

Abstract
In the carnivorous plant family Lentibulariaceae, all three genome compartments (nuclear, chloroplast, and mitochondria) have some of the highest rates of nucleotide substitutions across angiosperms. While the genera Genlisea and Utricularia have the smallest known flowering plant nuclear genomes, the chloroplast genomes (cpDNA) are mostly structurally conserved except for deletion and/or pseudogenization of the NAD(P)H-dehydrogenase complex (ndh) genes known to be involved in stress conditions of low light or CO2 concentrations. In order to determine how the cpDNA are changing, and to better understand the evolutionary history within the Genlisea genus, we sequenced, assembled and analyzed complete cpDNA from six species (G. aurea, G. filiformis, G. pygmaea, G. repens, G. tuberosa and G. violacea) together with the publicly available G. margaretae cpDNA. In general, the cpDNA structure among the analyzed Genlisea species is highly similar. However, we found that the plastidial ndh genes underwent a progressive process of degradation similar to the other terrestrial Lentibulariaceae cpDNA analyzed to date, but in contrast to the aquatic species. Contrary to current thinking that the terrestrial environment is a more stressful environment and thus requiring the ndh genes, we provide evidence that in the Lentibulariaceae the terrestrial forms have progressive loss while the aquatic forms have the eleven plastidial ndh genes intact. Therefore, the Lentibulariaceae system provides an important opportunity to understand the evolutionary forces that govern the transition to an aquatic environment and may provide insight into how plants manage water stress at a genome scale.
Introduction
The carnivorous plant Genlisea has astonished scientists for many years. Charles Darwin was seduced by this “remarkable genus” which he described at the end of his book Insectivorous Plants [1]. The genus Genlisea A.St.-Hil. belongs to the carnivorous family Lentibulariaceae together with genera Utricularia and Pinguicula [2]. Genlisea encompass about 30 species that inhabit open areas with nutrient-poor soil distributed in tropical Africa and the Neotropics (eight of twenty species are endemic to Brazil) [3–6]. Genlisea are small, rootless, terrestrial herbs commonly known as “corkscrew plants” due to Y-shaped-underground leaves that are twisted helically and have the ability to capture, digest and absorb prey [7,8]. It is difficult to distinguish different species based solely on the vegetative forms due to Genlisea having a diverse set of intraspecific phenotypes. Despite Darwin’s early interest however, Genlisea remains poorly studied due to cultivation challenges, and being found in isolated and remote habitats [9].
Genlisea and Utricularia have one of the highest nucleotide substitution rates across all three genome compartments (nucleus, chloroplast, mitochondria) in comparison to other angiosperms [10–12] with previous studies revealing that both genera have an exclusive mutation in the mitochondrial cytochrome c oxidase gene (cox1) [13]. These mutations lead to a proton pumping change and, during oxidative phosphorylation, cause electrons to leak into the mitochondria, generating reactive oxygen species (ROS). It is proposed that the ROS can damage DNA, which produces breaks in the double helix structure, leading to point mutations [14–16]. On an evolutionary timescale this potential increase in ROS could explain the high nucleotide substitution rate, the process of genome miniaturization [17], and a high diversification of morphological traits [14].
Previous systematic studies were carried out using morphological traits, mainly based on capsule dehiscence together with trap, pollen, flower characteristics [4,18,19] and molecular markers from the three plastidial loci: trnK/matK, rps16 and trnQ-rps16. Phylogenies based on these markers suggested two major groups within Genlisea: the subgenus Genlisea, comprising the sections Genlisea, Africanae and Recurvatae, and the subgenus Tayloria. However, due to the recent discovery of new species, unresolved clades and possible cryptic species, the evolutionary history of Genlisea requires further investigation [4,18].
Chloroplast genome (cpDNA) sequencing and analysis of different species provides a powerful tool to dissect out the evolutionary history of plant genera. The highly conserved structure and gene content of the cpDNA enable plant evolution and phylogeny studies [20]. Structural rearrangements, gene decay and loss are often observed in cpDNA and inform a plethora of evolutionary relationships among different taxa. For example, plastid gene loss in the most extreme cases is linked to lineages with heterotrophic nutrition, such as parasitic [21] and mycoheterotrophic plants [22].
One of the gene losses that occur in such plants is related to the NAD(P)H-dehydrogenase complex (ndh) genes. The ndh genes consist of eleven (11) subunits in the cpDNA (ndhA, B, C, D, E, F, G, H, I, J and K) that encodes, along with nuclear genes, the thylakoid NAD(P)H dehydrogenase complex [23]. This complex is involved in photosynthesis, the photosynthetic response and stress acclimation [24], and has been hypothesized to be related to the transition to terrestrial habitats [14,16]. The eleven ndh subunit genes are present in the aquatic Lentibulariaceae species, but are lost in the terrestrial Utricularia species, suggesting that the evolutive history of the ndh genes among the Utricularia lineages followed an opposite trend, and that the ndh function may be dispensable in terrestrial forms [25]. However the presence and absence of the ndh genes remain to be established in Genlisea species. Therefore, the ndh genes in the cpDNA can provide a valuable resource for the understanding of Genlisea evolution and how these genes can be associated to the habitats.
To better understand the evolutionary history of the Genlisea genus and explore the role of ndh gene loss, we sequenced, assembled six chloroplast genomes and, together with the published G. margaretae cpDNA, carried out a full analysis. These seven Genlisea species represent both subgenera Tayloria (G. violacea) and Genlisea (G. aurea, G. filiformis, G. pygmaea, G. repens, G. tuberosa and G. margaretae). We found that the chloroplast genome is highly similar across species, but unlike their aquatic relatives, in the terrestrial Genlisea species the ndh genes are deleted, fragmented or pseudogenized. These findings not only add to the understanding of terrestrial heterotrophic plants, and their cpDNA evolution, but also provide an important opportunity to understand the evolutionary forces that govern the transition to an aquatic environment at a genome scale.
Material and methods
Plant samples, preparation and sequencing
Fresh photosynthetic leaves of Genlisea species were sampled from natural populations and also cultivated and stored in silica gel. Total DNA was extracted using modified CTAB protocol and concentration, integrity and purity was assessed using NanodropTM spectrophotometer (Thermo Scientific) and Agilent 2100 Bioanalyzer (Agilent Genomics). Herbarium vouchers are deposited at the Herbarium JABU at Universidade Estadual Paulista (UNESP/FCA; ICMBio/ MMA for collecting permits SISBIO #26938 and #48516) (S1 Table).
The paired-end libraries were prepared using Illumina library preparation manufacturer’s protocol and genomic DNA was sequenced using Illumina Miseq Platform (Illumina, San Diego, CA).
The publicly available Genlisea aurea DNA sequencing data was obtained from raw genome database SRA (accession number SRR916071) that was previously used for nuclear genome assembly [26].
Assembly and annotation
The quality of raw reads was assessed by FastQC [27]. Removal of adapters from both ends and trimming to obtain high quality reads were performed using the Platanus_trim (v.1.0.7) [28] with Phred quality score of >30 and length cutoff of 150bp for 300bp reads, 100bp for 150bp reads, 80bp for 100bp reads and 50bp for 75bp reads (see S1 Table). In addition, to exclude nuclear and mitochondrial genomes, the Genlisea species chloroplast genome paired end reads were extracted by mapping all raw reads to the reference cpDNA Utricularia gibba (NC021449) with Bowtie2 (v.2.2.3) [29] (i.e.–very-sensitive-local with–N 1 modification). Then this selected set of reads was assembled using Spades (v.3.7.1) [30] software with default parameters. Uncertain regions, such as IR junctions, were picked out from published Lentibulariaceae species (U. gibba and Genlisea margaretae [NC025652.1]) to extend the length using iteration method with MITObim (v.1.8) [31]. As the assembly usually collapses the inverted repeats in one single contig, the IR region of some species were manually inverted and duplicated to integrate the whole chloroplast genome using BioEdit (v.7.2) [32]. High quality filtered reads were mapped back using Bowtie2 (i.e.–very-sensitive; end-to-end) in Geneious Pro (v.10.2.3) [33] to each assembled chloroplast genome to confirm assembly accuracy quality and repeat region junctions (S1 Table; S1 Fig).
The annotation of the chloroplast genomes were performed using Dual Organellar GenoMe Annotator (DOGMA) [34] with manual corrections for start and stop codons and intron boundaries by comparison to homologous genes from sequenced chloroplast of Utricularia gibba, U. reniformis (NC029719.2) and Genlisea margaretae. The tRNA genes were also verified with ARAGORN [35] and tRNAscan-SE [36]. The codon usage was calculated using CodonW (v1.4.4) [37].The circular chloroplast genome maps were drawn using OrganellarGenome DRAW tool (OGDRAW) [38].
To determine whether a gene was a pseudogene, fragmented or deleted gene, Blastn and Blastx searches were performed using other chloroplasts as reference, such as U. gibba, and a pseudogene was characterized according to the absence of start and/or stop codon, frameshift and genes with more than 20% of the coding region in comparison to other related species. The genes that are considered as fragmented were any group of nucleotides that had at least >25bp and had correspondence to position and blastn and tblastx alignment with the complete gene.
Repeat identification
REPuter [39] was used to search both direct and palindrome sequences, with a minimum repeat size of > 30bp and a sequence identity greater than 90% (parameters: repfind–f–p–l 30 –h 3 –best 10,000). Microsatellites for mono-, di-, tri-, penta- and hexanucleotides were detected using the Perl script MISA [40]. The established parameters were performed according with Silva et al. [25].
Identity and variation analyses
The chloroplast genomes were aligned using MAFFT (v.7) [41] with FFT-NS-2 parameters and identity comparisons between chloroplasts were conducted with mVISTA program [42].
Average p-distances were calculated to determine genetic divergence between Genlisea species and the number of phylogenetically informative characters (PICs) for each plastome gene, intergenic spacers, introns and pseudogenes using PAUP (v.4b10) [43]. Nonparametric Spearman test was used to test for correlation between PICs and average p-distances between sequences of Genlisea species.
Phylogenomic analyses
Phylogenetic analyses were performed to different partitions by using the whole chloroplast genome sequence, protein coding genes, intergenic spacers, LSC (Large Single Copy), SSC (Small Single Copy), IR (Inverted Repeat) and ndh genes. For ndh phylogenetic tree, pseudogenes and fragments of deleted genes of at least 25bp were considered (S2 and S3 Tables). Previously published Lentibulariaceae chloroplast genomes were included (Utricularia foliosa [KY025562], U. gibba [NC021449], U. macrorhiza [NC025653], U. reniformis [NC029719.2] and Pinguicula ehlersiae [NC023463]) and Tectona grandis (Lamiaceae) [NC020098], Sesamum indicum (Pedaliaceae) [NC016433] and Tanaecium tetranolobum (Bignoniaceae) [NC027955] cpDNA used as outgroup.
The alignments were conducted using MAFFT (v.7) [41] and the evolutionary model (best-of-fit) that was most appropriate for all the data according with corrected Akaike Information Criterion (AICc), calculated using jModelTest [44].
Maximum parsimony criterion was performed using PAUP (v.4b10) [43] with heuristic searches of 2,000 replicates and bootstrap analysis with 1,000 pseudoreplicates, both using the tree bisection-reconnection branch swapping (TBR) and random addition of sequences. The probabilistic analysis was conducted using RAxML (v.8) [45] for maximum likelihood (ML) using the default parameters with bootstrap support of 1,000 pseudoreplicates and MrBayes (v.3) [46] for Bayesian inference with 5×105 generations with two runs and four chains following the substitution matrix assessed as mentioned above. Both analyses were performed on CIPRES Science Gateway website [47] and cladograms were edited with the program TreeGraph2 (beta v.2.0) [48].
In an attempt to test also the phylogenetic signal of ndh genes in Genlisea lineages, we created a matrix with 22 characters. The characters 1 to 11 we codified if each ndh gene (ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, and ndhK) was absent (0) or present (1) and the characters 12 to 22 if each gene was pseudogenized (0), decayed (1) or complete (2) for each of the eleven ndh genes and carried out a parsimony analysis (S2–S4 Tables). The consensual tree (strict consensus) of most parsimonious trees was presented and evolution of ndh genes was traced using both matrix and chloroplast phylogenomic tree described above with PAUP (v.4b10) with ACCTRAN optimization [43].
Results
Genome content and organization of the six Genlisea chloroplast genomes
The cpDNA of Genlisea ranged from 140,010 bp (G. aurea) to the largest plastome of the sequenced species with 143,416 bp (G. violacea) (Fig 1, Table 1). All six chloroplast genomes display a quadripartite structure, which consists of a pair of inverted repeats (IR) separated by a Large Single Copy (LSC) and a Small Single Copy (SSC) region. The plastomes contain 103 unique genes, including 69 protein-coding genes, 30 tRNAs, 4 rRNAs and the average GC content was 38.57±0.08%. Fourteen genes contain a single intron, such as atpF, petB, petD, rpl16, rpl2, rpoC1, rps12, rps16, trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA and trnV-UAC, while clpP and ycf3 have two introns. The orf42, orf56, and ycf68 genes of the IR region are pseudogenes due to lack of start and/or stop codons (Table 2).
Fig 1. Physical chloroplast genome maps of six assembled Genlisea species.
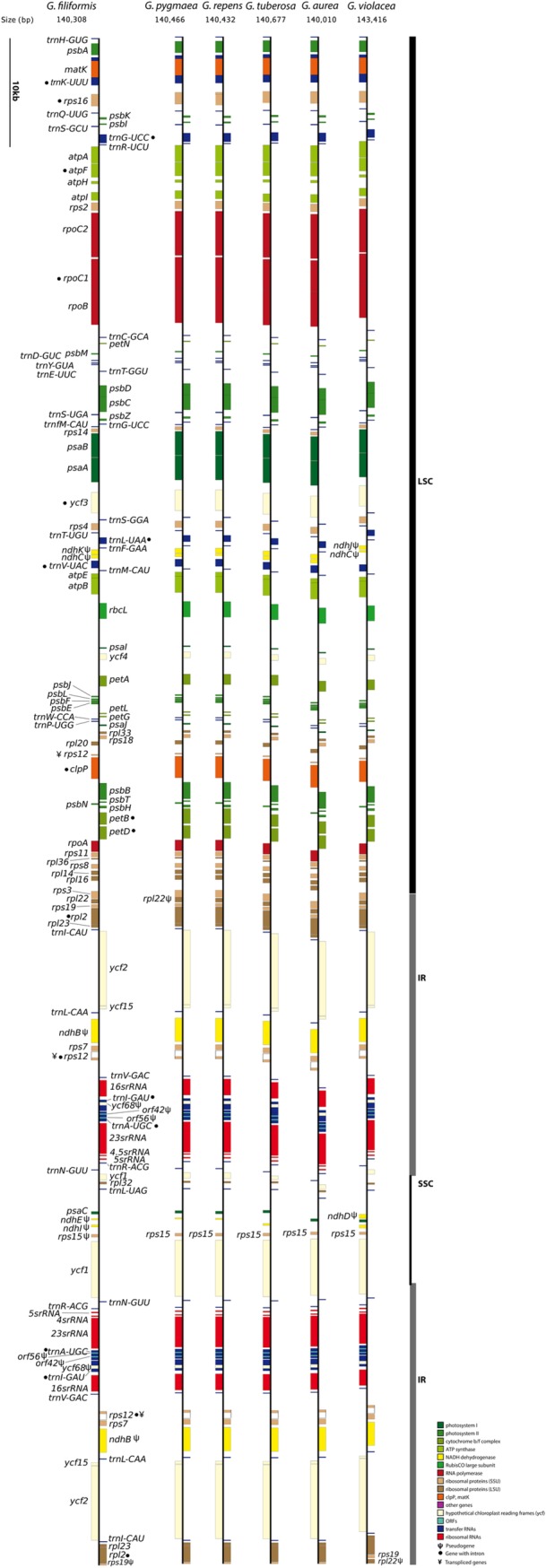
The chloroplast genome is showed with the genes colorized according to the functional classes for each species. The genes shown on the right side of each cpDNA map are transcribed clockwise, whereas gene on the left side are transcribed counter clockwise. The symbol Ψ after the gene name indicates that is a pseudogene, • the presence of introns and ¥ denotes transpliced genes. Large single copy (LSC), inverted repeats (IR) and single copy repeat (SSC) are represented by the black and grey bars.
Table 1. Summary of assembly data for Genlisea plastomes (for details about sequencing data see S1 Table).
| Species | cpDNA size (bp) | LSC size (bp) |
SSC size (bp) |
IRs size (bp) |
GC content (%) |
GenBank accession number |
|---|---|---|---|---|---|---|
| Genlisea aurea | 140,010 | 80,653 | 9,419 | 24,969 | 38.5 | MF593121 |
| G. filiformis | 140,308 | 79,754 | 10,316 | 25,119 | 38.7 | MF593122 |
| G. pygmaea | 140,466 | 79,888 | 10,346 | 25,116 | 38.6 | MF593123 |
| G. repens | 140,432 | 79,875 | 10,325 | 25,116 | 38.5 | MF593124 |
| G. tuberosa | 140,677 | 80,347 | 10,462 | 24,934 | 38.5 | MF593125 |
| G. violacea | 143,416 | 81,089 | 10,969 | 25,679 | 38.6 | MF593126 |
Table 2. Genes in the six Genlisea chloroplast genomes (except G. margaretae).
| Category of genes | Group of gene | Name of the gene |
|---|---|---|
| Self-replication | Ribosomal RNA genes (rRNAs) | 4.5S rRNA (2x), 5S rRNA (2x), 16S rRNA (2x), 23S rRNA (2x) |
| Transfer RNA genes (tRNAs) | trnH-GUG, trnK-UUU●, trnQ-UUG, trnS-GCU, trnG-UCC●, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCC●, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAA●, trnF-GAA, trnV-UAC●, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnL-CAA (2x), trnV-GAC (2x), trnI-GAU ● (2x), trnA-UGC ● (2x), trnR-ACG (2x), trnN-GUU (2x), trnL-UAG | |
| Small subunit of ribosomal protein | rps2, rps3, rps4, rps7 (2x), rps8, rps11, rps12● (2x) ¥, rps14, rps15**, rps16●, rps18, rps19*** | |
| Large subunit of ribosomal protein | rpl2● (2x), rpl14, rpl16●, rpl20, rpl22*, rpl23 (2x), rpl32, rpl33, rpl36 | |
| RNA polymerase subunit | rpoA, rpoB, rpoC1●, rpoC2 | |
| Photosynthesis | NADH dehydrogenase | All are ψ or deleted (see Figs 1 and 4 for each Genlisea species) |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ, ycf3●, ycf4 | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Cytochrome b/f complex | petA, petB●, petD●, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpF●, atpH, atpI | |
| Rubisco large subunit | rbcL | |
| Other genes | Translation initiation factor | infA |
| Maturase | matK | |
| Protease | clpP● | |
| Envelope membrane protein | cemA | |
| Subunit of acetyl-CoA-carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Unknown function | Conserved hypothetical protein | ycf1, ycf2 (2x), ycf15 (x2), ycf68 ψ (2x), orf56 ψ (2x), orf42 ψ (2x) |
● Gene with intron
ψ Pseudogenes
¥ Transpliced genes.
* One of duplicated gene is partial in G. violacea and is pseudogene in G. pygmaea
**Pseudogene in G. filiformis
*** Duplicated gene in G. violacea.
Overall, all the Genlisea cpDNAs are highly conserved in organization and structure (Fig 1), except for the ndh genes that are pseudogenized, fragmented or deleted in all Genlisea plastomes. In addition, G. violacea has slightly expanded IR/LSC boundary genes with the duplication of intact rps19 gene and rpl22 as pseudogene (see S2 Fig), and the rps15 and rpl22 are present as pseudogenes in G. filiformis and G pygmaea, respectively (Fig 1).
Repeats in the Genlisea plastomes
Repeats were divided in three categories: tandem, direct and palindromic (Fig 2). The great majority of the repeats across the chloroplast genomes were simple sequence repeats (SSRs) of lengths between 7 and 20 bp. An average of 210 repeats were detected in the six chloroplast genomes, 6.80% (69 repeats) of which are direct repeats, 5.80% (59 repeats) were palindromic repeats, and 87.40% (888 repeats) tandem repeats (Fig 2; S5 Table). Moreover, most of the repeats are located in the intergenic regions (39.40%), followed by coding (36.60%) and intronic regions (14.90%). Few repeats were found in tRNA, rRNA and pseudogenes regions (9.10%). The majority of microsatellites in all species are A/T mono- and dinucleotides. There are few tetra- and pentanucleotide and one hexanucleotide in G. pygmaea. Among all chloroplast genomes, 41 repeat regions (4%) were shared by all analyzed Genlisea species (S5 Table).
Fig 2. Analysis of repeats in Genlisea chloroplast genomes.

(A) Quantity of tandem, direct and palindromic repeats of each species. (B) Quantity of repeats by length.
Molecular markers identification
Genome wide comparison allowed the identification of genomic regions that could be used as possible phylogenetic markers to reconstruct the evolutionary history of the genus. A positive correlation between the percentage of variable sites, given by p-distance, and phylogenetically informative characters (PICs) (ρ = 0.583, P<0.001; S3 Fig) were identified. Thus, the PICs of each coding and non-coding alignment region were used to identify potential regions for phylogenetics and population studies.
The divergence hotspot analysis given by p-distance and phylogenetically informative characters (S6 Table) revealed that the most informative regions for phylogenetic analyses were non-coding DNA regions such as intergenic spacers and introns (Fig 3; S4 Fig). Moreover, the p-distance between Genlisea and Pinguicula was 0.043, Genlisea and Utricularia 0.057 and between Genlisea species was of 0.032. The overall p-distance between G. repens and G. pygmaea, the most related species in this study, was 0.001. Phylogenetically informative characters suggest that the top ten regions with the greatest number of PICs are three genes (ycf1, matK and rpoC2), two introns (rpl16-intron, trnK-intron) and five intergenic regions (trnK-rps16, rps12-clpP, petA-psbJ, rpl20-rps12, rps12-trnV) (S4 Fig; S6 Table).
Fig 3. Sequence identity plots for the six assembled Genlisea species and previously published G. margaretae.

Phylogenomic analysis
Regarding the Lentibulariaceae, the topologies were totally congruent for all chloroplast dataset partitions (LSC, IR, SSC, coding regions, intergenic spacers and introns; S5 Fig). The whole chloroplast alignment resulted in 178,161 characters of which 21,687 are informative sites (Table 3). The most parsimony, Bayesian (BS) and maximum likelihood (ML) trees are highly congruent with very high support (ML bootstraps and posterior probabilities mostly 100) and support Lentibulariaceae as a monophyletic group, and Genlisea–Utricularia as sister clade with Pinguicula. When all branch lengths for each cladogram are visualized, the IR tree depicts very short branches (S5 Fig), resulting from the lowest proportion of variable sites (9%; Table 3). These results support that the Genlisea genus is monophyletic and its topology follows previous phylogenetic studies [18]: subgenus Tayloria (represented by G. violacea) as a sister clade to subgenus Genlisea (G. margaretae, G. filiformis, G. pygmaea, G. repens, G. tuberosa and G. aurea) (Fig 4). Moreover, the phylogenetic analyses based on the ndh genes partition, which treated each nucleotide ordinarily as a character, reveals a topology totally congruent to the trees resulting from other partitions and whole plastomes (Fig 4; S5 Fig). Also, when the processes that could be involved in the ndh degeneration (pseudogenization and decay) were codified in a multistate character matrix (see S2–S4 Tables); the resultant tree (Fig 5B) was mostly congruent with the nucleotide-by-nucleotide tree (Figs 4 and 5A).
Table 3. Datasets and phylogenetic statistics for each Genlisea cpDNA partition.
| Whole chloroplast | LSC | SSC | IR | Protein coding | Intergenic spacers | Introns | ndh genes | |
|---|---|---|---|---|---|---|---|---|
| Alignment (bp) | 178,161 | 99,235 | 20,156 | 28,636 | 67,437 | 46,068 | 15,090 | 9,462 |
| Overall GC content (%)–Only Genlisea species | 38.5 | 36.4 | 30.5 | 43.5 | 40.4 | 32.5 | 36.1 | 35.7 |
| Overall GC content (%)–Genlisea + outgroup | 38.1 | 36.1 | 31.3 | 43.1 | 40.4 | 32 | 35.9 | 35.2 |
| Variable sites (%) | 40,427 (22%) | 27,508 (27%) | 7,753 (38%) | 2,752 (9%) | 12,817 (19%) | 15,502 (33%) | 4,057 (26%) |
1,944 (20%) |
| Informative sites (%) | 21,687 (12%) | 15,218 (15%) | 4,275 (21%) | 1,140 (4%) | 6,909 (10%) | 8,616 (18%) | 2,360 (15%) | 535 (6%) |
| Consistency index (CI) | 0.856 | 0.852 | 0.837 | 0.922 | 0.845 | 0.855 | 0.847 | 0.976 |
| Retention index (RI) | 0.875 | 0.876 | 0.847 | 0.919 | 0.868 | 0.875 | 0.881 | 0.948 |
| Model of substitution (AICc) | GTR+G+I | GTR+G+I | TVM+G+I | TVM+G+I | GTR+G+I | TVM+G+I | GTR+G+I | TVM+G |
Fig 4. Phylogenomics of whole chloroplasts of Genlisea species and ndh genes evolution.

The boxes indicate the ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ and ndhK genes. Black boxes denote intact genes, yellow boxes pseudogenized genes, red boxes fragmented and white boxes indicate deleted genes. Blue lines indicate the aquatic Utricularia species with complete ndh repertoire. Numbers of support values are all 100% for Bayesian inference, maximum likelihood and maximum parsimony bootstrap, except for outgroup clade S. indicum and T. tetranolobum with parsimony bootstrap value of 85%.
Fig 5. Phylogenetic hypothesis based on ndh sequences.

A. Analyses based on ndh sites (nucleotide-by-nucleotide). In this analysis, each nucleotide was used as one character (e.g. char1, char2, char3) B. Strict consensus of the two most parsimonious trees (33 steps; IC = 0.70; IR = 0.78) based on the matrix codified for ndh patterns. In this analysis, each ndh gene was applied to two characters: one codified as absent (state 0) or present (state 1) (characters 1 to 11) and other codified as pseudogenized (state 0), decayed (state 1), complete (state 2) and inapplicable (state “-”, when the gene is deleted) (characters 12 to 22). For details see S2, S3 and S4 Tables.
Discussion
Chloroplast genomes are a powerful tool to understand the evolutionary forces acting on a species because their structure and sequence are highly constrained across flowering plants. The carnivorous plant family Lentibulariaceae has been shown to have a high rate of nucleotide substitution in all three genome compartments, including the chloroplast genome [10]. In this study we describe seven Genlisea cpDNA including both subgenera within carnivorous plant Genlisea: subgen. Tayloria (G. violacea) and subgen. Genlisea (G. aurea, G. filiformis, G. pygmaea, G. repens, G. tuberosa and G. margaretae).
The Genlisea cpDNA have typical quadripartite structure with a similar gene repertoire, as previously described for other Lentibulariaceae [25,49,50]. However, we do find that the ndh genes are deleted, fragmented or pseudogenized, which provides new insight into the evolutionary trajectory of Genlisea as well as the terrestrial forms of the Lentibulariaceae.
Even though cpDNAs are structurally conserved, changes in genome composition have been identified in many species of angiosperms [51] and also in some gnetophytes [52]. These variations are principally due to the expansion and contraction of IR and SSC regions [53] and gene loss and duplicated genes in IR/SC or IR/LSC boundaries [54]. Among the six cpDNAs described in this study and the previously published Genlisea margaretae cpDNA [50], G. violacea proved to be the most divergent from the other Genlisea species with possible IR expansion that includes duplication of rps19 gene and partial duplication of rpl22 gene. In addition, G. filiformis and G. pygmaea showed pseudogenization of rps15 and rpl22 genes, respectively. However, the absence of these genes is observed in other angiosperms. For instance, the rpl22 gene was loss in several cpDNA, such as legumes [55,56], Gossypium [57], Citrus [58], Castanea [59], Quercus [60] and Passiflora species [61]. Moreover, some studies suggest that there is strong evidence that the rpl22 gene has been transferred to the nucleus in some angiosperms [56,60].
The GC content among seed plant plastomes ranges between 34–40% and, comparing each cpDNA region, the SSC is the one with the lowest GC content [51]. For the Genlisea cpDNAs, we also found that the SSC had the lowest GC content (31.3%). One explanation for the SSC having the lowest GC content is that this region is susceptible to nucleotide substitutions, which is consistent with the high level of nucleotide variation (38%) we observed, compared to other cpDNA regions (Table 3).
The codon usage in Genlisea plastomes is similar to that reported for other Lentibulariaceae family cpDNA. Approximately 19,268 codons represent the coding repertoire of the protein coding regions (S7 Table). Codons frequency that ends with A and T have higher usage than G and C ending codons. For all plastomes the most frequent codon was Leucine (with approximately 1,989; 10.35%), whereas the least frequent was Cysteine (approximately 210–1.10%).
The identification of phylogenetically informative characters (PICs; including the parsimony informative characters) is an important procedure for evaluating characters with phylogenetic signal. Indeed, the PICs are represented by the synapomorphies [62,63] rather than nucleotide changes lacking phylogenetic signal. In this context, the results presented in this study support that the cpDNA is a powerful source of information for phylogenetic inferences. For Genlisea, two previous phylogenetic studies employed the cpDNA loci trnK/matK and rps16 [4,18]. Our study suggests that other cpDNA regions (such as ycf1, rpl20-rps12, rpoC2) have more PICs and consequently have higher phylogenetic signal than previously considered sequences used to assess phylogenies and populations studies.
According to the Consortium for the Barcode of Life’s (CBOL), further studies are necessary to define the best DNA sequences for DNA barcoding of plants [64,65]. As many plants have poor resolution at the population level, previous studies have proposed using combinations of loci (as matK, rbcL, trnH-psbA), suggesting that no unique region exists [65,66]. However, a recent study suggested a single region in ycf1 gene [67] could be used as a better barcode. Our PIC and divergence analysis corroborate usage of ycf1 and/or matK for barcoding purposes, since ycf1 is the first PIC classification and matK is the sixth (S6 Table).
Widely used in plant genotyping [68,69], SSRs are an important source of genetic variation that can be used for species discrimination, population structure and genetic diversity [69]. Similarly to our findings for Genlisea species, previous studies on cpDNA SSRs of Lentibulariaceae [70], reported that the chloroplast genomes have a large number of SSRs [25,50]; similarly, we find many SSRs across Genlisea species. Long repeats, represented by direct repeats and palindromic repeats can cause hairpin structures, which are associated with recombination, and can contribute significantly to rearranged gene order and addition of polymorphism [71,72]. In the evaluated Genlisea species, the long repeats were mainly found in non-coding regions, which is consistent with most angiosperms [73]. And, although long repeats are rare in Lentibulariaceae [50], both the smallest (G. aurea) and the largest chloroplast genomes (G. violacea) have a high number of direct repeats and palindromic repeats. In the G. violacea chloroplast genome, regions with palindromic repeats are found near the LSC/IR junctions, suggesting they could be contributing to IR expansion.
In Utricularia reniformis [25], repeat hotspots seem to be associated with ndh gene degradation, since some repeats regions are close to ndh genes. However, in Genlisea the repeats are dispersed over the cpDNA indicating that, for this genus, there is no relationship between the repeats and ndh pseudogenization. This observation suggests that, unlike Utricularia, different evolutionary processes are acting in the Genlisea ndh loci.
Different dataset partitions (IR, LSC, SSC, coding regions, intergenic spacers and introns; S5 Fig), recovered the same tree topology for Lentibulariaceae with high clade support. Indeed, all datasets contained a considerable percentage of informative characters, thus phylogenetic signal can be found along the whole Genlisea cpDNA.
The eleven ndh genes present in all Genlisea species are pseudogenized, decayed or even deleted (Fig 5; S4 Table). ndh genes losses have been found a few times in other taxa and are attributed to heterotrophic plants [23], some conifers [52], orchids [74], and other species of Lentibulariaceae [25,49,50]. And even with the remarkable degradation of ndh genes, the nucleotide composition of ndh still provides sufficient signal for a phylogenetic analysis (Figs 4 and 5). As such, the topology of ndh phylogenetic tree reveals the cladogenetic separation of different subgenera (Tayloria and Genlisea) and resolution of all Genlisea species (Fig 5). While in some orchids [74] the ndh losses seem to have no relation with taxonomy, and environment where these species are found, in Lentibulariaceae the ndh genes appear to have been maintained in aquatic taxa [25,49,50].
When the ndh gene events (arisen, pseudogenization, decay or deletion) are traced in the total evidence (entire plastomes) phylogenetic analysis (Fig 4), we can verify a different scenario when comparing Genlisea lineages to Pinguicula and Utricularia lineages. The terrestrial taxa Pinguicula (represented by the P. ehlersiae) and U. reniformis have most ndh genes as pseudogenized or deleted. Interestingly, the clade represented by the aquatic species of Utricularia (U. foliosa, U. macrorhiza, and U. gibba) has gained, probably as independent (or not) reversion events (Fig 4; clade denoted with blue lines), the almost entire ndh repertoire. We have previously shown that aquatic species of Utricularia have maintained and conserved ndh genes [25]. The ndh genes activity appears dispensable under favorable conditions, as pointed out by transcriptomic studies [75] and verified in knock-out mutants [76–78]. But, episodes of abiotic stress can impact terrestrial habitats and, according to Ruhlman et al. [75], appear to be the cause of retention of ndh genes. Nonetheless, our phylogenetic hypothesis shows that Lentibulariaceae follows an opposite trend, since terrestrial species of Pinguicula, U. reniformis and all seven Genlisea possess degenerated ndh genes and the aquatic species of Utricularia, on the other hand, display a conserved ndh repertoire. Moreover, it is important to emphasize that the aquatic environment also provides a stressful habitat for plants, since these habitats can present low carbon and light availability, anoxia, wave exposure, significant restrictions to sexual reproduction, and sometimes also osmotic stress and limited nutrient supply [79]. Thus, the complete recovery of all eleven genes for the aquatic Utricularia supports the hypothesis that the ndh genes are conserved in stressful habitats.
The trend of decay and deletion of the ndh genes, represented within the different lineages of Genlisea is remarkable. The Genlisea clade presented the highest concentration of fragmented and deleted ndh genes, when compared to Utricularia and Pinguicula species (Fig 4). In an attempt to phylogenetically test this tendency of ndh genes to degrade in Genlisea lineages, we codified the state (present, pseudogenized, decayed or deleted; S2–S4 Tables) for each of eleven plastidial ndh genes and carried out a parsimony analysis. The consensual topology of both most parsimonious trees (Fig 5A and 5B) also supports this hypothesis when compared with the total evidence tree (Fig 4).
As in most Lentibulariaceae cpDNA, the loss of ndh genes does not seem to affect plant fitness despite the harsh environmental conditions common for the carnivorous habit [25,50]. However, as seen in the present study, it has been reported that in terrestrial species most of ndh genes were lost for Lentibulariaceae species (Fig 1).
Silva et al. [80] identified several pseudogenes of plastid origin in U. reniformis mtDNA. For instance, the presence of the ndhJ-ndhK-ndhC loci in the mtDNA supports the hypothesis of lateral transfer since these genes are absent in the cpDNA [80]. Similar translocation of ndh genes from the plastome to the mitochondrial genome was also suggested to the Epidendroideae orchid Erycina pusilla [81]. According to this study, other than the ndh genes could be transferred to mtDNA, since more than 76% of the cpDNA genome was transferred into the mtDNA genome of E. pusilla and the largest cpDNA insertion into the mtDNA genome in this species was 12kb.
In addition to the transfer of plastid genes to the mtDNA, cpDNA genes can also be transferred to the nuclear genome [82]. With the G. aurea nuclear genome published [26], we performed blastn and tblastn searches of all plastidial ndh genes subunits and none of these genes were also found in nuclear assembled scaffolds. However, one cannot discard the idea that these genes are present in the mtDNA. This hypothesis has to be further investigated since the mitochondrial genome is not available [26].
Studies have pointed out the function of ndh genes for modulating ROS in chloroplasts [23]. Plants with high expression of ndh genes also have an increasing concentration of ROS, which can lead to the cell death [83]. Assuming that terrestrial environments are less stressful than aquatic ones [79], the presence of complete ndh repertoire is understandable for aquatic species of Lentibulariaceae, since these genes are important for ROS modulation in the presence of their high respiratory rates. However, only the aquatic Lentibulariaceae species of Utricularia have had their respiration rates measured [84]. More chloroplast genomes from the Genlisea and Utricularia lineages are required to test this hypothesis. But the oxidant activities of ROS are well known for DNA [14,85,16] and it is not difficult to suppose their deleterious action even in genomes from different compartments.
Conclusions
Here we report the chloroplast genome of six Genlisea species of both subgenera: Tayloria and Genlisea. These genomes were compared with the previously published G. margaretae cpDNA, showing that they are very similar in content and have the same gene order and quadripartite structure. Phylogenomic analysis showed that using coding regions, non-coding regions and even decayed ndh sequences it is possible to obtain the evolutionary history with great congruence, recovering with high support the position of assessed taxa in Genlisea genus and Lentibulariaceae family. Importantly, we corroborate previous observations that distinct from the aquatic taxa of Lentibulariaceae, the terrestrial Genlisea chloroplast genomes showed a pseudogenization and a progressive degradation of ndh genes, as reported for other Lentibulariaceae. In summary, we propose that the Lentibulariaceae system provides an important opportunity to understand the evolutionary forces that govern the transition to an aquatic environment, and may provide insight into how plants manage water stress at a genome scale. These findings may have implications for engineering crop species for better water stress tolerance, both too much and too little water.
Supporting information
All quality-trimmed reads from sequencing data sets have been mapped back to the reconstructed plastid supercontig. The upper plot indicates the identity per site and the lower plot shows the coverage plot per species.
(DOCX)
Note the product of G. violacea cpDNA that presents the duplication of rps19 gene and rpl22 as pseudogene (amplicon with 1,194 bp), while the other species present an expected product with ~490 bp. (Amplification reactions of the rpl2-trnH(GUG) marker were conducted in 25 μL of the solution containing 20 mM of MgCl2, 100 mM of dNTPs, 10 mM of each primer, 1 U of Dream Taq Polymerase–Fermentas, and 50 ng of DNA template. The thermal profile for amplification was 1min at 94°C; 35 cycles of 40s at 94°C, 20s at 64°C, 90s at 72°C, and 5min of final extension at 72°C. Forward primer = 5’-AGT CGG ACA AGT GGG GAA TG-3’; reverse primer = 5’-GGA TGT GGC CAA GTG GAT CA-3’).
(DOCX)
Statistics from Spearman correlation tests are given near the corresponding trend lines.
(DOCX)
PIC values are represented as bars and cpDNA region is marked by colors. Black dots represent p-distance. Only PIC of ndhs were not calculated to avoid p-distance alignment artefact (see S6 Table).
(DOCX)
Numbers above are parsimony bootstrap (left), maximum likelihood bootstrap (right) and posterior probability values are represented below. Lamiales species were used as outgroup.
(DOCX)
(DOCX)
(DOCX)
The characters were codified according the S2 Table. (G. = Genlisea; P. = Pinguicula; U. = Utricularia).
(DOCX)
Numbers within table refer to sequence length (bp). Colors refer to the state of character: white–deleted gene; yellow–pseudogenized; pink–decayed gene; grey–complete gene; n/a–absent.
(DOCX)
F–Direct repeats; P–Palindromic repeats; T–Tandem repeats (inside parenthesis the repeated nucleotide). Common genes with repeats between the six species are highlighted with yellow background color in G. aurea table.
(DOCX)
Deleted ndh genes in all Genlisea species and boundaries between ndh pseudogenes are uncertain and were not included in this analysis (represented as n/a).
(DOCX)
(DOCX)
Acknowledgments
The authors thank Dr. T.S. Balbuena and Dr. J.A.M. de Souza for their careful reading of our manuscript and their helpful comments and suggestions.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
SRS was supported with fellowship by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). VFOM and AMV thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp – Proc. #2013/05144-0 and #2013/25164-6) and VFOM the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for the fellowship (Bolsa de Produtividade - Proc. #309040/2014-0). The funder (J. Craig Venter Institute and 10X Genomics) provided support in the form of salaries for authors TPM and EJM, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section. The other funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Darwin C. Insectivorous plants D. Appleton and Company, London; 1899. [Google Scholar]
- 2.Jobson RW, Playford J, Cameron KM, Albert VA. Molecular Phylogenetics of Lentibulariaceae Inferred from Plastid rps16 Intron and trnL-F DNA Sequences: Implications for Character Evolution and Biogeography. Syst. Bot. 2003;28: 157–171. [Google Scholar]
- 3.BFG—The Brazil Flora Group. Growing knowledge: an overview of Seed Plant diversity in Brazil. Rodriguésia, 2015;66: 1085–1113. [Google Scholar]
- 4.Fleischmann A, Schäferhoff B, Heubl G, Rivadavia F, Barthlott W, Müller KF. Phylogenetics and character evolution in the carnivorous plant genus Genlisea A. St.-Hil. (Lentibulariaceae). Mol. Phylogenet. Evol. 2010;56: 768–83. doi: 10.1016/j.ympev.2010.03.009 [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann A, Rivadavia F, Gonella PM, Heubl G. A revision of Genlisea subgenus Tayloria (Lentibulariaceae). Syst. Bot. 2011;40: 1–40. [Google Scholar]
- 6.Miranda VFO, Menezes CG, Silva SR, Díaz YCA., Rivadavia F. Lentibulariaceae in Lista de Espécies da Flora do Brasil. Jard. Botânico do Rio Janeiro 12 Set 2015. Available from: http://floradobrasil.jbrj.gov.br/reflora/floradobrasil/FB146. [Google Scholar]
- 7.Barthlott W, Porembski S, Fischer E, Gemmel B. First protozoa-trapping plant found. Nature 1998; 392:447 doi: 10.1038/330379548248 [Google Scholar]
- 8.Płachno BJ., Adamus K, Faber J, Kozłowski J. Feeding behaviour of carnivorous Genlisea plants in the laboratory. Acta Bot. Gall. 2005;152: 159–164. [Google Scholar]
- 9.Fleischmann A. Monograph of the Genus Genlisea Poole, Dorset, England: Redfern Natural History Productions; 2012. [Google Scholar]
- 10.Jobson R, Albert VA. Molecular Rates Parallel Diversification Contrasts between Carnivorous Plant Sister Lineages. Cladistics 2002;18: 127–136. [DOI] [PubMed] [Google Scholar]
- 11.Müller K, Borsch T, Legendre L, Porembski S, Theisen I, Barthlott W. Evolution of Carnivory in Lentibulariaceae and the Lamiales. Plant Biol. 2004;6: 477–490. doi: 10.1055/s-2004-817909 [DOI] [PubMed] [Google Scholar]
- 12.Müller KF, Borsch T, Legendre L, Porembski S, Barthlott W. Recent progress in understanding the evolution of carnivorous Lentibulariaceae (Lamiales). Plant Biol. (Stuttg). 2006;8: 748–57. [DOI] [PubMed] [Google Scholar]
- 13.Jobson RW, Nielsen R, Laakkonen L, Wikström M, Albert VA. Adaptive evolution of cytochrome c oxidase: Infrastructure for a carnivorous plant radiation. Proc. Natl. Acad. Sci. U.S.A. 2004;101: 18064–18068. doi: 10.1073/pnas.0408092101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albert VA, Jobson RW, Michael TP, Taylor DJ. The carnivorous bladderwort (Utricularia, Lentibulariaceae): A system inflates. J. Exp. Bot. 2010;61: 5–9. doi: 10.1093/jxb/erp349 [DOI] [PubMed] [Google Scholar]
- 15.Ibarra-Laclette E, Albert VA, Pérez-Torres CA, Zamudio-Hernández F, Ortega-Estrada MDJ, Herrera-estrella A, et al. Transcriptomics and molecular evolutionary rate analysis of the bladderwort (Utricularia), a carnivorous plant with a minimal genome. BMC Plant Biol. 2011;11: 101 doi: 10.1186/1471-2229-11-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laakkonen L, Jobson RW, Albert VA. A new model for the evolution of carnivory in the bladderwort plant (Utricularia): Adaptive changes in cytochrome c oxidase (COX) provide respiratory power. Plant Biol. 2006;8: 758–764. doi: 10.1055/s-2006-924459 [DOI] [PubMed] [Google Scholar]
- 17.Greilhuber J, Borsch T, Müller K, Worberg A, Porembski S, Barthlott W. Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biol. 2006;8: 770–777. doi: 10.1055/s-2006-924101 [DOI] [PubMed] [Google Scholar]
- 18.Fleischmann A, Michael TP, Rivadavia F, Sousa A, Wang W, Temsch EM, et al. Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms. Ann. Bot. 2014;114: 1651–1663. doi: 10.1093/aob/mcu189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fromm-Trinta E. Revisão das espécies do gênero Genlisea St.-Hil. -Lentibulariaceae- das regiões sudeste e sul do Brasil. Rodriguésia 1979;31: 17–139. [Google Scholar]
- 20.Daniell H, Lin C-S, Yu M, Chang W-J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016;17: 134 doi: 10.1186/s13059-016-1004-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bungard RA. Photosynthetic evolution in parasitic plants: Insight from the chloroplast genome. BioEssays 2004;26: 235–247. doi: 10.1002/bies.10405 [DOI] [PubMed] [Google Scholar]
- 22.Schelkunov MI, Shtratnikova VY, Nuraliev MS, Selosse MA, Penin AA, Logacheva MD. Exploring the limits for reduction of plastid genomes: A case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evol. 2015;7: 1179–1191. doi: 10.1093/gbe/evv019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martín M, Sabater B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010;48: 636–645. doi: 10.1016/j.plaphy.2010.04.009 [DOI] [PubMed] [Google Scholar]
- 24.Peng L, Yamamoto H, Shikanai T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. Biochim. Biophys. Acta—Bioenerg. 2011;1807: 945–953. doi: 10.1016/j.bbabio.2010.10.015 [DOI] [PubMed] [Google Scholar]
- 25.Silva SR, Diaz YCA, Penha HA, Pinheiro DG, Fernandes CC, Miranda VFO, et al. The chloroplast genome of Utricularia reniformis sheds light on the evolution of the ndh gene complex of terrestrial carnivorous plants from the lentibulariaceae family. PLoS One 2016;11: 1–29. doi: 10.1371/journal.pone.0165176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leushkin EV, Sutormin RA, Nabieva ER, Penin AA, Kondrashov AS., Logacheva MD The miniature genome of a carnivorous plant Genlisea aurea contains a low number of genes and short non-coding sequences. BMC Genomics 2013;14: 476 doi: 10.1186/1471-2164-14-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrews S. FastQC: A quality control tool for high throughput sequence data. 2010. Available from: http://www.bioinformatics.babraham.ac.uk/projects/Fastqc/. [Google Scholar]
- 28.Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24: 1384–1395. doi: 10.1101/gr.170720.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9: 357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012;19: 455–477. doi: 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013;41: e129 doi: 10.1093/nar/gkt371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41: 95–98. [Google Scholar]
- 33.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012;28: 1647–1649. doi: 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004;20: 3252–3255. doi: 10.1093/bioinformatics/bth352 [DOI] [PubMed] [Google Scholar]
- 35.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32: 11–16. doi: 10.1093/nar/gkh152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowe TM, Eddy SR. TRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1996;25: 955–964. doi: 10.1093/nar/25.5.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peden JF. CodonW. 2005 Available from: http://codonw.sourceforge.net/
- 38.Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW- a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41: W575–81. doi: 10.1093/nar/gkt289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurtz S, Schleiermacher C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999;15: 426–427. doi: 10.1093/bioinformatics/15.5.426 [DOI] [PubMed] [Google Scholar]
- 40.Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003;106: 411–422. doi: 10.1007/s00122-002-1031-0 [DOI] [PubMed] [Google Scholar]
- 41.Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability Article Fast Track. 2013;30: 772–780. doi: 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004;32: W273–W279. doi: 10.1093/nar/gkh458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swofford DL. PAUP* Phylogenetic Analysis Using Parsimony * (and other methods). version 4.0 2002. Sinauer Assoc; Sunderland, Massachusetts. [Google Scholar]
- 44.Santorum JM, Darriba D, Taboada GL, Posada D. Jmodeltest.Org: Selection of Nucleotide Substitution Models on the Cloud. Bioinformatics, 2014;30: 1310–1311. doi: 10.1093/bioinformatics/btu032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30: 1312–1313. doi: 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogeny. Bioinformatics 2001;17: 754–755. doi: 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- 47.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. in 2010 Gateway Computing Environments Workshop, GCE 2010. 2010. doi: 10.1109/GCE.2010.5676129
- 48.Stöver BC, Müller KF TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics 2010;11: 7 doi: 10.1186/1471-2105-11-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva SR, Pinheiro DG, Meer EJ, Michael TP, Varani AM, Miranda VFO. The complete chloroplast genome sequence of the leafy bladderwort, Utricularia foliosa L. (Lentibulariaceae). Conserv. Genet. Resour. 2017;9: 213–216. doi: 10.1007/s12686-016-0653-5 [Google Scholar]
- 50.Wicke S, Schaferhoff B, dePamphilis CW, Muller KF Disproportional Plastome-Wide Increase of Substitution Rates and Relaxed Purifying Selection in Genes of Carnivorous Lentibulariaceae. Mol. Biol. Evol. 2014;31: 529–545. doi: 10.1093/molbev/mst261 [DOI] [PubMed] [Google Scholar]
- 51.Jansen RK, Ruhlman TA. Genomics of Chloroplasts and Mitochondria In: Bock R, Knoop V. Advances in Photosynthesis and Respiration. Netherlands:Springer; 2012. 475. [Google Scholar]
- 52.Wu CS, Wang YN, Hsu CY, Lin CP, Chaw SM. Loss of different inverted repeat copies from the chloroplast genomes of pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011;3: 1284–1295. doi: 10.1093/gbe/evr095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dugas DV, Hernandez D, Koenen EJM, Schwarz E, Straub S, Hughes CE, et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015;5: 16958 doi: 10.1038/srep16958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goulding SE, Olmstead RG, Morden CW, Wolfe KH. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996;252: 195–206. doi: 10.1007/s004389670022 [DOI] [PubMed] [Google Scholar]
- 55.Doyle JJ, Doyle JL, Palmer JD. Multiple Independent Losses of Two Genes and One Intron from Legume Chloroplast Genomes. Syst. Bot. 1995;20: 272–294. doi: 10.2307/2419496 [Google Scholar]
- 56.Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991;10: 3073–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee S-B, Kaittanis C, Jansen RK, Hostetler JB, Tallon LJ, Town CD, et al. The complete chloroplast genome sequence of Gossypium hirsutum: organization and phylogenetic relationships to other angiosperms. BMC Genomics 2006;7: 61 doi: 10.1186/1471-2164-7-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bausher MG, Singh ND, Lee S-B, Jansen RK, Daniell H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var “Ridge Pineapple”: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006;6: 21 doi: 10.1186/1471-2229-6-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jansen RK, Saski C, Lee SB, Hansen AK, Daniell H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011;28: 835–847. doi: 10.1093/molbev/msq261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Y, Zhou T, Duan D, Yang J, Feng L, Zhao G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016;7: 959 doi: 10.3389/fpls.2016.00959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cauz-Santos LA, Munhoz CF, Rodde N, Cauet S, Santos AA, Penha HA, et al. The Chloroplast Genome of Passiflora edulis (Passifloraceae) Assembled from Long Sequence Reads: Structural Organization and Phylogenomic Studies in Malpighiales. Front. Plant Sci. 2017;8: 334 doi: 10.3389/fpls.2017.00334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fitch WM. On the Problem of Discovering the Most Parsimonious Tree. The American Naturalist 1977;111: 223–57. [Google Scholar]
- 63.Saitou N, Nei M. The number of nucleotides required to determine the branching order of three species, with special reference to the human-chimpanzee-gorilla divergence. J. Mol. Evol. 1986;24: 189–204. doi: 10.1007/BF02099966 [DOI] [PubMed] [Google Scholar]
- 64.Hollingsworth PM Refining the DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 2011;108: 19451–2. doi: 10.1073/pnas.1116812108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.CBOL Plant Working. A DNA Barcode for Land Plants. Proc. Natl. Acad. Sci. U.S.A. 2009;106: 12794–97. doi: 10.1073/pnas.0905845106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pang X, Liu C, Shi L, Liu R, Liang D, Li H, et al. Utility of the trnH-psbA Intergenic Spacer Region and Its Combinations as Plant DNA Barcodes: A Meta-Analysis. PLoS One 2012;7: e48833 doi: 10.1371/journal.pone.0048833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong W, Xu C, Li C, Su J, Zuo Y, Shi S, et al. ycf1, the most promising plastid DNA barcode of land plants. 2015:1–5. doi: 10.1038/srep08348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Batley J. Plant Genotyping. Methods Mol. Biol. 2015;1245: 101–18. doi: 10.1007/978-1-4939-1966-6_8 [DOI] [PubMed] [Google Scholar]
- 69.Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012;99: 193–208. doi: 10.3732/ajb.1100394 [DOI] [PubMed] [Google Scholar]
- 70.Clivati D, Gitzendanner MA, Hilsdorf AWS, Araújo WL, Miranda VFO. Microsatellite markers developed for Utricularia reniformis (Lentibulariaceae). Am. J. Bot. 2012;99: E375–378. doi: 10.3732/ajb.1200080 [DOI] [PubMed] [Google Scholar]
- 71.Maul JE, Lilly JW, Cui L, dePamphilis CW, Miller W, Harris EH, et al. The Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats. Plant Cell 2002;14: 2659–79. doi: 10.1105/tpc.006155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quandt D, Müller K, Huttunen S. Characterisation of the Chloroplast DNA psbT-H Region and the Influence of Dyad Symmetrical Elements on Phylogenetic Reconstructions. Plant Biol. 2003;5: 400–410. doi: 10.1055/s-2003-42715 [Google Scholar]
- 73.Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 2011;76: 273–97. doi: 10.1007/s11103-011-9762-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo J, Hou BW, Niu ZT, Liu W, Xue QY, Ding XY. Comparative chloroplast genomes of photosynthetic orchids: Insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PLoS One 2014;9 doi: 10.1371/journal.pone.0099016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruhlman TA, Chang W-J, Chen JJW, Huang Y-T, Chan M-T, Zhang J, et al. NDH expression marks major transitions in plant evolution and reveals coordinate intracellular gene loss. BMC Plant Biol. 2015;15: 100 doi: 10.1186/s12870-015-0484-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rumeau D, Bécuwe-Linka N, Beyly A, Louwagie M, Garin J, Peltier G. New subunits NDH-M, -N, and -O, encoded by nuclear genes, are essential for plastid Ndh complex functioning in higher plants. Plant Cell 2005;17: 219–32. doi: 10.1105/tpc.104.028282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ueda M, Kuniyoshi T, Yamamoto H, Sugimoto K, Ishizaki K, Kohchi T, et al. Composition and physiological function of the chloroplast NADH dehydrogenase-like complex in Marchantia polymorpha. Plant J. 2012;72: 683–693. doi: 10.1111/j.1365-313X.2012.05115.x [DOI] [PubMed] [Google Scholar]
- 78.Yamori W, Sakata N, Suzuki Y, Shikanai T, Makino A. Cyclic electron flow around photosystem I via chloroplast NAD(P)H dehydrogenase (NDH) complex performs a significant physiological role during photosynthesis and plant growth at low temperature in rice. Plant J. 2011;68: 966–976. doi: 10.1111/j.1365-313X.2011.04747.x [DOI] [PubMed] [Google Scholar]
- 79.Santamaría L. Why are most aquatic plants widely distributed? Dispersal, clonal growth and small-scale heterogeneity in a stressful environment. Acta Oecologica 2002;23: 137–154. doi: 10.1016/S1146-609X(02)01146-3 [Google Scholar]
- 80.Silva SR, Alvarenga DO, Aranguren Y, Penha HA, Fernandes C, Pinheiro DG, et al. The mitochondrial genome of the terrestrial carnivorous plant Utricularia reniformis (Lentibulariaceae): Structure, comparative analysis and evolutionary landmarks. 2017; 1–26. doi: 10.1371/journal.pone.0180484 [DOI] [PMC free article] [PubMed]
- 81.Lin C-S, Chen JJW, Huang Y, Chan M, Daniell H, Chang W, et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015;5: 9040 doi: 10.1038/srep09040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sanderson MJ, Copetti D, Búrquez A, Bustamante E, Charboneau JLM, Eguiarte LE, et al. Exceptional reduction of the plastid genome of saguaro cactus (Carnegiea gigantea): Loss of the ndh gene suite and inverted repeat 1. Am. J. Bot. 2015;102: 1115–1127. doi: 10.3732/ajb.1500184 [DOI] [PubMed] [Google Scholar]
- 83.Petrov V, Hille J, Mueller-Roeber B, Gechev TS. ROS-mediated abiotic stress-induced programmed cell death in plants. Front. Plant Sci. 2015;6: 69 doi: 10.3389/fpls.2015.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adamec L. Respiration and photosynthesis of bladders and leaves of aquatic Utricularia species. Plant Biology, 2006;8: 765–769. doi: 10.1055/s-2006-924540 [DOI] [PubMed] [Google Scholar]
- 85.Ibarra-Laclette E, Albert VA, Herrera-Estrella A, Herrera-Estrella L. Is GC bias in the nuclear genome of the carnivorous plant Utricularia driven by ROS-based mutation and biased gene conversion? Plant Signal. Behav. 2011;6: 1631–1634. doi: 10.4161/psb.6.11.17657 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All quality-trimmed reads from sequencing data sets have been mapped back to the reconstructed plastid supercontig. The upper plot indicates the identity per site and the lower plot shows the coverage plot per species.
(DOCX)
Note the product of G. violacea cpDNA that presents the duplication of rps19 gene and rpl22 as pseudogene (amplicon with 1,194 bp), while the other species present an expected product with ~490 bp. (Amplification reactions of the rpl2-trnH(GUG) marker were conducted in 25 μL of the solution containing 20 mM of MgCl2, 100 mM of dNTPs, 10 mM of each primer, 1 U of Dream Taq Polymerase–Fermentas, and 50 ng of DNA template. The thermal profile for amplification was 1min at 94°C; 35 cycles of 40s at 94°C, 20s at 64°C, 90s at 72°C, and 5min of final extension at 72°C. Forward primer = 5’-AGT CGG ACA AGT GGG GAA TG-3’; reverse primer = 5’-GGA TGT GGC CAA GTG GAT CA-3’).
(DOCX)
Statistics from Spearman correlation tests are given near the corresponding trend lines.
(DOCX)
PIC values are represented as bars and cpDNA region is marked by colors. Black dots represent p-distance. Only PIC of ndhs were not calculated to avoid p-distance alignment artefact (see S6 Table).
(DOCX)
Numbers above are parsimony bootstrap (left), maximum likelihood bootstrap (right) and posterior probability values are represented below. Lamiales species were used as outgroup.
(DOCX)
(DOCX)
(DOCX)
The characters were codified according the S2 Table. (G. = Genlisea; P. = Pinguicula; U. = Utricularia).
(DOCX)
Numbers within table refer to sequence length (bp). Colors refer to the state of character: white–deleted gene; yellow–pseudogenized; pink–decayed gene; grey–complete gene; n/a–absent.
(DOCX)
F–Direct repeats; P–Palindromic repeats; T–Tandem repeats (inside parenthesis the repeated nucleotide). Common genes with repeats between the six species are highlighted with yellow background color in G. aurea table.
(DOCX)
Deleted ndh genes in all Genlisea species and boundaries between ndh pseudogenes are uncertain and were not included in this analysis (represented as n/a).
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.