

Abstract
The circadian rhythm is a fundamental physiological process present in all organisms that regulates biological processes ranging from gene expression to sleep behavior. In vertebrates, circadian rhythm is controlled by a molecular oscillator that functions in both the suprachiasmatic nucleus (SCN; central pacemaker) and individual cells comprising most peripheral tissues. More importantly, disruption of circadian rhythm by exposure to light-at-night, environmental stressors and/or toxicants is associated with increased risk of chronic diseases and aging. The ability to identify agents that can disrupt central and/or peripheral biological clocks, and agents that can prevent or mitigate the effects of circadian disruption, has significant implications for prevention of chronic diseases. Although rodent models can be used to identify exposures and agents that induce or prevent/mitigate circadian disruption, these experiments require large numbers of animals. In vivo studies also require significant resources and infrastructure, and require researchers to work all night. Thus, there is an urgent need for a cell-type appropriate in vitro system to screen for environmental circadian disruptors and enhancers in cell types from different organs and disease states. We constructed a vector that drives transcription of the destabilized luciferase in eukaryotic cells under the control of the human PERIOD 2 gene promoter. This circadian reporter construct was stably transfected into human mammary epithelial cells, and circadian responsive reporter cells were selected to develop the in vitro bioluminescence assay. Here, we present a detailed protocol to establish and validate the assay. We further provide details for proof of concept experiments demonstrating the ability of our in vitro assay to recapitulate the in vivo effects of various chemicals on the cellular biological clock. The results indicate that the assay can be adapted to a variety of cell types to screen for both environmental disruptors and chemopreventive enhancers of circadian clocks.
Keywords: Genetics, Issue 127, Circadian rhythm, PERIOD 2, promoter activity, in vitro real-time bioluminescence assay, luciferase expression vector, destabilized firefly luciferase, plasmid transfection.
Introduction
The circadian clock regulates a wide range of biological processes from diurnal expression of genes to sleep behavior in a predictable rhythm with a periodicity of approximately 24 h. Epidemiological studies strongly suggest that chronic disruption of circadian rhythm increases the risk of breast and prostate cancer in shift workers, including nurses and flight crews1,2,3. These findings are corroborated by rodent studies, demonstrating that exposure to constant light, light-at-night, or light cycles that mimic jet-lag increase tumor incidence and accelerates tumor growth4,5. Based on data from both human and rodent studies, the International Agency for Research on Cancer classified shift-work as a probable human carcinogen (Type 2A) in 20106.
Previously, we demonstrated that a single carcinogenic dose of the mammary tumor specific carcinogen, N-nitroso-N-methylurea (NMU), disrupted the circadian expression of major circadian genes (CGs) (e.g., Period 2, Per2) and several circadian-controlled genes (CCGs), including key DNA damage responsive and repair (DDRR) genes in the target mammary gland (but not in the liver). Moreover, resetting the circadian expression of both Per2 and DDRR genes towards the normal by a chemopreventive regimen of dietary L-methyl-selenocysteine (MSC) reduced the incidence of tumor by 63%. These findings were the first to show a mechanistic link between circadian rhythm, chemical carcinogenesis and chemoprevention7,8. Exposures to other environmental toxicants shown to disrupt circadian gene expression in vivo are also associated with increased risk of environmental diseases9,10. Understanding the mechanisms that link circadian disruption by environmental toxicants and pathogenesis may lead to mechanistically-based approaches to disease prevention. However, studies aimed at defining the interactions between the exposures and circadian rhythm are usually performed in vivo. A typical in vivo experiment investigating the impact on circadian rhythm requires large numbers of animals, as tissues from at least three control and three exposed animals must be collected every 3-4 h over a 24 or 48 h period. Development of a validated in vitro system that recapitulates in vivo observations and mechanisms would therefore not only reduce the number of animals required, but also dramatically reduce experimental costs and the requirement that researchers work continuously over a 24-48 h period. Moreover, a validated in vitro system could be used for high throughput screening of compounds and/or genetic alteration that affect circadian rhythm, or its response to environmental stressors or toxicants. Therefore, the strategical combination of in vitro and in vivo models and experiments are needed to obtain different insights with different focus.
In mammals, circadian oscillators exist not only in specialized neurons of the SCN, but also in most peripheral cell types. These molecular clocks are similar to those in established fibroblast cell lines and in primary fibroblasts from embryos or adult animals; however, there is a need for tissue type-specific cellular models11. Consequently, traditional studies of locomotor activity in vivo, SCN explants ex vivo, and cell-based in vitro assays in immortalized fibroblast cells are widely used to study cell-autonomous circadian defects. However, there is no evidence indicating that an in vitro fibroblast cell-based assay can recapitulate circadian mechanisms and responses present in cells of other peripheral organs in vivo. Different cell types can have distinct patterns of gene expression, xenobiotic metabolism, and DDRR, and the links between toxicity and circadian gene expression may be cell-type specific and/or modulated by different physiological parameters. In addition, circadian oscillators in fibroblast-based systems have not been fully assessed for responses to environmental toxicants, stressors and preventive agents that link exposures to mechanisms of disease development and prevention. Thus, there is a need for facile, validated cell-type specific, in vitro bioluminescence assays to study organ specific environmental circadian disruptors. Although a variety of cellular clock models (e.g., in liver, keratinocytes, and fat cells, as well as an osteosarcoma cell line) have been developed in recent years12,13,14,15, the assay described here is the first cellular clock model in breast epithelial cells, and the first demonstration to recapitulate in vivo responses to environmental stressors, toxicants, drugs, and chemopreventive agents.
Renilla luciferase (rLuc) and firefly luciferase are 30-61 kDa monomeric proteins that do not require posttranslational processing for enzymatic activity and can function as a genetic reporter immediately upon translation. Once the substrate associates with the luciferase enzyme, the biochemical reaction catalyzed generates a flash of light. Thus, luciferase constructs are widely used as a gene expression reporter system in vitro and in vivo. However, in circadian rhythm studies, the utility of the luciferase reporter is limited by the relatively long half-life of the luciferase protein (T1/2 = 3.68 h) relative to the period (especially to the short period) for changes in circadian gene expression; however, numerous studies over the years have successfully used the luciferase gene in the pGL3 vector, indicating that the rapidly degradable luciferase may not be necessary for reporting circadian rhythms, especially for the rhythms with a longer period, such as 24 h. Therefore, a reporter plasmid using destabilized luciferase vector, pGL[Luc2P/Neo], that contains hPEST (a protein destabilization sequence) has been developed, allowing us to use it as a circadian reporter vector for our current in vitro bioluminescence assay. The protein encoded by Luc2P has a much shorter half-life (T1/2 = 0.84 h) and hence, responds more quickly and with a greater magnitude to changes in transcriptional activity than wild-type, indicating that it can be used to monitor the rhythmic expression of luciferase regulated by the PER2 promoter accurately in real-time16.
Protocol
1. Construction of PER2 Promoter Driven Destabilized Luciferase Reporter Vector
Purchase a customized pLS[hPER2P/rLuc/Puro] vector that contains cDNA encoding rLuc and human PER2 promoter fragment (hPER2P, 941 bp) at the site between Sac I and Hind III in the multiple cloning region17.
- Cut the human PER2 promoter fragment (hPER2P, 941 bp) out from the vector.
- Add 2.5 µL restriction enzyme buffer, 10 µL (180 ng) pLS[hPER2P/rLuc/Puro] vector, and 1 µL each of the restriction enzymes, Sac I (10 unit/µL) and Hind III (10 unit/µL), into a DNA/RNA/nucleotide-free microcentrifuge tube. Add ultrapure water (10.5 µL) up to 25 µL and mix gently by pipetting.
- Incubate at 37 °C for 90 min in a heating block.
- Run the whole volume (25 µL) of the restriction enzyme reaction on 0.7% agarose gel containing 0.0001% ethidium bromide (EtBr) to separate the hPER2P from the vector. NOTE: EtBr, 3,8-diamino-1-ethyl-6-phenylphenantridinium bromide (CAS registration number: 1239-45-8), is an odorless red liquid. It is an intercalating agent that is commonly used as a fluorescent tag (nucleic acid stain) in agarose gel electrophoresis and visible as an orange color under UV light. Possible risks of irreversible mutagenic effects have occurred in experimental animals, although none of the regulatory agencies categorized it as a carcinogen18. Therefore, we collected used agarose gel and electrophoresis buffer containing EtBr as chemical hazard waste, and requested Rutgers Environmental Health & Safety (REHS) to pick up and dispose safely.
- Cut a piece of agarose gel containing a EtBr fluorescence band at 941 bp, and purify the hPER2P fragments from the gel with a DNA gel extraction kit, followed by quantification on a spectrophotometer by measuring absorption density (OD) at 260 nm wavelength.
Linearize 1.0 µL (1 µg) of the destabilized firefly luciferase expression vector, pGL[Luc2P/Neo] with the same method as described in steps 1.2.1-1.2.2. Extract the vector with a DNA clean-up kit followed by quantification as described above using a spectrophotometer.
- Ligate the hPER2P into the linearized vector pGL[Luc2P/Neo]. NOTE: Per the manufacturer's instruction, the ideal molar ratio of insert and vector is 2:1. For 50 ng vector, the ideal amount of insert is calculated as 23.6 ng with a formula, [(50 ng vector x 1.0 kb insert)/4.242 kb vector] x (2/1) = 23.6 ng insert]. Based on the concentrations of hPER2P (7.26 ng/µL) and pGL[Luc2P/Neo] (50 ng/µL), the volumes of 50 ng pGL[Luc2P/Neo] and 23.6 ng hPER2P are calculated as 1 µL and 3.26 µL, respectively.
- Add 2 µL T4 DNA ligase reaction buffer (10X) (50 mM Tris-HCl, 10 mM MgCl2, 1 mM ATP, and 10 mM DTT), 3.26 µL (23.6 ng) hPER2P, 1 µL (50 ng) pGL[Luc2P/Neo], and 13.74 µL water up to 20 µL, mix gently.
- Add 1 µL T4 DNA ligase (400 unit/µL), mix gently and incubate at 16 °C overnight in a thermocycler, and then chill the ligated vector on ice per the manufacturer's instruction.
- Transform the ligated vector pGL[hPer2P/Luc2P/Neo] to chemically competent E. coli19.
- Set the water bath at 42 °C, warm up Super Optimal Broth with catabolite repression (S.O.C.) medium (2% Tryptone, 0.5% Yeast Extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose) at room temperature, warm up Luria-Bertani (LB) agar plate (containing 100 µg/mL ampicillin, 80 µg/mL X-gal, and 0.5 µM IPTG) in 37 °C incubator, and thaw on ice one vial of competent E. coli cells for each transformation.
- Add 6 µL (21 ng) ligated vector or 1 µL (30 ng) empty vector (negative control) to one tube of chemically competent E. coli (50 µL) and then gently flip the tube to mix. Incubate on ice for 30 min.
- Heat shock in a 42 °C water bath for 30 s without shaking and then return to ice.
- Select an E. coli colony transformed with a ligated vector using blue/white screening.
- Add 250 µL S.O.C. medium in the transformed E. coli tube and incubate it for 1 h at 37 °C with shaking at 200 rpm.
- Spread 10-50 µL of transformed E. coli on the surface of the LB agar plate evenly. Put the plate upside down in 37 °C incubator overnight. NOTE: An efficient cloning reaction should produce several hundred colonies. Plating two different volumes is recommended to ensure that at least one plate will have big, clear white or blue color, and well-spaced colonies. When the colonies are too small and the color is not distinguishable, using a microscope (10X or 50X) is helpful.
- Pick 3-6 white colonies from the plates with sterilized tooth sticks, and release each colony into one culture tube containing 4 mL of LB culture medium with 50 µg/mL ampicillin.
- Incubate the tubes at 37 °C with shaking at 200 rpm overnight. Extract the DNA using 2 mL of the 4 mL cultured E. coli with a plasmid DNA extraction mini prep kit per the manufacturer's instruction.
- Verify and analyze the insert (hPER2P, 941 bp)
- Digest the plasmid DNA with restriction enzymes and run electrophoresis as described in steps 1.2.1-1.2.3 to confirm if there is an insert. NOTE: Positive control (hPER2P purified at step 1.2.4) and negative control (E. coli transformed with an empty vector) were included.
- Sequence the selected plasmid DNA using M13 forward and M13 reverse primers to confirm the orientation and sequence of insert in the plasmid19. NOTE: Only one colony that had an insert with the correct size of hPER2P in electrophoresis and correct orientation and sequence in the sequencing result was selected.
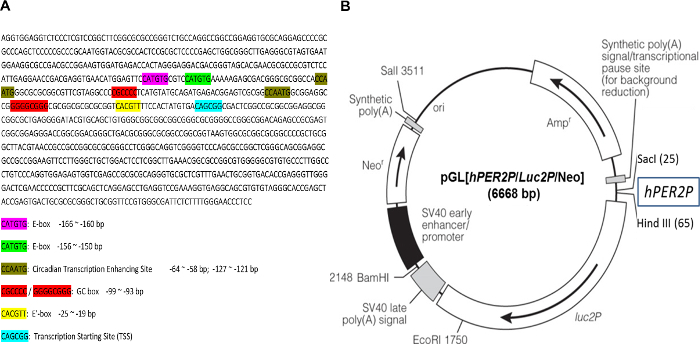
- Analyze the human PER2 promoter fragment (hPER2P, 941 bp) sequence for the circadian regulatory elements, including E-box motif (Bmal1 binding site, CAT/CGTG), CCAATC, GC box, and transcription start site (CAGCGG)20.
Amplify the selected E. coli by adding the leftover 2 mL cultured E. coli into 200 mL LB culture medium containing 50 µg/mL ampicillin and then incubating it overnight at 37 °C with shaking at 200 rpm.
Extract DNA with an endotoxin-free plasmid maxi prep kit per the manufacturer's instruction, and then quantify it by measuring OD at 260 nm wavelength with a spectrophotometer. Aliquot and store the plasmid DNA, pGL[hPer2P/Luc2P/Neo], at -80 °C freezer for future use.
2. Transient Transfection
- Purchase and culture MCF10A cells
- Purchase an immortalized, non-transformed normal human mammary epithelial cell line (MCF10A) from a commercial cell bank, where cells are cytogenetically tested and authenticated with short tandem repeat analysis before freezing. Thaw and maintain each vial of frozen cells in culture for a maximum of 8 weeks. NOTE: Unlike other cells, these cells have contact inhibition once they get to ~70% confluence. It requires that subculture is conducted at ~70%.
- Culture the cells with mammary epithelial cell growth medium (MEGM), containing mammary epithelial basal medium (MEBM), growth supplements, and cholera toxin in a cell culture incubator at 37 °C, 95% humidity, and 5% CO2. NOTE: Purchase ready-to-use growth factors provided in SingleQuot (SQ) and obtain cholera toxin separately. Final concentrations of growth supplements are 0.4% bovine pituitary extract, 0.1% insulin, 0.1% hydrocortisone, 0.1% human epidermal growth factors, 100 ng/mL cholera toxin, and 0.05% gentamycin sulfate and 0.05% amphotericin B.
- Dissociate the cells in 10-cm culture dish by incubation with 10 mL trypsin/EDTA for ~ 20 min and then neutralize the trypsin by adding 10 mL trypsin neutralizing solution. Collect all cells with HEPES buffered saline solution (HBSS).
- Count the cells with a hemocytometer under an inverted microscope and calculate the concentration of the single cell suspension, and then seed a certain number of cells as needed. Swirl the plates thoroughly to obtain an even distribution of cells in each plate. Incubate cells in the cell culture incubator at 37 °C, 95% humidity, and 5% CO2. NOTE: Purchase a subculture reagent pack containing trypsin/EDTA, trypsin neutralizing solution, and HBSS to use.
- Transient transfection of cells with the circadian vector constructed.
- Seed 2 x 105 MCF10A cells evenly in 35 mm culture dish with MEGM and grow to 20-30% confluence (next day).
- Warm up the reduced serum media (e.g., Opti-MEM) in 37 °C water bath and transfection reagent at room temperature for 30 min. Dilute the plasmid DNA constructed (pGL[hPer2P/Luc2P/Neo]) to 30 ng/µL with endo-toxin free Tris-EDTA (TE) buffer and keep at room temperature.
- Prepare the plasmid transfection mixture in a 1.5 mL microcentrifuge tube by adding 63.6 µL warm reduced serum media first, then adding 33.4 µL (0.1 µg) plasmid DNA, and mix gently by pipetting, and then lastly adding 3 µL of transfection reagent directly into the center of the tube without touching the wall. After mixing gently by pipetting, keep at room temperature for 30 min.
- Drop the entire plasmid mixture (100 µL) directly onto the center surface of the medium in the plate and then mix gently by shaking the dish. Culture the cells in a CO2 incubator for additional 48-72 h. NOTE: It is predicted that the highest transfection efficiency would be at 40-70% confluence of these cells due to their contact inhibition at ~ 70%. Therefore, transfection works best starting at ~ 20% and stop at ~ 70% confluence.
3. Establishment of the In Vitro Bioluminescence Assay in Transiently Transfected MCF10A Cells
Starve the cultured cells at ~70% confluence (after transfection for 48-72 h) in MEBM without growth factors for 24 h.
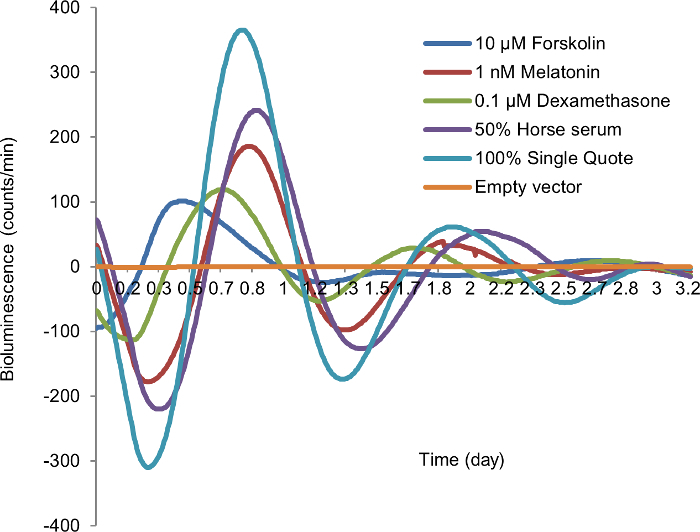
Treat the cells with synchronization agent (e.g., 50% horse serum (HS)) in MEBM for 1.5 h in a CO2 incubator. NOTE: For selection of an ideal synchronization agent, 10 µM forskolin, 1 nM melatonin, 0.1 µM dexamethasone, 50% HS, and 100% SQ were compared in terms of circadian amplitude and period in the circadian vector transfected cells. Empty vector transfected cells are treated with 100% SQ as a negative control.
Pre-prepare 20 mL recording medium (for 10 of the 30-mm culture dishes) by adding 5 mL of MEGM, 15 mL of MEBM, 150 µL sodium bicarbonate, 200 µL HEPES, and 40 µL luciferin stock solution (50 mM) in a 50 mL sterilized centrifuge tube. NOTE: The final concentrations of each components: 20% SQ and 20 ng/mL cholera toxin, 0.06% sodium bicarbonate, 1% penicillin/streptomycin, and 10 mM HEPES. Warm up pre-prepared recording medium and sterilized 1x PBS for 30 min in 37 °C water bath. Add 100 µM luciferin immediately before use.
Wash the cells with warm 1x Dulbecco's Phosphate-Buffered Saline (D-PBS) for 3 times after the incubation with the synchronization agent, and then add 2 mL recording medium.
Seal the dish with a sterilized cover class using silicon grease to prevent evaporation and then label it on the side.
Place the sealed dish in the seat inside the luminometer, and turn on the option for saving the file location in the folder (for the detail, see section 6).
4. Selection of Stably Transfected Cells
Optimize an ideal G418 concentration (800 µg/mL) with a kill curve assay according to the manufacturer's instruction for selection of the stably transfected cells.
After transient transfection for 48 h or 72 h, subculture and seed the cells with MEGM in larger dishes at 5,000 cells/dish (100-mm). After 24 h culture in the cell culture incubator (37 °C, 95% humidity, and 5% CO2), treat the cells with 800 µg/mL of G418 by replacing the old medium with new medium containing 800 µg/mL G418 every 3-4 days for 2 weeks.
Subculture the surviving stably transfected colonies for 1-3 passages with MEGM containing 500µg/mL G418 to maintain the stable expression of hPer2P/Luc2P, and freeze in liquid nitrogen for future use.
Treat 500µg/mL G418 after general subculture to re-select the stably transfected population of cells, if the bioluminescence intensity in the result of in vitro bioluminescence assay becomes progressively lower after use in many passages.
5. Treatment with Chemicals
At 48 h or 72 h post-transfection, starve the cells from growth factors in MEBM for 24 h.
After synchronization with 50% HS for 2 h, treat cells with 0.25 mM or 0.5 mM nitrosomethylurea (NMU), 20 nM EX527, or 1 µM cambinol for 1 h in MEBM containing 20% SQ.
After washing with 1x D-PBS, add recording medium containing 12.5 µM MSC alone, or in combination with 20 nM EX527 or 1 µM cambinol, to the cells. Monitoring the bioluminescence with the luminometer.
Treat the stably transfected MCF10A/PER2-dLuc cells with NMU at 0.5, 1, or 2 mM, EX527 at 40 nM, or Cambinol at 2.0 µM, for 1 h after synchronization with 50% HS, and then incubate the cells in recording medium containing 12.5 µM of MSC or different doses (5, 10, or 20 µM) of n-acetylcysteine (NAC). Place the plates in the luminometer, record and save the bioluminescence by luminometer for 4-8 days as described in section 3.6. NOTE: NMU is a methylated nitrosourea compound with mutagenic, carcinogenic, and teratogenic properties. NMU is a direct-acting alkylating agent and has a short half-life (T1/2 = ~ 30 min). It is stable at acidic condition (pH 4-5), but unstable at alkaline condition (pH 9-10) and at temperatures beyond 20 °C. Therefore, bleach at 10% can be used as an efficient deactivating agent for cleaning bench surfaces and lab wares potentially contaminated by NMU solution. Leftover stock solution is picked-up and safely disposed of by REHS. Based on the mode of action of chemicals, synchronized cells can be treated for shorter times before culturing in recording medium. Cells can also be treated in the recording medium for a longer time (several days).
6. Data Collection, Analysis, and Presentation
NOTE: Cell viability needs to be determined using standard methods (e.g., MTT assay) after treatment of cells at the same concentrations for the same times indicated. All treatment concentrations used in the in vitro bioluminescence assay were lower than the 30% lethal concentration (LC30). All experiments were conducted in triplicate and representative results were presented.
After sealing the dish (step 3.5), load the dishes onto the seats in the luminometer, which is kept inside an incubator set at 37 °C without H2O and CO2, and connected to a computer.
Click "save" and enter the names for the folder and file where the recorded luminescence signal from the corresponding dish will be saved. NOTE: The system detects the luminescence signal in real-time from the cells in the dish at individual positions. The signals are transferred to the computer through 4-photon-counting photomultiplier tubes, and saved and displayed in the computer by the data collection software.
Analyze the signals after recording for 4-7 days, which could be followed by medium change and continuous recording for a second week if necessary. NOTE: To obtain circadian parameters, including phase, period length, rhythm amplitude, and damping rate, we used the luminometer analysis software to analyze the bioluminescence data.
Detrend raw data with a running average, and analyze the best-fits to a sine wave to get period, phase, amplitude, and damping rate21,22.
Due to the high transient bioluminescence upon treatment and medium change, exclude the first cycle of data from analysis.
For data presentation, plot raw data (bioluminescence, count/s) against time (h or day). When necessary, baseline-subtracted data can be plotted to compare amplitude and phase.
Representative Results
Circadian bioluminescence reporter vector: human PER2 promoter-driven expression of destabilized luciferase variant
The DNA sequence comprising a 941 bp fragment derived from the human PER2 promoter used to construct the circadian reporter vector, pGL[hPer2P/Luc2P/Neo, was first analyzed for the presence of regulatory elements known to regulate circadian gene expression. Bioinformatics analysis showed that within this promoter fragment, there are three different Bmal1 binding sites (E-box) (CAT/CGTG), two circadian transcription enhancing sites (CCAATG), two GC boxes, and a transcription start site (Figure 1). Hence this reporter vector was anticipated to reflect circadian gene expression.
Optimization of synchronization agent
Several agents have been used to synchronize or affect the autonomous circadian rhythm of fibroblast cells for in vitro bioluminescence studies. To select an efficient and cost-effective cell-type specific synchronization agent in mammary epithelial cells, we compared 10 µM forskolin, 1 nM melatonin, 0.1 µM dexamethasone, 50% HS, and 100% SQ in mammary epithelial cells (MCF10A) transiently transfected with the circadian reporter vector. Cells synchronized with 50% HS showed the best circadian rhythm, with 3 peaks of circadian induced luminescence, compared to only one or two peaks in cells synchronized with other agents. In addition, the bioluminescence profiles of cells synchronized with 50% HS produced acceptable period, amplitude, and best-fit value during data analysis (Figure 2). Based on these findings, we selected this cost-effective method as the cell synchronization agent for all subsequent experiments.
Disruption and restoration of cellular circadian rhythm by chemical exposures
In this in vitro model, untreated, transiently transfected cells (control group) showed at least two complete cycles of luminescence signaling after synchronization (Figure 3A). Cells exposed to the direct acting mutagen, NMU, showed a dose-dependent disruption of circadian gene expression as reflected by the loss of luminescence peaks. While treatment with 0.25 mM NMU did not disrupt the cellular circadian rhythm, a dose of 0.5 mM NMU initially delayed and later abolished circadian rhythm, as indicated by the disappearance of the second peak of luminescence at ~72 h post-treatment. Inhibition of SIRT1 activity with 20 nM Ex257 or 1 µM cambinol similarly disrupted circadian rhythm by dampening the subsequent circadian cycle (Figure 3A-1). Importantly, the addition of MSC (12.5 µM) to the culture medium restored towards normal first and second cycles of circadian gene expression in NMU-treated cells. MSC not only restored the rhythm disrupted by NMU, but also prevented the disruptive effects of SIRT1 inhibitors, Ex257 and cambinol (Figure 3A-2).
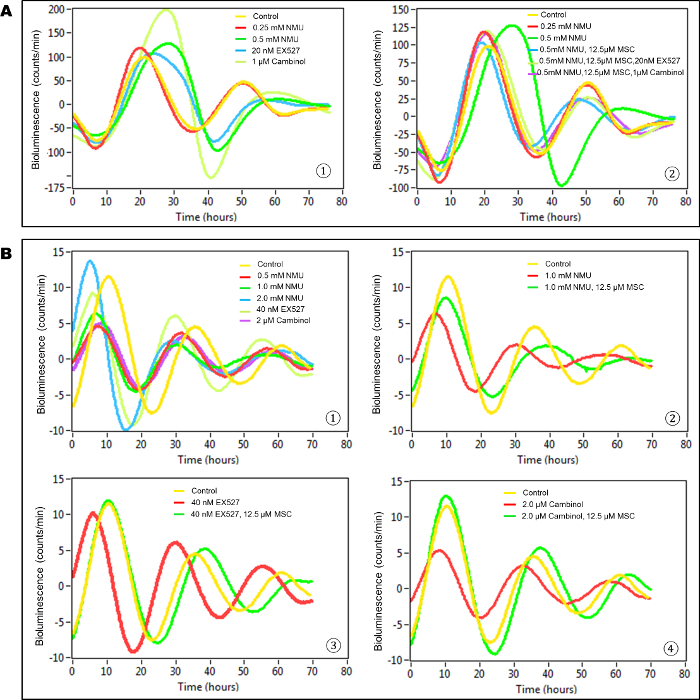
In stably transfected cells, both NMU and SIRT1 inhibitors disrupted circadian rhythm, albeit at higher concentrations than that observed in transient transfectants (Figure 3B-1). MSC (12.5 µM) restored circadian rhythms in the cells pretreated with 1 mM NMU (Figure 3B-2). More importantly, MSC also restored circadian rhythms in the cells treated with SIRT1 inhibitors, including EX257 (40 nM) (Figure 3B-3) and cambinol (2 µM) (Figure 3B(4)), respectively.
In comparison between Figure 3A and Figure 3B, the bioluminescence intensity was much higher but the rhythm was sustained for a shorter time in transiently transfected cells vs stably transfected cells (~ 10 times higher, 2 times shorter) in general, even after treatment of cells with NMU or MSC. In addition, the cellular circadian rhythm was 2-fold more sensitive to exposure to chemicals in transiently transfected cells compared to the stably transfected cells.
Quantitative dose-dependent change of cellular circadian rhythm by chemicals
Similarly, the disrupted cellular circadian rhythm of the stably transfected cells treated with 1 mM NMU was restored by the treatment of NAC in a dose-dependent manner (0-20 µM) (Figure 4A) as observed in the changes of circadian period (Figure 4B) and phase (Figure 4C).
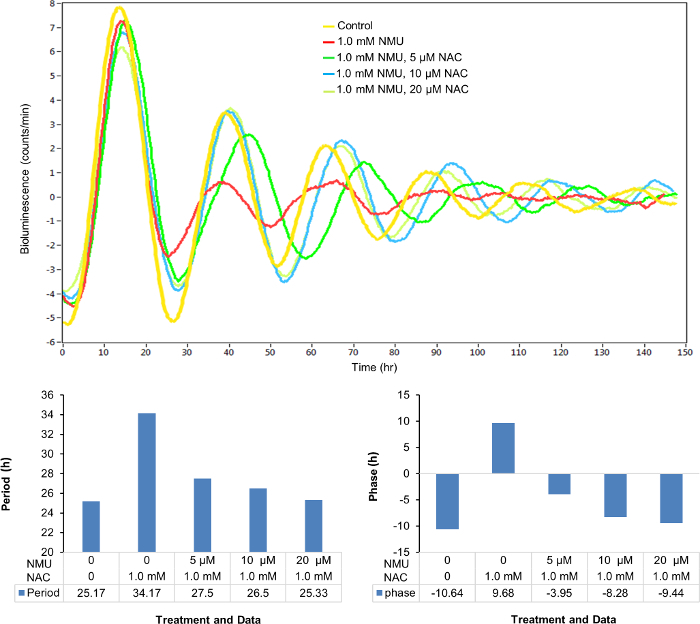
Figure 1: Sequence of human PER2 promoter fragment and the circadian reporter vector. (A) Human PER2 promoter fragment (941 bp) was sequenced, and the sequence was analyzed for the elements of functional circadian promoter. E-box, CACGTT / CATGTG; circadian transcription enhancing site, CCAATG; GC box, CGCCCC / GGGGCGGG; transcription starting site (TSS): CAGCGG. (B) Schematic diagram of destabilized luciferase reporter vector, pGL[hPer2P/Luc2P/Neo]. The transcription of destabilized luciferase (dLuc) is under direct control of the hPER2 promoter. A co-expressed neomycin resistance gene (Neo) facilitates selection of infected cells by G418. Please click here to view a larger version of this figure.
Figure 2: Synchronization of in vitro cellular circadian rhythm by different synchronization agents. After 24 h starvation, MCF10A cells transiently transfected by circadian reporter vector were treated with each synchronization agent for 2 h, followed by recording luminescence signal. Blue, 10 µM forskolin; red, 1 nM melatonin; green, 0.1 µM dexamethasone; purple; 50% horse serum (HS); teal, 100% SingleQuot (SQ); yellow, empty vector (negative control). X-axis, time (day); Y-axis, bioluminescence (count/min). Please click here to view a larger version of this figure.
Figure 3: MSC restored cellular circadian rhythms disrupted by NMU and SIRT1 inhibitors in mammary epithelial cells in vitro. Bioluminescence assays were performed on MCF10A/PER2-dLuc reporter cells after synchronization with 50% HS. X-axis, time (h) (post-NMU treatment time); Y-axis, bioluminescence (count/min). (A) Results from transiently transfected cells. (1) Cells were treated with 0, 0.25 or 0.5 mM NMU, 20 nM EX527, or 1 µM cambinol for 1 h following synchronization. Yellow: control; red: 0.25 mM NMU; green: 0.5 mM NMU; blue: 20 nM EX527; yellow green: 1µM cambinol. (2) Cells were treated with 12.5 µM MSC alone, or in combination with 20 nM EX527 or 1 µM cambinol in recording medium following exposure to 0.5 mM NMU. Yellow: Control; red: 0.25 mM NMU; green: 0.5 mM NMU; blue: 0.5 mM NMU + 12.5 µM MSC; yellow green: 0.5 mM NMU + 12.5 µM MSC + 20 nM EX527; purple: 0.5 mM NMU + 12.5 µM MSC + 1 µM cambinol. (B) Results from stably transfected cells. The 3rd- 5th peaks which showed clean and clear differences among groups are presented as a representative result. (1) Cells were treated with 0, 0.5, 1.0 or 2.0 mM NMU, 40 nM EX527, or 2 µM cambinol for 1 h after synchronization. Yellow: Control; red: 0.5 mM NMU; green: 1.0 mM NMU; blue: 2.0 mM NMU; yellow green: 40 nM EX527; purple: 2 µM cambinol. (2) Cells were treated with 12.5 µM MSC in recording medium following exposure to 1.0 mM NMU. Yellow: Control; red: 1.0 mM NMU; green: 1.0 mM NMU + 12.5 µM MSC. (3) Cells were treated with 12.5 µM MSC following exposure to 40 nM EX257. Yellow: Control; red: 40 nM EX257; green: 40 nM EX257 + 12.5 µM MSC. (4) Cells were treated with 12.5 µM MSC following exposure to 2 µM cambinol. Yellow: Control; red: 2 µM cambinol; green: 2 µM cambinol + 25 µM MSC. This result was previously reported and is licensed under CC BY 2.022. Please click here to view a larger version of this figure.
Figure 4: NAC dose-dependently restored the cellular circadian rhythm disrupted by NMU. Cells were treated with 1.0 mM NMU or vehicle control for 1 h after synchronization. (A) Representative result of bioluminescence (count/min) against time (h). Yellow: Control; red: 1.0 mM NMU; green: 1.0 mM NMU, 5 µM NAC; blue: 1.0 mM NMU, 10 µM NAC; yellow green: 1.0 mM NMU, 20 µM NAC. (B) Period (hours). (C) Phase (hours). Please click here to view a larger version of this figure.
Discussion
In mammalian cells, periodicity of the circadian clock is regulated by interconnected transcriptional/translational feedback loops. Heterodimers of Bmal1 and either Clock or Npas2 regulate circadian transcription by binding to E-box elements in the promoters of core CGs, including Per2 and Cry, and numerous CCGs4. As they accumulate in the cell, heterodimers of Per:Cry are post-translationally modified and transported to the nucleus to repress Clock:Bmal1 transcriptional activity. This negative feedback loop allows core CGs to regulate their own transcription and set up the rhythmic expression of CGs and CCGs23. Although most mammalian cells have an intrinsic circadian clock, the clocks in individual cells can be synchronized by both intrinsic conditions (e.g., redox potential) and external environmental stimuli24. Circadian gene expression in vivo can be synchronized by melatonin, a hormone produced by the pineal gland in response to signals received from the central pacemaker (SCN). The SCN integrates signals from light impinging on specialized melanospin-expressing photosensitive retinal ganglion cells in the retina. Melatonin released from the pineal gland enters the circulation and regulates circadian rhythm of SCN and most peripheral cells in vitro and in vivo25,26. Circadian rhythm can also be regulated by stress hormones (e.g., cortisol and catecholamines)27,28 and growth factors29,30. Deregulation of growth factors is known to be associated with circadian disruption of cell growth and differentiation31.
The circadian reporter vector we constructed exploits this negative biochemical feedback mechanism. The core circadian negative feedback loop consists of transcriptional activators, BMAL1 and CLOCK, which bind to the circadian E/E´-box binding motifs in PER2 promoter, and repressors PERs and CRYs, which negatively regulate BMAL1 expression. In the present reporter system, the rhythmic activation of the PER2 promoter by BMAL1 and CLOCK facilitates circadian expression of destabilized luciferase, allowing for a more accurate circadian periodicity of luminescence signals. The construction of a reporter plasmid includes the BMAL1 binding site, the circadian rhythm enhancing site, GC box, and the transcription start site of the hPER2 promoter. The vector allows for real-time readout for cellular autonomic circadian rhythm in transfected cells and can be used to identify conditions or specific agents that alter this endogenous circadian pattern of gene expression. However, when cells are cultured in vitro or ex vivo, their circadian oscillators quickly become desynchronized. It is therefore necessary to resynchronize their rhythms before any circadian assays can be performed in vitro. In mammary epithelial cells, we found that 50% HS, melatonin, and 100% SQ were each able to induce efficient synchronization of the circadian rhythm in vitro. Dexamethasone and forskolin produced only partial synchronization. Dose-dependent tests would be necessary for the development and selection of new synchronizing agents in future studies. Given the low cost, efficiency and its wide use as the synchronization agent, 50% HS was found to be an effective synchronization agent that produced robust and reproducible cellular circadian rhythm in mammary epithelial cells and neural cells, as previously reported in many other cell types and different experimental settings24.
To validate the in vitro model of circadian regulation, we next asked if the response of the circadian rhythm to environmental stressors in vitro would recapitulate the effects we observed in vivo. Previously, we demonstrated that a single carcinogenic dose of NMU disrupted the circadian expression of major CGs (e.g., Per2) and several CCGs in the target mammary gland. Moreover, a chemopreventive regimen of MSC reset the circadian expression of both Per2 and CCGs genes towards the normal. We further demonstrated that exposure of cells to toxicants and stressors can alter circadian gene expression through their effects on NAD+-dependent SIRT1 activity7,8,22. The results obtained using the in vitro circadian reporter assay showed that exposure to NMU caused disruption of cellular circadian gene expression, and that more interestingly, MSC is not only able to reset the cellular circadian rhythm disrupted by NMU, but also reset the circadian rhythm disrupted by the SIRT1 inhibitors. These results indicated that the in vitro assay not only reproduces the effects of NMU on circadian gene expression, but also reproduces the effects of MSC on the disrupted circadian rhythm in vivo via SIRT1-dependent mechanisms. The in vitro circadian reporter system thus has the potential to detect a variety of toxicants and stressors that alter NAD+/NADH levels in the cell, including direct inhibitors and modulators of cellular redox cycling as well as genotoxic agents that can deplete NAD+ via Poly ADP-ribose dependent DNA repair32. These results suggest that the in vitro reporter system provides a useful tool to screen for cell conditions and chemical agents that can disrupt or restore circadian gene regulation in vivo.
Although the in vitro assay can be set up using cells transiently or stably transfected with the reporter construct, some differences were noted in the response to the treatment of chemicals between transiently and stably transfected cells. The stably transfected cells were more resistant to the treatment of chemicals, particularly NMU and SIRT1 inhibitors, than transiently transfected cells. These differences could reflect subtle differences in regulation of the vector promoter in stable transfectants due to the influences from the sequences surrounding specific integration sites. Therefore, selection of stable transfectants by antibiotic treatment may also select clones that are more resistant to circadian disruption. Thus, individual in vitro cell models using transient or stable transfectants must be validated for their ability to recapitulate in vivo responses. Different transfection methods also induce varying levels of genotoxic stress and toxicity on cells and result in their different susceptibility to toxic effects of chemicals. Therefore, cytotoxicity test and pilot experiments are required for the selection of transfection and treatment conditions. Despite the difference in their sensitivity, the overall results obtained with our mammary epithelial circadian reporter cells for disruption and restoration of cellular circadian rhythm by chemicals were consistent and comparable between transiently transfected and stably transfected cells, and the results recapitulated the effects we observed in vivo.
To determine if the circadian reporter vector could be used in other cell types, human glioblastoma/astrocytoma cells (U-87 MG) were stably transfected with this circadian vector. The preliminary results using the in vitro cellular bioluminescence assay showed that after synchronization, these neural-derived tumor cells have a regular and strong cellular circadian rhythm comparable to that observed in human mammary epithelial cells. Cells treated with IC261, a well-known inhibitor of casein kinase 1 δ (CK1δ) and CK1 ε, showed a prolonged circadian period, but lower amplitude as reported by others (data not shown)33. These results indicate that the circadian luciferase reporter vector and in vitro cellular bioluminescence assay are applicable in other organ specific cells, including neural cells.
The cellular reporter system developed here could therefore be used broadly as an in vitro assay for determination of circadian gene expression in different cell types in general. The experimental procedure is simple, fast, and safe. However, this cellular reporter system cannot be readily adapted in cells that are difficult to transfect, non-dividing cells (e.g., neuronal cells), since they have notoriously low transfection efficiencies. Virus-based transfection methods (e.g., lentivirus transfection) may offer a higher efficiency solution for these cells34. However, the time and difficulty associated with lentiviral production makes this method difficult and time consuming. Moreover, a proper safety-level lab is necessary for production and use of the virus. In addition, high resolution single cell luminescence detectors could be used for the determination of persistent, independently-phased rhythms of circadian gene expression in non-dividing cells35.
Studies of mammalian circadian rhythm in vivo are not only labor intensive, and costly, but they also require the use of large numbers of animals. The availability of in vitro systems could significantly reduce the number of animals required to test associations between disruption of circadian gene expression and the development of chronic diseases, including carcinogenesis and metabolic syndrome. Quantitative data on circadian parameters, including period, amplitude, and phase, can inform dose- and/or time-dependent effects of chemicals on cellular circadian rhythm. For example, we observed that antioxidant NAC can dose-dependently restore the period and phase of cellular circadian rhythm disrupted by NMU. The in vitro model could also be used to screen and classify environmental circadian disruptors and to investigate the impact of disrupted circadian rhythm on the impairment of cellular function, providing mechanistic insights for the development of intervention strategies for cohorts at increased risk to breast cancer, metabolic syndromes, and neurophysiological disorders, because of abnormal work schedules.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the 2012 Society of Toxicology (SOT)-Colgate Palmolive Grant for Alterative Research (M. Fang) and the international collaboration research fund from Animal and Plant Quarantine Agency, Republic of Korea (M. Fang), and the NIEHS grant P30ES005022 (H. Zarbl). We would like to thank Dr. Zheng Chen (McGovern Medical School at The University of Texas Health Science Center at Houston) for his helpful discussion, Mr. Shao-An Juan for his experimental assistance, and Ms. Kimi Nakata for her proof reading.
References
- Akerstedt T, et al. Night work and breast cancer in women: a Swedish cohort study. BMJ Open. 2015;5(4):e008127. doi: 10.1136/bmjopen-2015-008127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent ME, El-Zein M, Rousseau MC, Pintos J, Siemiatycki J. Night work and the risk of cancer among men. Am J Epidemiol. 2012;176(9):751–759. doi: 10.1093/aje/kws318. [DOI] [PubMed] [Google Scholar]
- Knutsson A, et al. Breast cancer among shift workers: results of the WOLF longitudinal cohort study. Scand J Work Environ Health. 2013;39(2):170–177. doi: 10.5271/sjweh.3323. [DOI] [PubMed] [Google Scholar]
- Fu L, Lee CC. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer. 2003;3(5):350–361. doi: 10.1038/nrc1072. [DOI] [PubMed] [Google Scholar]
- Fu L, Kettner NM. The circadian clock in cancer development and therapy. Prog Mol Biol Transl Sci. 2013;119:221–282. doi: 10.1016/B978-0-12-396971-2.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. Painting, Firefighting, and Shiftwork. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 2010;98:563–764. [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zarbl H. Chemopreventive doses of methylselenocysteine alter circadian rhythm in rat mammary tissue. Cancer Prev Res (Phila Pa) 2008;1(2):119–127. doi: 10.1158/1940-6207.CAPR-08-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang MZ, Zhang X, Zarbl H. Methylselenocysteine resets the rhythmic expression of circadian and growth-regulatory genes disrupted by nitrosomethylurea in vivo. Cancer Prev Res (Phila) 2010;3(5):640–652. doi: 10.1158/1940-6207.CAPR-09-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla LF, Kruger R. Converging environmental and genetic pathways in the pathogenesis of Parkinson's disease. J Neurol Sci. 2011;306(1-2):1–8. doi: 10.1016/j.jns.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Aitlhadj L, Avila DS, Benedetto A, Aschner M, Sturzenbaum SR. Environmental exposure, obesity, and Parkinson's disease: lessons from fat and old worms. Environ Health Perspect. 2011;119(1):20–28. doi: 10.1289/ehp.1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagoshi E, Brown SA, Dibner C, Kornmann B, Schibler U. Circadian gene expression in cultured cells. Methods Enzymol. 2005;393:543–557. doi: 10.1016/S0076-6879(05)93028-0. [DOI] [PubMed] [Google Scholar]
- Ramanathan C, et al. Cell type-specific functions of period genes revealed by novel adipocyte and hepatocyte circadian clock models. PLoS Genet. 2014;10(4):e1004244. doi: 10.1371/journal.pgen.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporl F, et al. A circadian clock in HaCaT keratinocytes. J Invest Dermatol. 2011;131(2):338–348. doi: 10.1038/jid.2010.315. [DOI] [PubMed] [Google Scholar]
- Zhang EE, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139(1):199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101(15):5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc GM, Boockfor FR, Faught WJ, Frawley LS. Development of a destabilized firefly luciferase enzyme for measurement of gene expression. Biotechniques. 2000;29(3) doi: 10.2144/00293rr02. [DOI] [PubMed] [Google Scholar]
- Genomics S. Technical Note: SwitchGear methods for gene model construction and transcription start site prediction. 2009.
- Scientific F. Safety Data Sheet: Ethidium bromide, 1% Solution/Molecular Biology. 2010.
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. New York: Greene Publishing Associates and Wiley-Interscience; 1994. [Google Scholar]
- Yoo SH, et al. A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc Natl Acad Sci U S A. 2005;102(7):2608–2613. doi: 10.1073/pnas.0409763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, et al. Identification of diverse modulators of central and peripheral circadian clocks by high-throughput chemical screening. Proc Natl Acad Sci U S A. 2012;109(1):101–106. doi: 10.1073/pnas.1118034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Guo WR, Park Y, Kang HG, Zarbl H. Enhancement of NAD+-dependent SIRT1 deacetylase activity by methylselenocysteine resets the circadian clock in carcinogen-treated mammary epithelial cells. Oncotarget. 2015. [DOI] [PMC free article] [PubMed]
- Chen-Goodspeed M, Lee CC. Tumor suppression and circadian function. J Biol Rhythms. 2007;22(4):291–298. doi: 10.1177/0748730407303387. [DOI] [PubMed] [Google Scholar]
- Balsalobre A. Clock genes in mammalian peripheral tissues. Cell Tissue Res. 2002;309(1):193–199. doi: 10.1007/s00441-002-0585-0. [DOI] [PubMed] [Google Scholar]
- Jung-Hynes B, Huang W, Reiter RJ, Ahmad N. Melatonin resynchronizes dysregulated circadian rhythm circuitry in human prostate cancer cells. J Pineal Res. 2010;49(1):60–68. doi: 10.1111/j.1600-079X.2010.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gall C, et al. Melatonin plays a crucial role in the regulation of rhythmic clock gene expression in the mouse pars tuberalis. Ann N Y Acad Sci. 2005;1040:508–511. doi: 10.1196/annals.1327.105. [DOI] [PubMed] [Google Scholar]
- Vujovic N, Davidson AJ, Menaker M. Sympathetic input modulates, but does not determine, phase of peripheral circadian oscillators. Am J Physiol Regul Integr Comp Physiol. 2008;295(1):R355–R360. doi: 10.1152/ajpregu.00498.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofl C, Becker C, Prank K, von zur Muhlen A, Brabant G. Twenty-four-hour rhythms of plasma catecholamines and their relation to cardiovascular parameters in healthy young men. Eur J Endocrinol. 1997;137(6):675–683. doi: 10.1530/eje.0.1370675. [DOI] [PubMed] [Google Scholar]
- Yu H, et al. Circadian rhythm of circulating fibroblast growth factor 21 is related to diurnal changes in fatty acids in humans. Clin Chem. 2011;57(5):691–700. doi: 10.1373/clinchem.2010.155184. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, et al. Modulation of circadian rhythm of DNA synthesis in tumor cells by inhibiting platelet-derived growth factor signaling. J Pharmacol Sci. 2008;107(4):401–407. doi: 10.1254/jphs.08080fp. [DOI] [PubMed] [Google Scholar]
- Yang X, Guo M, Wan YJ. Deregulation of growth factor, circadian clock, and cell cycle signaling in regenerating hepatocyte RXRalpha-deficient mouse livers. Am J Pathol. 2010;176(2):733–743. doi: 10.2353/ajpath.2010.090524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag L. Structure and function of poly(ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Curr Vasc Pharmacol. 2005;3(3):209–214. doi: 10.2174/1570161054368625. [DOI] [PubMed] [Google Scholar]
- Kon N, Sugiyama Y, Yoshitane H, Kameshita I, Fukada Y. Cell-based inhibitor screening identifies multiple protein kinases important for circadian clock oscillations. Commun Integr Biol. 2015;8(4):e982405. doi: 10.4161/19420889.2014.982405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan C, Khan SK, Kathale ND, Xu H, Liu AC. Monitoring Cell-autonomous Circadian Clock Rhythms of Gene Expression Using Luciferase Bioluminescence Reporters. J Vis Exp. 2012. p. e4234. [DOI] [PMC free article] [PubMed]
- Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol. 2004;14(24):2289–2295. doi: 10.1016/j.cub.2004.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]