

Abstract
Human tumor growth depends on rapidly dividing cancer cells undergoing population expansion. Even advanced tumors, however, contain slowly proliferating cancer cells for reasons that remain unclear. Here, we selectively disrupt the ability of rapidly proliferating cancer cells to spawn AKT1low daughter cells that are rare, slowly proliferating, tumor-initiating, and chemotherapy-resistant using β1-integrin activation and the AKT1-E17K mutant oncoprotein as experimental tools in vivo. We find that selective depletion of AKT1low slow proliferators actually reduces the growth of a molecularly diverse panel of human cancer cell xenograft models without altering global cell proliferation or survival in vivo. Moreover, we find that unusual cancer patients with AKT1-E17K-mutant solid tumors also fail to produce AKT1low quiescent cancer cells and that this correlates with significantly prolonged survival after initial treatment compared to other patients. These findings support a model whereby human solid tumor growth depends on not only rapidly proliferating cancer cells but also on the continuous production of AKT1low slow proliferators.
Keywords: Quiescent cancer cells, Slow proliferators, AKT1, β1-Integrin, cell heterogeneity
INTRODUCTION
Human tumors are admixtures of rapidly and slowly dividing cancer cells. Rapid proliferators clearly dictate growth, progression, and response to chemotherapy in all cancer types, but most cancer cells proliferate very slowly or not at all within human tumors (1). Moreover, clinical evidence suggests that some slow proliferators may actually remain quiescent for prolonged periods, where they are highly resistant to treatment of various types, only to eventually re-enter the cell cycle, replicate, and cause disease relapse (2). While rapid proliferators evolve through clonal selection over time, we do not understand why human tumors produce so many slow proliferators (3). Generally, the proliferative rate of a cancer cell depends on its time spent in G1 of the cell cycle. Oncogenic changes accelerate G1 transit, but a suboptimal tumor microenvironment with imbalances in growth factors, nutrients, or oxygen can slow transit through G1, or induce autophagy, senescence, or apoptosis where cells stop proliferating altogether (4). Rapidly proliferating cancer cells may also experience random aberrancies in DNA replication or cell division that activate checkpoints to temporarily arrest cell cycle progression. In addition, host cells can stunt proliferating cancer cells through both direct and indirect effects. Against this complex backdrop, individual cancer cells within tumors will proliferate at different rates depending on the precise balance of these extrinsic and intrinsic factors. Therefore, many believe that quiescent cancer cells within tumors simply represent extreme examples of these stochastic forces, which complicate the diagnosis and treatment of cancer patients, and are difficult to tackle.
A wide variety of human cancer cell lines growing in culture produce quiescent cells continuously, and at low frequency, regardless of cancer type or oncogenomic profile (i.e., 1 – 2% of the overall population) (5). Using these lines, we recently discovered that proliferating cancer cells sensing an irregular extracellular collagen matrix (ECM) via their β1-integrin receptor during mitosis will divide asymmetrically to spawn quiescent daughter cells (5, 6). A conserved signaling mechanism regulates this special type of cell division – specifically, sub-optimal β1-integrin ligation reduces focal adhesion kinase activity, which induces the mTORC2 kinase complex to partially activate the AKT1 protein kinase, resulting in TTC3-mediated ubiquitination and partial degradation of AKT1 by the proteasome (6). AKT1low progeny produced in this way are not autophagic, senescent, or apoptotic but, rather, temporarily quiescent in a unique “G0-like” state marked by the MCM2low/H3K9me2low/HES1high profile (5–7). With β1-integrin re-engagement, however, these AKT1low quiescent cancer cells (QCCs) reactivate their AKT signaling to immediately resume cycling, divide normally, and produce progeny that are indistinguishable from other actively dividing cells within the population (6).
AKT1low QCCs intrinsically resist cytotoxic stress of various types and also extrude CD63+ exosomes that can promote the stress-resistance of proliferating neighbors (5, 7). Additionally, these QCCs secrete WNT-, TNF-, and VEGF-related proteins, suggesting they communicate with their microenvironment to modulate local inflammation in complex ways (7). QCCs do not express markers typically associated with “cancer stem cells” (CSCs), but they do manifest characteristics definitional of these difficult to study subpopulations, including an increased tumor-initiating capacity compared to AKT1high rapid proliferators when injected into immune-compromised mice in vivo (7–10). Curiously, QCCs can also up-regulate the JARID1B histone demethylase to differentiate into more rapidly cycling AKT1high/JARID1Bhigh CSCs that promote experimental tumor growth, suggesting that AKT1low QCCs and AKT1high/JARID1Bhigh cancer cells may in fact represent distinct states within a dynamic CSC pool (10–15). AKT1low QCCs are not, however, a fixed, discrete subpopulation from which all other cancer cells arise. Rather, any proliferating cancer cell may dynamically produce AKT1low QCCs depending on local micro-environmental conditions within tumors (5, 6). Importantly, we have also found that human tumors actually contain small QCC fractions prior to treatment (i.e., ~ 1 – 2% of malignant cells), which can survive combination chemotherapy given to patients over 4 – 6 months, suggesting QCCs may in fact be able to remain quiescent over long periods of time to mediate clinically relevant treatment resistance (5, 16). Given these remarkable observations, here, we asked whether solid tumor growth might actually depend on rapidly proliferating cancer cells producing AKT1low cancer cells that are rare, quiescent, tumor initiating, and treatment-resistant.
MATERIALS AND METHODS
A detailed description of methods and computational analysis is provided in a Supplementary file.
Cell lines
A375 melanoma, PC9 lung, MDA-MB-231 breast, HCT116 colon, and MCF7 breast human cancer cell lines were purchased from ATCC, where they were validated. HCT116 AKT1/2−/− was purchased from Horizon Discovery, where it was validated. A375, MDA-MB-231, and MCF7 were maintained in DMEM, 10% FCS, 4 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin; HCT116 and HCT116 AKT1/2−/− in McCoy’s 5α medium supplemented with 10% FCS, 100 U/mL penicillin, and 100 μg/mL streptomycin; PC9 in RPMI, 10% FCS, 25% glucose, 1% sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin. All the cells were grown at 37°C and 5% CO2.
DNA constructs and viral infection
The double-strand DNA sequence of AKT1 (NM_001014431.1) was synthesized and cloned into pLVX-One by GenScript. The AKT1 sequence was then amplified by PCR and cloned into plasmid pTRIPZ. Virus carrying the desired fusion gene was produced using established protocols.
Cell immunofluorescence
Cells were grown directly on collagen IV–coated coverslips (Sigma). Cells were fixed in 3.7% formaldehyde, permeabilized using 0.1% Triton X-100, and treated with 0.1% SDS. They were blocked in 1% BSA and then incubated with primary antibody diluted in blocking solution, washed, and incubated with the respective secondary antibody. QCCs were identified using the previously validated markers H3K9me2 (Abcam), Hes1 (Abnova), and MCM2 (Cell Signaling), as described in Dey-Guha, 2015 (6). All secondary antibodies were Alexa Fluor conjugates (488, 555, 568, 633, and 647; Invitrogen).
Flow cytometry
Cells were fixed with cold methanol for 30 minutes at −20°C followed by PBS wash. AKT1 antibody incubation was performed in PBS containing 10% FBS for blocking. After 3 hours, cells were washed 3x with PBS and incubated with NucBlue Fixed Cell ReadyProbes Reagent (Invitrogen) for DNA content. Flow cytometry analysis was performed in a Becton Dickinson FACSAria II. Akt1 Alexa Fluor647 Conjugate was used (Cell Signaling).
Western blots
We used standard protocols for SDS-PAGE electrophoresis and used the following primary antibodies: AKT1, phospho-AKT1 (S473), S6, phospho-S6 (S235) from Cell Signaling and GAPDH (Sigma).
Xenograft tumors in vivo
Animal experiments were carried out under Massachusetts General Hospital Institutional Review Board–approved protocol. 5×105 cells were injected subcutaneously into the flanks of 8-week-old, female immunocompromised NU/NU mice (Charles River Laboratories), and growing tumors were measured by caliper. For genetic experiments inducing AKT1-WT and AKT1-E17K, mice were given water containing 2 μg/mL of doxycycline continuously two days after cell injection. For antibody/chemotherapy treatment, once the tumors were palpable, mice were treated with TS2/16 antibody - 18 mg/kg IP, every week for 5 weeks - or Paclitaxel (Sigma T7191 5mg) - 20 mg/kg IP, every week for five weeks. For production of TS2/16 antibody, the hybridoma clone HB243 was acquired from ATCC and antibody production/isolation was performed by the DFCI Monoclonal Antibody Core.
For tissue immunofluorescence, 5 μm sections of formalin-fixed paraffin-embedded (FFPE) tissues were de-waxed with xylene and rehydrated. Antigen retrieval was achieved by microwaving in unmasking solution (Vector Laboratories). After washing, sections were blocked in 5% FCS and incubated with primary antibody, washed, and incubated with the respective secondary antibody. H&E slides were used to assess the morphological integrity and geographical variation in morphology of the tissue samples. The slides were analyzed under the LEICA DC500 microscope.
RESULTS
To address this question, we first employed a pharmacologic approach to reduce QCCs in vivo. We previously found that the production of AKT1low QCCs, which arise in cancer cell populations through loss of β1-integrin signaling, can be eliminated through activation ofβ1-integrin on the cancer cell surface with a monoclonal antibody in vitro (6). Therefore, we injected parental A375, PC9, MDA-MB-231, HCT-116, and MCF-7 cells subcutaneously into nude mice to form palpable tumors. We then treated tumor-bearing mice with TS2/16 (a monoclonal antibody that activates human β1-integrin allosterically through a ligand-independent mechanism), in order to similarly reduce QCCs in vivo (17). We found that TS2/16 treatment indeed reduced QCCs while consistently impeding tumor growth in all five xenograft models in vivo (Figure 1A,B). Experimental and control bulk tumors were not different in proliferating (MKI67+) cell fractions, even though integrin activation has been associated with the increased replication and survival of cancer cells. A trend towards a higher percentage of apoptotic cells (CASP3+) was noted in TS2/16-treated tumors, however, which was only statistically significant in the A375 and MDA-MB-231 models. (Figure 1B) (18). In contrast, paclitaxel treatment reduced active proliferators (MKI67+) and impeded tumor growth in these same xenografts (Figure 1A,B). However, this intensive chemotherapy did not affect (and in some cases actually enriched for) QCCs, functionally confirming their slow-cycling, treatment-resistant nature in vivo (Figure 1B). As expected, the magnitude of tumor growth inhibition with paclitaxel treatment (which reduced rapid proliferators) was directly proportional to maximal growth rate (Vmax) across models (Figure 1C). Interestingly, we also noted a similar correlation with TS2/16 treatment that selectively reduced QCCs (Figure 1C). These surprising results were consistent with rare, quiescent, chemo-resistant malignant cells contributing to the growth of xenograft tumors independent of MKI67+ active proliferators in vivo.
Fig. 1. Activation of β1-integrin reduces QCCs and tumor growth in vivo.
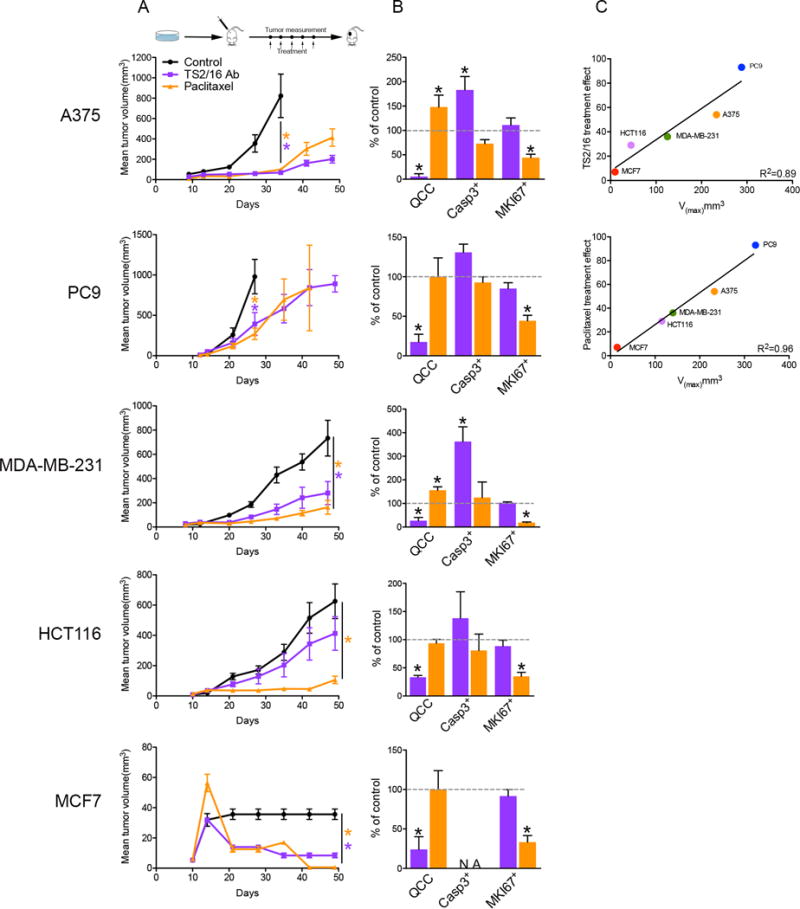
Mice with subcutaneous tumors were treated with either TS2/16 monoclonal antibody (18 mg/kg, i.p.) (purple) or paclitaxel (20 mg/kg, i.p.) (gold) once a week for 5 weeks versus untreated control (black) in 5 different xenograft models (n = 8 mice/arm). Tumor growth (A) and bar graph percentages of QCCs, cleaved-Caspase3+, and MKI67+ cells in TS2/16-treated (purple) or paclitaxel-treated (gold) versus untreated control tumors (B) in different xenograft models is shown. Error bars indicate mean ± SEM, * = p < 0.05. (C) Correlation of maximum velocity of tumor growth (Vmax) versus treatment effect for TS2/16 and paclitaxel.
To further confirm these results, we also designed a second, genetic approach to disrupt the ability of highly proliferative cancer cells to continuously produce AKT1low QCCs, based on detailed molecular knowledge of their genesis. AKT1low cancer cells are produced via degradation of the AKT1 kinase protein (6). In addition, allosteric AKT small-molecule inhibitors bind the AKT1 pleckstrin homology domain, disrupt AKT1 localization at the cell membrane, and induce proteasome-mediated AKT1 degradation (19). At sub-lethal doses, these allosteric inhibitors dramatically increase the QCC fraction in cancer cell lines, unlike catalytic inhibitors that directly interfere with kinase activity (5–7). In contrast, HCT116-AKT1/2−/− cells (an HCT116 variant with adeno-associated virus (AAV)-mediated disruption of the AKT1 and AKT2 genes) fail to produce QCCs, but lentiviral-mediated over-expression of a cDNA encoding wild-type AKT1 completely restores production of these MCM2low/H3K9me2low/HES1high cells in this AKT knockout line (Figure 2A) (6). Moreover, over-expression of a cDNA for myr-AKT1 (an artificial, mutant, kinase-active protein that constitutively localizes to the cell membrane) failed to rescue the production of QCCs in HCT116-AKT1/2−/− cells, in contrast to wild-type AKT1 (Figure 2A) (20).
Fig. 2. AKT1-E17K disrupts QCCs.
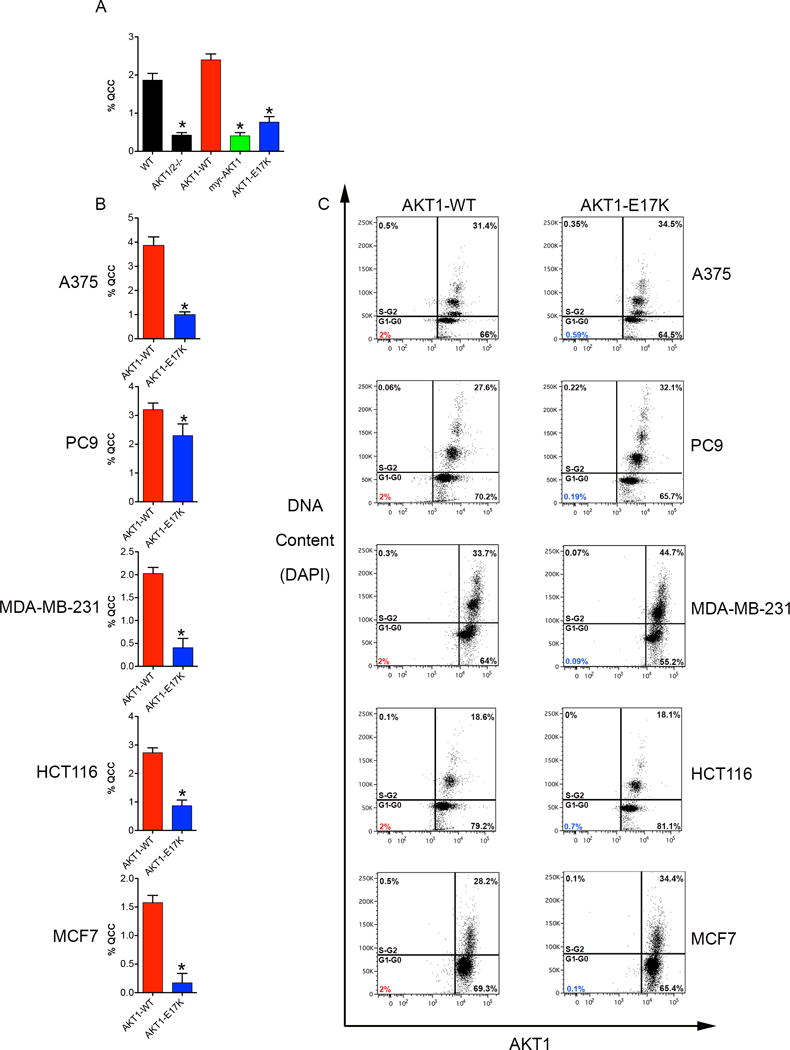
(A) Bar graph percentages of QCCs in the HCT116-AKT1/2−/− human cancer cell line with cDNAs for AKT1-WT or AKT1 mutants. (B) Bar graph percentages of QCCs in human cancer cell lines ectopically expressing inducible AKT1-WT or AKT1-E17K. Error bars indicate mean ± SEM for three independent experiments, * = p < 0.05. (C) Flow cytometry analysis of cell cycle (DAPI) and AKT1 levels.
Based on these experimental observations, we also tested a naturally occurring analogue of myr-AKT1. AKT1-E17K is a mutant protein, encoded by a heterozygous point mutation, which is found recurrently (but at very low frequency) in human tumors of many different types (E17K; c.49G>A) (21). It results from a glutamic acid-to-lysine substitution at amino acid 17, in the lipid-binding pocket of the AKT1 pleckstrin homology domain, and constitutively localizes to the cell membrane to pathologically activate AKT1 kinase, consistent with oncogenic function. Similar to myr-AKT1, we found that AKT1-E17K also failed to restore QCC production in the HCT116-AKT1/2−/− line (Figure 2A). This finding raised the possibility that AKT1-E17K might be a useful tool to disrupt QCCs in wild-type cancer cells.
To test this hypothesis, we engineered doxycycline-inducible lentiviral vectors expressing either AKT1-E17K or AKT1-wild type and transduced these constructs into five different, molecularly diverse human cancer cell lines (i.e., A375 (melanoma), PC9 (lung adenocarcinoma), MDA-MB-231 (breast “ER-”), HCT116 (colorectal), and MCF7 (breast “ER+”) cells). Indeed, we found that induction of AKT1-E17K reduced the QCC fraction in each of these parental cell lines compared to AKT1-WT (Figure 2B). We further confirmed this finding using flow cytometry with AKT1low QCCs defined as the bottom 2% of AKT1-expressors in the G0/G1 cell cycle fraction (Figure 2C) (5, 6). These surprising results suggested that AKT1-E17K acted in a dominant-negative manner to disrupt the production of QCCs.
Importantly, doxycycline induced equivalent levels of AKT1-E17K and AKT1-WT protein in cell line pairs, but did not alter their overall cell cycle profile compared to AKT1-WT (based on DNA content) (Figure 2C, Supp. Figure 1). In addition, AKT1-E17K and AKT1-WT overexpression did not increase AKT pathway activation beyond the high baseline already present in wild-type cells (as evidenced by equivalent levels of phospho-AKT1 and phospho-S6RP, a canonical downstream signaling target, in parental, AKT1-E17K, and AKT1-WT cells) (Supp. Figure 1) (22). Compared to AKT1-WT, AKT1-E17K also did not alter cancer cell: 1) proliferation (i.e., in normal fetal calf serum (10%), low serum (1%), or low oxygen (4%) conditions), 2) colony formation (i.e., on plastic or soft-agar), 3) translocation (i.e., migration or invasion), or 4) growth as multicellular tumor spheroids (Supp. Figure 2,3). These findings demonstrated that exogenous AKT1-E17K selectively reduced QCCs without appreciably altering the overall behavior of molecularly diverse human cancer cell lines growing under standard 2-D and 3-D assay conditions in vitro.
We therefore asked whether AKT1-E17K-mediated reduction in QCCs would affect the growth of experimental xenograft tumors, since QCCs have a greater tumor-initiating capacity than rapid proliferators when injected into immune-compromised mice in vivo (7, 10). We injected A375, PC9, MDA-MB-231, HCT-116, and MCF-7 cells carrying either inducible AKT1-E17K or AKT1-WT subcutaneously into immune-compromised nude mice. After waiting 48 hours, we fed these mice doxycycline to induce the expression of AKT1-E17K or AKT1-WT protein in growing xenografts. Indeed, we found that AKT1-E17K expression both reduced the 1 – 2% fraction of QCCs and also consistently impeded tumor growth in all five xenograft models (Figure 3A,B). Overall, however, AKT1-E17K tumors did not show consistently significant differences in apoptotic (CASP3+) or proliferating (MKI67+) cell fractions compared to AKT1-WT tumors (Figure 3B). Also, we did not detect consistent alteration in the frequency of senescent cells, as accessed by β-Gal staining (Supp. Figure 4) AKT1-E17K tumors (without small QCC fractions) also did not show consistent differences with AKT1-WT tumors in global sensitivity to paclitaxel chemotherapy, which kills actively dividing cells (Supp. Figure 5). These findings demonstrated that AKT1-E17K selectively depleted rare QCCs, while impeding the growth of cancer cell populations as xenograft tumors, without globally affecting their overall proliferation, survival, or resistance to iatrogenic stress in vivo.
Fig. 3. AKT1-E17K reduces QCCs and tumor growth in vivo.
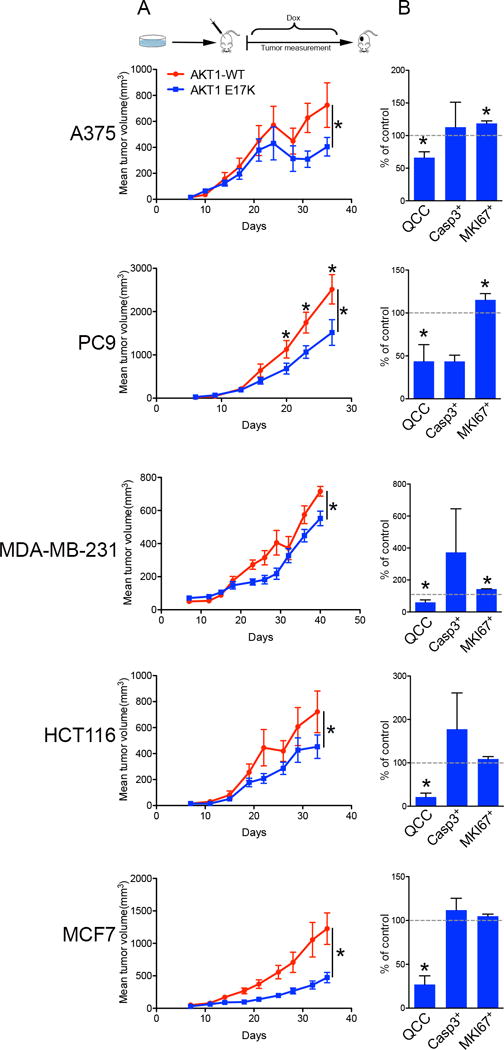
Human cancer cells ectopically expressing inducible AKT1-WT or AKT1-E17K were injected into mice, with induction two days after injection, in 5 different xenograft models (n = 10 mice/arm). Tumor growth (A) and bar graph percentages of QCCs, cleaved-Caspase3+, and MKI67+ cells in AKT1-E17K versus AKT1-WT tumors (B) in different xenograft models is shown. Error bars indicate mean ± SEM, * = p < 0.05.
In light of these experimental results, we asked if QCCs were also associated with clinical disease progression in cancer patients. We analyzed rare breast tumors with heterozygous, somatic, AKT1-E17K mutation for QCCs (23). Interestingly, AKT1-E17K breast tumors had a significantly lower AKT1low/H3K9me2low/HES1high QCC fraction than wild-type AKT1 tumors (p = 0.027) (Figure 4A,B, Supp. Table 1A). This finding raised the intriguing possibility that patients with AKT1-E17K breast tumors, which produce fewer QCC, might also have a less aggressive disease course than patients with AKT1-WT breast tumors. To test this hypothesis, we turned to The Cancer Genome Atlas (TCGA) database. Since the AKT1-E17K mutant is observed most frequently in the ER+ breast cancer subset, we identified unusual patients with primary ER+ breast tumors expressing either AKT1-WT (n = 662) or heterozygous AKT1-E17K mutation with at least six months clinical follow-up or a death event (n = 20) (i.e., ~2% of total) (Supp. Table 1B) (23, 24). Indeed, we found that ER+/AKT1-E17K patients had a significantly better overall survival compared to ER+/AKT1-AKT1-WT patients in this retrospective analysis (p = 0.025) (Figure 4C). In fact, ER+/AKT1-E17K patients followed a distinctly indolent clinical course compared to patients with ER+/AKT-WT tumors from other known breast cancer molecular subsets (i.e., ER+/Luminal A, ER+/Luminal B, or ER-) (p = 0.010) (Figure 4D, Supp. Table 1C).
Fig. 4. AKT1-E17K, QCCs, and clinical outcome in human cancers.
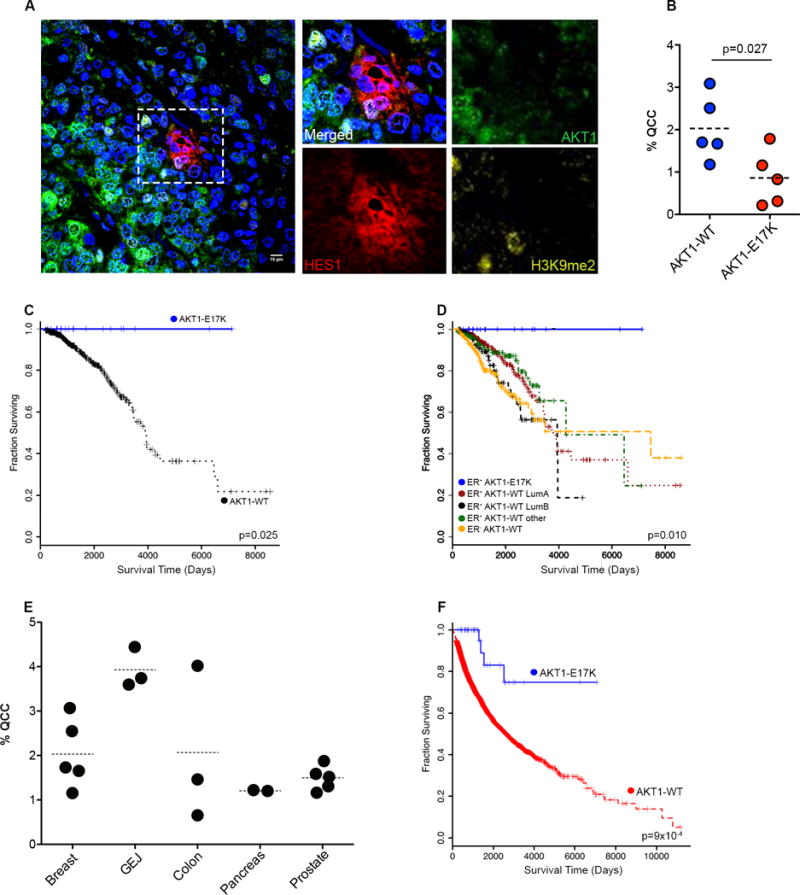
(A) Illustrative image of QCC in single section from a human ER+ primary breast tumor (Blue = DAPI nuclear stain). (B) percentage of QCCs in individual human breast tumors. (C) Kaplan–Meier plots of overall survival in ER+/AKT1-E17K breast cancer patients versus patients with ER+/AKT1- WT breast tumors. (D) ER+/AKT1-WT breast tumors subdivided by known molecular subtypes (from TCGA). (E) Percentage of QCCs in individual primary tumors from a variety of epithelial cancer types (i.e., breast, gastro-esophageal junction, colon, pancreas, prostate). (F) Kaplan–Meier plot of overall survival in all cancer patients with AKT1-E17K mutation versus AKT1-WT (from TCGA).
Given these striking findings, we analyzed available clinico-patho-genetic information searching for confounding factors beyond disruption of QCC that might explain this correlation. AKT1-E17K patients were enriched for younger, pre-menopausal individuals with ER+PR+HER2-, luminal A or B subtype, invasive ductal carcinomas compared to AKT1-WT patients (Supp. Table 1D). AKT1-E17K tumors were slightly smaller at diagnosis than AKT-WT tumors, but did not differ significantly in degree of nodal involvement, presence of distant metastases, or overall TNM stage, consistent with a primary growth deficit without major differences in spread of disease prior to treatment (Supp. Table 1D). Moreover, AKT1-E17K and AKT1-WT patients were treated similarly with surgery +/− local radiation and standard adjuvant hormonal and/or chemotherapy to suppress incipient micro-metastatic disease, without systematic differences in the drugs they received (Supp. Table 1B,C). Notably, AKT1-E17K and AKT1-WT tumors did not differ significantly in recurrent cancer mutations (beyond AKT1-E17K itself), transcript expression, gene-set enrichments, or expression of key cancer proteins, demonstrating a global similarity in molecular profiles (Supp. Table 1D–H). Overall, these observations were consistent with AKT1-E17K patients that produce fewer QCCs following a more indolent clinical course due to reduced outgrowth and/or increased treatment sensitivity of their disseminated disease.
Finally, we obtained a random set of epithelial tumors from different anatomical sites. We scored these tumors for AKT1low/H3K9me2low/HES1high QCC and confirmed their existence at low frequency in additional human tumor types beyond ER+ breast cancers (Figure 4E). We therefore considered all AKT1-E17K patients in the TCGA database irrespective of tumor type or treatment (i.e., ~0.5% of total) (Supp. Table 1I). Again, we found that AKT1-E17K patients as a group (n = 39) had a significantly better overall survival than AKT1-WT patients (n = 7793) (p = 9×10−4) (Figure 4F, Supp. Table 1J). While the majority of AKT1-E17K patients in this pooled analysis had ER+ breast tumors (n = 20), a number of patients with other solid tumor types contributed to this correlation (n = 19; head and neck, thyroid, lung, stomach, colorectal, prostate, cervical, uterine, and melanoma). This finding corroborated an independent report describing 3 other unusual patients with AKT1-E17K prostate adenocarcinomas and aggressive clinical features also following very indolent clinical courses over two decades after initial diagnosis (25).
DISCUSSION
AKT1-E17K is a somatic “hotspot” mutant that occurs recurrently in human cancers. Biochemical evidence supports an oncogenic function for this mutant protein as a constitutively active kinase that experimentally increases cell survival, proliferation, and migration (21, 22). Its very low frequency in human tumors (e.g., ~0.5% of TCGA), however, has been difficult to understand given the central role that PTEN/PI3K/AKT signaling plays in human tumor initiation, progression, and maintenance, and the high frequency of PTEN loss-of-function and PI3 kinase activating mutations seen in many different cancers (24). Here, we find that the AKT1-E17K mutant oncoprotein selectively disrupts the ability of highly proliferative cancer cells to spawn AKT1low daughter cells that are rare, quiescent, chemotherapy resistant, and pro-tumorigenic. Although AKT1low QCCs may resemble slow cyclers within the “cancer stem cell” pool by many criteria, they are not a discrete subpopulation from which all other cancer cells arise (10, 13–15). Rather, any dividing cancer cell may spawn AKT1low daughter cells depending on loss of interaction with the extracellular matrix. Collectively, our findings support a model in which AKT1low QCCs may represent an adaptive cellular behavior elicited by suboptimal β1-integrin activation that results from irregularities in the extracellular collagen matrix. Thus, it appears that continuous and facultative production of AKT1low slow proliferators in necessary for optimal tumor growth in a variety of malignant cell contexts. Interestingly, decreased AKT signaling has been reported in a melanoma cell sub-population – so called Rh123low - that is enriched for stem cell-like activities, including the ability to self-renew, produce non-stem progeny and form melanospheres that are relatively quiescent and chemo-resistant (26). It remains unclear, however, whether Rh123low cells relate to AKT1low QCCs or represent a different cell state and future work will be required to clarify this point.
In addition, we also find that therapeutic β1-integrin activation retards the production of QCCs and reduces tumor growth in multiple xenograft models. This is associated with a trend towards a higher percentage of apoptotic (CASP3+) cells in TS2/16-treated tumors, only reaching statistical significance in the A375 and MDA-MB-231 models. We previously reported that AKT1low QCCs can increase local stress-resistance, communicate with neighboring cells, secrete inflammatory proteins, and epigenetically differentiate into more rapidly cycling JARID1Bhigh tumor-initiating cancer stem cells to produce cellular heterogeneity (5, 7, 10, 11). All of these functions may plausibly contribute to the ability of QCCs to preferentially initiate tumors, promote tumor growth, and mediate resistance to both naturally-arising and iatrogenic stresses in both cancer and their surrounding stromal cells cross diverse tumor contexts, although further work will be required to fully explore these complex possibilities in vivo (7, 10, 16). In addition, a number of reports suggest that dormant/slowly proliferating cells may play a role in metastatic progression and the minimal residual disease state (27, 28). The properties associated to the AKT1low cells that we describe here and in our previous works also support this notion (6, 7). However, additional studies are required to more directly associate AKT1low cells with the metastatic cascade.
Interestingly, we also find that rare patients with AKT1-E17K-mutant tumors also produce fewer QCCs and survive significantly longer with treatment, but otherwise are remarkably similar in clinical, pathologic, and molecular features to patients with AKT1 wild-type tumors. Notably, AKT1-E17K somatic mutation is found at very high frequency in human breast papillomas (up to 50%), which are benign epithelial clonal expansions, but at very low frequency in breast ductal carcinoma in situ (DCIS) precursor lesions and invasive breast cancers (~2 – 4%), suggesting a negative selection for this allele during tumor progression (29, 30). Furthermore, conditional expression of AKT1-E17K in the mouse mammary gland, 1) produces mammary epithelial hyperplasia that does not progress to carcinoma, and 2) blocks HER2-driven mammary tumor formation in genetically engineered mice (31). In aggregate, these data are consistent with AKT1-E17K mutant protein actually having two important but opposing effects in human tumorigenesis: 1) promoting early clonal expansion by virtue of increased kinase activity (explaining its high frequency in pre-malignancy and recurring presence in tumors of various types), while 2) simultaneously impeding tumor outgrowth by disrupting the production of QCCs that promote growth and chemo-resistance (explaining both its low overall frequency in human cancers that come to clinical attention and the relatively good clinical prognosis for patients with these tumors that receive standard adjuvant treatment). They also suggest that expression of AKT1-E17K may powerfully predict solid tumor indolence and that therapeutically reducing AKT1low QCCs, particularly in the minimal residual disease setting, might prove useful for impeding human solid tumor growth.
Supplementary Material
Acknowledgments
We acknowledge OSBREAC and the patients donating tissue to this consortium. We thank Gordon Mills (MD Anderson Cancer Center) for genotype verification of AKT1-E17K patient samples. We thank the MGH/DFHCC Histopathology and DFCI/DFHCC Monoclonal Antibody Cores for support. The results shown in Figure 4 are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
FINANCIAL INFORMATION
S. Ramaswamy was supported by a Stand Up To Cancer Innovative Research Grant (Grant Number SU2C-AACR-IRG0911). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. Also by National Cancer Institute (R01 CA185086, C06 CA05926), Susan G. Komen for the Cure (IIR 12223648), and Harvard-Ludwig Center for Cancer Research. C.P. Alves received fellowship from CNPq “Ciência sem Fronteiras”— Brazil (202620/2012-3), S. Kabraji was supported by the Santander/MGH Fellowship, A.C. Yeh was supported by an HHMI Medical Student Research Fellowship, X. Sole was supported by a “Bolsa de Ampliacion de Estudios, Instituto de Salud Carlos III, Ministerio de Economía y Competitividad (BA12/00021)”. A-L Børresen-Dale and H. G. Russnes are supported by the Norwegian Cancer Association, the Norwegian South-East Regional Health Authorities, the Radiumhospital Hospital Foundation.
Abbreviation
- QCC
quiescent cancer cells
References
- 1.Loddo M, Kingsbury SR, Rashid M, Proctor I, Holt C, Young J, et al. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. British journal of cancer. 2009;100:959–70. doi: 10.1038/sj.bjc.6604924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeh AC, Ramaswamy S. Mechanisms of cancer cell dormancy - Another hallmark of cancer? Cancer research. 2015;75:5014–22. doi: 10.1158/0008-5472.CAN-15-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 5.Dey-Guha I, Wolfer A, Yeh AC, J GA, Darp R, Leon E, et al. Asymmetric cancer cell division regulated by AKT. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12845–50. doi: 10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dey-Guha I, Alves CP, Yeh AC, Salony, Sole X, Darp R, et al. A mechanism for asymmetric cell division resulting in proliferative asynchronicity. Molecular cancer research : MCR. 2015;13:223–30. doi: 10.1158/1541-7786.MCR-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salony, Sole X, Alves CP, Dey-Guha I, Ritsma L, Boukhali M, et al. AKT Inhibition promotes non-autonomous cancer cell survival. Molecular cancer therapeutics. 2016;15:142–53. doi: 10.1158/1535-7163.MCT-15-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 10.Facompre NDHKMS, X. Kabraji S, Belden Z, Sahu V, Whelen K, Tanaka K, Weinstein GS, Montone KT, Roesch A, Gimotty PA, Herlyn M, Rustgi AK, Nakagawa H, Ramaswamy S, Basu D. JARID1B mediates transition between distinct states within the oral cancer stem cell pool. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-15-3377. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–94. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto S, Wu Z, Russnes HG, Takagi S, Peluffo G, Vaske C, et al. JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer cell. 2014;25:762–77. doi: 10.1016/j.ccr.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nature medicine. 2009;15:1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 14.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014;14:275–91. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 16.Kabraji S, Sole X, Huang Y, Bango C, Bowden M, Bardia A, et al. AKT1low quiescent cancer cells persist after neoadjuvant chemotherapy in triple negative breast cancer. Breast cancer research : BCR. 2017;19:88. doi: 10.1186/s13058-017-0877-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byron A, Humphries JD, Askari JA, Craig SE, Mould AP, Humphries MJ. Anti-integrin monoclonal antibodies. Journal of cell science. 2009;122:4009–11. doi: 10.1242/jcs.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 19.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10581–6. doi: 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, et al. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. The American journal of pathology. 2001;159:431–7. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 22.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bleeker FE, Felicioni L, Buttitta F, Lamba S, Cardone L, Rodolfo M, et al. AKT1(E17K) in human solid tumours. Oncogene. 2008;27:5648–50. doi: 10.1038/onc.2008.170. [DOI] [PubMed] [Google Scholar]
- 24.Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer cell. 2007;12:104–7. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Boormans JL, Korsten H, Ziel-van der Made AC, van Leenders GJ, Verhagen PC, Trapman J. E17K substitution in AKT1 in prostate cancer. British journal of cancer. 2010;102:1491–4. doi: 10.1038/sj.bjc.6605673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Touil Y, Zuliani T, Wolowczuk I, Kuranda K, Prochazkova J, Andrieux J, et al. The PI3K/AKT signaling pathway controls the quiescence of the low-Rhodamine123-retention cell compartment enriched for melanoma stem cell activity. Stem cells. 2013;31:641–51. doi: 10.1002/stem.1333. [DOI] [PubMed] [Google Scholar]
- 27.Ghajar CM. Metastasis prevention by targeting the dormant niche. Nature reviews Cancer. 2015;15:238–47. doi: 10.1038/nrc3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nature reviews Cancer. 2014;14:611–22. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunlap J, Le C, Shukla A, Patterson J, Presnell A, Heinrich MC, et al. Phosphatidylinositol-3-kinase and AKT1 mutations occur early in breast carcinoma. Breast cancer research and treatment. 2010;120:409–18. doi: 10.1007/s10549-009-0406-1. [DOI] [PubMed] [Google Scholar]
- 30.Troxell ML, Levine J, Beadling C, Warrick A, Dunlap J, Presnell A, et al. High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23:27–37. doi: 10.1038/modpathol.2009.142. [DOI] [PubMed] [Google Scholar]
- 31.Mancini ML, Lien EC, Toker A. Oncogenic AKT1(E17K) mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis. Oncotarget. 2016 doi: 10.18632/oncotarget.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.