dawson J, Pierce D, Dixit A, et al. Safety, feasibility, and efficacy of vagus nerve stimulation paired with upper-limb rehabilitation after ischemic stroke. Stroke. 2016;47(1):143–50. doi: 10.1161/STROKEAHA.115.010477. [DOI] [PMC free article] [PubMed] [Google Scholar]