

Abstract
Background
Cyclin-dependent kinase 5 (Cdk5) belongs to the family of proline-directed serine/threonine kinases and plays a critical role in neuronal differentiation, migration, synaptogenesis, plasticity, neurotransmission and apoptosis. The deregulation of Cdk5 activity was observed in post mortem analysis of brain tissue of Alzheimer’s disease (AD) patients, suggesting the involvement of Cdk5 in the pathomechanism of this neurodegenerative disease. However, our recent study demonstrated the important function of Cdk5 in regulating inflammatory reaction.
Methods
Since the role of Cdk5 in regulation of inflammatory signalling in AD is unknown, we investigated the involvement of Cdk5 in neuroinflammation induced by single intracerebroventricular (icv) injection of amyloid beta protein (Aβ) oligomers in mouse. The brain tissue was analysed up to 35 days post injection. Roscovitine (intraperitoneal administration) was used as a potent Cdk5 inhibitor. The experiments were also performed on human neuroblastoma SH-SY5Y as well as mouse BV2 cell lines treated with exogenous oligomeric Aβ.
Results
Our results demonstrated that single injection of Aβ oligomers induces long-lasting activation of microglia and astrocytes in the hippocampus. We observed also profound, early inflammatory response in the mice hippocampus, leading to the significant elevation of pro-inflammatory cytokines expression (e.g. TNF-α, IL-1β, IL-6). Moreover, Aβ oligomers elevated the formation of truncated protein p25 in mouse hippocampus and induced overactivation of Cdk5 in neuronal cells. Importantly, administration of roscovitine reduced the inflammatory processes evoked by Aβ in the hippocampus, leading to the significant decrease of cytokines level.
Conclusions
These studies clearly show the involvement of Cdk5 in modulation of brain inflammatory response induced by Aβ and may indicate this kinase as a novel target for pharmacological intervention in AD.
Electronic supplementary material
The online version of this article (10.1186/s12974-017-1027-y) contains supplementary material, which is available to authorized users.
Keywords: Neuroinflammation, Amyloid beta, Alzheimer’s disease, Cdk5, Cytokines, Gene expression
Background
The development of Alzheimer’s disease (AD) has been attributed to excessive accumulation of amyloid β (Aβ) and hyperphosphorylated MAP tau protein causing the formation of senile plaques and neurofibrillary tangles, respectively. Although these molecular mechanisms are recognised as fundamental to almost all aspects of AD pathogenesis [1], recently, various intracellular signaling molecules, such as cyclin-dependent kinase 5 (Cdk5, EC 2.7.11.22), glycogen synthase 3β, and mammalian target of rapamycin, have also been implicated in the development of AD [2–4]. Among these, Cdk5 has been identified as a key mediator of AD pathogenesis [5, 6].
Cdk5 belongs to the group of proline-directed serine-threonine cyclin-dependent kinases (Cdks). Contrary to cell cycle-associated Cdks, Cdk5 plays a critical role in regulation of mammalian central nervous system development, as well as synaptic plasticity and neurotransmission [7–9]. The main mechanism responsible for pathological overactivation of Cdk5, which was observed in many diseases of the central nervous system, involves calpain-mediated cleavage of activator proteins p35 and p39. Although complexes of Cdk5 with truncated peptides p25 or p29 are not catalytically more active than Cdk5-p35 or Cdk5-p39 [10], the significantly longer half-life, due to greater stability of p25 and p29, prolongs Cdk5 activation. In cellular experimental models, it has been observed that Aβ stimulates the cleavage of p35 to p25, and the inhibition of Cdk5 reduces Aβ-evoked cell death [11]. Also, recent in vivo studies have demonstrated the calpain-dependent deregulation of Cdk5 activity in a mouse model of AD [12–14]. Moreover, post mortem analysis of brain preparations of AD patients indicates an accumulation of p25 and an increase of Cdk5 activity [15, 16]. Another mechanism that may modify Cdk5 activity is post-translational modification of Cdk5, p35 or p39: phosphorylation, S-nitrosylation, sumoylation, ubiquitylation and acetylation [5, 17, 18]. It was previously demonstrated that increased activity of Cdk5/p25 may be responsible for the hyperphosphorylation of MAP tau, the destabilisation of the cytoskeleton and neuronal death [19]. The overexpression of p25 or p35 induces the phosphorylation of APP at Thr668, which significantly increases the secretion of Aβ peptides [20]. The increased activity of Cdk5 may also be responsible for the transcriptional activation of the BACE1 promoter and in consequence for enhanced amyloidogenesis [21]. Therefore, the deregulation of Cdk5 activity may be a potentially important factor in the Aβ-evoked neurotoxic cascade.
A plethora of scientific reports posits inflammation as a key component of AD pathomechanism, not only in late phase when massive neurodegeneration and cognitive impairment occur, but also in early phase of the disease, when no symptoms are present. The presence of senile plaques induces inflammatory response in AD brain, leading to activation of microglia and astrocytes and in consequence to increased production of pro-inflammatory mediators [22]. However, also monomeric and oligomeric Aβ may affect the function of glial cells within the brain. Aβ could activate microglia and astrocytes to induce the production of inflammatory cytokines, including interleukin 1 (IL-1), tumour necrosis factor α (TNF-α), IL-6, IL-8 and reactive oxygen species, that may directly damage neurons [22, 23]. Increasing evidence suggests that different cytokines, including interleukins, TNF-α and transforming growth factor beta (TGF-β) or interferon-γ (IFN-γ), actively participate in AD pathomechanism and may serve as diagnostic or therapeutic targets [24]. In addition, several epidemiological studies have indicated that a long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs) may reduce the risk of developing AD [25, 26]. Some data suggest that inflammation may be an initiating factor of the cascade leading to overproduction of Aβ and to neurodegeneration [27–30]. The recent discoveries demonstrated that Cdk5 is involved in regulation of peripheral inflammatory processes, but little is known about the role of Cdk5 in regulating inflammation-related signalling in AD. It was demonstrated that chronic inflammatory reaction induces activation of Cdk5, leading to hyperphosphorylation of target proteins. In vitro studies showed that Cdk5 may be activated by interleukin-6 (IL-6) and TNF-α, and it plays an important role in inflammation-related signalling [31–33]. Moreover, our recent results indicate the activation of Cdk5 is an essential factor regulating inflammation-related gene expression in the hippocampus during systemic inflammatory response (SIR) [34]. The experiments on transgenic mouse model of AD suggested that there is interplay of inflammatory reaction with Cdk5 [35, 36]. Also, roscovitine, a potent Cdk5 inhibitor, has been found to exert anti-inflammatory actions in several in vivo models [37]. Based on previous reports, we aimed to study the involvement of Cdk5 in regulation of inflammatory signalling in the mouse brain after single intracerebroventricular (icv) injection of Aβ oligomers.
Methods
Materials
HFIP-pretreated amyloid β(1–42) and amyloid β(1–42) with scrambled sequence were obtained from rPeptide (Bogart, GA, USA). Anti-Cdk5, anti-p35/p25 antibodies and roscovitine were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-Iba1 antibody was from Abcam (Cambridge, UK). Anti-phospho-serine antibodies were from Cell Signaling Technology (Beverly, MA, USA). Anti-mouse IgG were from GE Health Care (Little Chalfont, Buckinghamshire, UK). Anti-rabbit IgG were from Sigma-Aldrich (St. Louis, MO, USA). 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) was from Cayman Chemical Company (Ann Arbor, MI, USA). Fluo-4 was from Thermo Fisher Scientific (Waltham, MA, USA). Histone H1 was from Millipore (Temecula, CA, USA). Chemiluminescent reagent Clarity Western ECL Substrate was from Bio-Rad Laboratories (Hercules, CA, USA). Protein G-Dynabeads were from (Novex, Life Technologies). The High Capacity cDNA Reverse Transcription Kit, Power SYBR Green PCR Master Mix, TaqMan Gene Expression Master Mix and TaqMan Gene Expression Assays were from Applied Biosystems (Foster City, CA, USA). Cytometric Bead Arrays (CBA) flex sets were from BD Biosciences (San Jose, CA, USA). Protease inhibitor cocktail Complete was from Roche Diagnostics GmbH (Mannheim, Germany). Acrylamide, APS, chloroform, dithiothreitol, DMSO, DNAse I, isopropanol, TEMED, Tri-reagent, anti-GFAP and anti-GAPDH antibodies and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of Aβ oligomers
Oligomerisation of Aβ1–42 was performed according to Stine et al. [38]. Amyloid β was dissolved (5 mM) in anhydrous DMSO and further diluted in cell culture medium Phenol Red-free Ham’s F-12 to 100 μM concentration. After 30-s vortexing, Aβ solution was incubated at 4 °C for 24 h. In accordance with previous data, 24-h incubation of Aβ1–42 monomers at physiological ionic strength and neutral pH at 4 °C yielded small-size oligomeric assemblies of Aβ, mainly trimers and tetramers (Fig. 1a). Additionally, conformation state of Aβ was confirmed by using Thioflavin T (ThT) [39]. The same protocol was applied for Aβ1–42 with scrambled sequence of amino acids (Aβscr). Freshly prepared solutions of Aβ1–42 were examined by atomic force microscopy (AFM) to confirm their structure. Typical result is shown in Fig. 1b. The spherical structures with a mean height of 2.5–3.5 nm determined by particle analysis method were observed. Such results are with good agreement to other works presenting low molecular oligomers [40, 41]. Aβ preparations were used directly after oligomerisation.
Fig. 1.
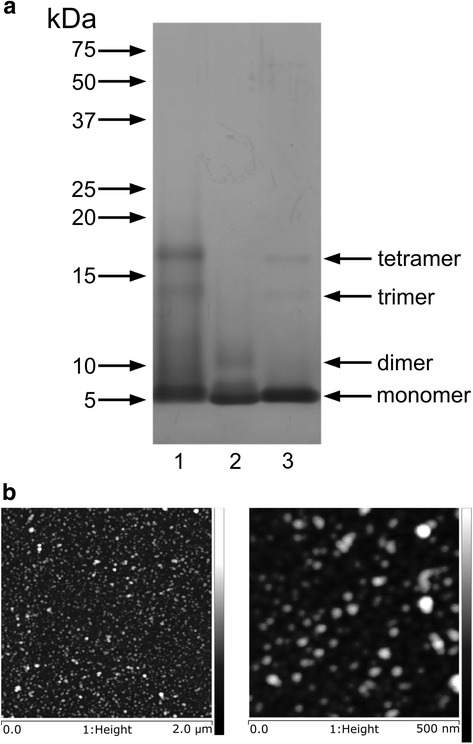
SDS-PAGE and morphological analysis of Aβ conformers. a Representative picture showing Aβ species after separation with 15% SDS-PAGE followed by silver staining. 1 Aβ1–42 after 24 h oligomerisation, 2 Aβscr after 24 h oligomerisation and 3 Aβ1–42 without oligomerisation. b Typical AFM images presenting spherical structures/oligomeric structures. Presented height scale is 5 nm
Atomic force microscopy
Multimode 8 Nanoscope atomic force microscope (AFM, Bruker, USA) was used to image the surfaces of the mica substrate and the freshly deposited oligomeric structures. Silicon cantilevers, ETALON series with a spring constant of ca. 3.5 Nm−1 (NT-MDT, Russia) or HQ:NSC19/No Al type with a spring constant of ca. 0.5 Nm−1 (Mikromasch, Bulgaria) were applied for imaging in PeakForce Tapping™ Force Microscopy Mode. Calibration of the microscope was achieved by the imaging of calibration gratings supplied by the manufacturer. The images presented in this work are height type images. The examination of surfaces for artefacts by AFM, and the reproducibility, was performed in the common way, i.e. by changing the AFM cantilever and moving the sample in the X or Y direction or by varying the scanning angle and scan rate. Oligomeric samples were prepared by applying a drop of 10 μl Aβ1–42 solution on freshly cleaved mica (Ted Pella Inc., USA). After incubation for 10 min, the sample was rinsed with deionised water (Millipore Inc., USA) and dried under a gentle stream of argon.
Animals
All the experiments were carried out on male C57BL/6 mice, 3 months old, supplied from the Animal House of Mossakowski Medical Research Centre PAS (Warsaw, Poland) which runs breeding of small rodents in SPF standard. The animals were maintained under controlled conditions of temperature and humidity with 12-h light/dark cycle. All of the experiments conducted on the animals were approved by the IV Local Ethics Committee for Animal Experimentation in Warsaw and were carried out in accordance with the EC Council Directive of November 24, 1986 (86/609/EEC), following the ARRIVE guidelines and guidelines published in the NIH Guide for the Care and Use of Laboratory Animals and the principles presented in the “Guidelines for the Use of Animals in Neuroscience Research” by the Society for Neuroscience. All efforts were made to minimise animal suffering and to reduce the number of animals used. Injections were performed between 9 a.m. and 1 p.m. All manipulations were performed gently and quickly to avoid stress-induced alterations.
Aβ 1–42 was administered intracerebroventricularly (icv) at the dose of 0.5 nmol per mice as previously described by Cakala and co-workers [42]. In brief, the mice were anesthetised by intraperitoneal (ip) injection of ketamine/xylazine cocktail (100/10 mg/kg b.w.) and placed in a stereotaxic frame (Stoeling Co., USA). A 1-mm hole was drilled 1 mm posterior to the bregma and 1.3 mm lateral. A microsyringe with a 26-gauge stainless steel needle (Hamilton) was inserted to a 2-mm depth, and 5 μl of Aβ solution was slowly injected for 5 min. The control animals received injection of the solvent. Separate groups of mice received additional ip injection of potent and selective Cdk5 inhibitor, roscovitine (seliciclib, CYC202). Roscovitine was dissolved in DMSO, diluted to the desired concentration with saline and administered intraperitoneally at a dose of 50 mg/kg b.w. as described previously by Czapski and co-workers [34]. The animals from the respective experimental groups received an appropriate volume of the solvent. Roscovitine was injected directly (30 min) before injection of Aβ. The animals were then returned to their home cage. Then, after the appropriate time (3 h or 1, 3, 7, 14, 21 and 35 days) after injection, the mice were decapitated, the brains were dissected and the hippocampi were isolated on ice-cold Petri dish. The tissue was used immediately or was frozen in liquid nitrogen and stored in −80 °C until analysis. Every effort has been made to minimise the number of animals used and reduce the amount of pain, distress and/or discomfort.
Cell culture and treatment
Human neuroblastoma SH-SY5Y cell line was obtained from Merck and was cultured in F12/MEM medium supplemented with 15% heat-inactivated foetal bovine serum (FBS), 1% non-essential amino acids, 50 units/ml penicillin and 50 μg/ml streptomycin and L-glutamine. BV2 microglia were maintained in RPMI supplemented with 5% heat-inactivated FBS, 50 units/ml penicillin and 50 μg/ml streptomycin and L-glutamine at 37 °C. Cell lines were cultured at 37 °C with 5% CO2 and 95% relative humidity. The cells were seeded into 60-mm and 35-mm culture dishes or 96-well plates, and the growth medium was changed into standard Hanks’ Balanced Salt Solution (HBSS). Then, the cells were treated with exogenous Aβ oligomers (10 μM) for 3 h. Suitable solvent was added to respective controls.
Fluorometric measurements of changes in [Ca2+]i
Changes in intracellular Ca2+ ([Ca2+]i) concentration in SH-SY5Y and BV2 cells were monitored using the fluorescent calcium-sensitive probe Fluo-4. Its acetoxymethyl ester derivative, Fluo-4 AM, easily penetrates plasma membranes, and inside the cells, it is cleaved by esterases to Fluo-4, which becomes highly fluorescent after binding with Ca2+. The experiment was performed as described previously by Wilkaniec et al. [43]. SH-SY5Y and BV2 cells were seeded onto 96-well dark plates at the density of 1.4 × 105 cells/ml. After 24 h, the cells were loaded with 10 μM Fluo-4 AM supplemented with 0.02% Pluronic® F-68 for 60 min at 37 °C in a HBSS. The cells were washed three times with HBSS and, to ensure complete AM ester hydrolysis, kept for 30 min at 37 °C in the dark. After a second washing, the fluorescence was measured using a microplate reader FLUOstar Omega (Ortenberg, Germany) set at 485-nm excitation and 538-nm emission wavelengths. After determining the baseline fluorescence of the cells incubated in HBSS, the changes in fluorescence after the addition of the test compounds were recorded every 15 s for 6 min. This 6-min treatment did not have any significant impact on cell viability. The results of fluorescence measurements are presented as percent changes in fluorescence intensity relative to the basal level versus duration of measurement (%F/F0). To quantify the change in the dynamics of the Ca2+ responses, the area under the curve (AUC) was calculated as a measure for the increase in intracellular Ca2+ [44].
Measurement of intracellular free radical level
Measurement of the free radical level was carried out using fluorescent indicator 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Cayman Chemical Company), as described previously [45]. DCFH-DA is intracellularly deacetylated to 2′,7′-dichlorodihydrofluorescein (DCFH) and then oxidised to a fluorescent compound, 2′,7′-dichlorofluorescein (DCF). SH-SY5Y and BV2 cells were incubated in DCFH-DA (10 μM) solution in HBSS with 20 mM Hepes (pH 7.4) and 0.02% Pluronic for 50 min at 37 °C in the dark. Then, the cells were washed three times, and the DCF fluorescence was measured using a microplate reader FLUOstar Omega (Ortenberg, Germany) at 485-nm excitation and 538-nm emission wavelengths. After determining the baseline fluorescence of the cells incubated in HBSS, the change in fluorescence after the addition of the test compounds was recorded 3 h after treatment. The results of fluorescence measurements are presented as percent of corresponding control.
Western blotting
Immunochemical analysis of protein level and phosphorylation was performed by Western blotting method in standard conditions. The sample was mixed with Laemmli buffer (2×) and denatured for 5 min at 95 °C. After standard SDS-PAGE on polyacrylamide gel, the proteins were transferred onto a nitrocellulose membrane and detected with specific antibodies. GAPDH level was analysed as a loading control. Densitometric analysis and size-marker-based verification was performed with TotalLab4 software.
Analysis of the mRNA level
RNA was isolated by using TRI-reagent according to the manufacturer’s protocol (Sigma Aldrich, St. Louis, MO, USA). Digestion of DNA contamination was performed by using DNase I according to the manufacturer’s protocol (Sigma Aldrich, St. Louis, MO, USA). RNA quantity and quality were controlled by spectrophotometric analysis and gel electrophoresis. A reverse transcription was performed by using the High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA, USA). Quantitative PCR was performed on an ABI PRISM 7500 apparatus using primers pair: Nos2 forward 5′- GGCAGCCTGTGAGACCTTTG-3′ and Nos2 reverse 5′-GCATTGGAAGTGAAGCGTTTC-3′ [46]. The levels of mRNA for Cdk5, Cdk5r1, Il1b, Il6, Il10, TNF-α and Actb were analysed by using the commercially available TaqMan Gene Expression Assays Mm00432437_m1, Mm00438148_m1, Mm00434228_m1, Mm00446190_m1, Mm00439614_m1, Mm00443258_m1 and ACTB_4352341E, respectively, according to the manufacturer’s instructions (Applied Biosystems). Actb was analysed as a reference gene. The relative levels of mRNA were calculated applying the ΔΔCt method.
Analysis of cytokine level
The level of cytokines was determined in tissue lysates by using Cytometric Bead Array (CBA) flex sets according to the producer’s (BD Biosciences) protocol. The method is a multiplexed bead-based immunoassay that allows simultaneous measuring of the levels of multiple proteins in one sample by flow cytometry. Briefly, tissue lysates were prepared according to the producer’s protocol and stored in −80 °C until analysis. Fifty microliters of each sample or standard were mixed with all capture beads (50 μl) and incubated in the dark for 1 h at room temperature (RT). Then, the phycoerythrin (PE)-conjugated detection reagent (50 μl) was added, and the tubes were incubated in the dark for 1 h at RT. After addition of wash buffer (1 ml), the samples were centrifuged at 200×g for 5 min and resuspended in 300 μl of wash buffer and immediately analysed by flow cytometry. Data acquisition (300 events for each cytokine) was performed by using a BD FACSCanto II flow cytometer with BD FACSDiva Software and FCAP Array software, version 3.0 (BD Biosciences, San Jose, CA, USA). Provided standards were used to build standard curves for each cytokine. Each cytokine’s concentration was indicated by their fluorescence intensity, calculated from a standard curve and normalised to protein level.
Cdk5 kinase assay
Activity of Cdk5 was determined with semi-quantitative method based on immunochemical measuring of phospho-serine level at recombinant histone H1 phosphorylated by immunoprecipitated Cdk5, according to Brooks [47] with modifications. SH-SY5Y cells were treated with 1–10 μM Aβ for 3 h, lysed in buffer containing 50 mM Tris-HCl (pH 7.4), 0.25 M NaCl, 0.1% v/v Nonidet P-40, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 1 mM Na4P2O7, 10 mM benzamidine, 50 μg/ml PMSF, 10 μg/ml TPCK, 10 μg/ml STI, 1 μg/ml aprotinin and 1 μg/ml leupeptin on ice. The protein extracts were combined with 25 μl Protein G-Dynabeads (Novex, Life Technologies), which were previously pre-incubated with mouse anti-Cdk5 (1 μg) antibody (Santa Cruz Biotechnology) overnight at 4 °C. Cell lysates were rotated with antibody-bound Dynabeads at 4 °C for 1 h, and the obtained complexes were sequentially washed three times with lysis buffer and then wash buffer (50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 1 mM dithiothreitol). A kinase reaction buffer consisting of 50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 1 mM dithiothreitol, 100 μM ATP, and 1.3 mg/ml histone H1 was added to the protein samples, followed by incubation at 23 °C for 60 min. The kinase reaction was then terminated by addition of 5× Laemmli buffer. The samples were boiled for 5 min and separated by standard SDS-PAGE, followed by transfer onto a nitrocellulose membrane and detection with anti-phospho-Ser antibody.
Statistical analysis
All experiments were carried out at least in triplicate. The presented data are means ± SEM. The distribution of data was tested by the Anderson-Darling normality test, and parametric and non-parametric tests were used for normal and non-normal distribution, respectively. Two group comparisons were done using Student’s t test. Multiple comparisons were analysed by one-way analysis of variance ANOVA with Bonferroni post hoc test or with Kruskal-Wallis nonparametric test with Dunn’s multiple comparisons post hoc test. Statistical significance was accepted at p < 0.05. The statistical analyses were performed by using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA).
Results
To assess the overall inflammatory status of the brain, murine hippocampi were collected during a time course: 3 h and 1, 3, 7, 14, 21 and 35 days after Aβ icv injection. In these samples, the protein levels of two markers associated with the identification of microglia (ionised calcium-binding adapter molecule 1, Iba-1) or astrocytes (glial fibrillary acidic protein, GFAP) were determined (Fig. 2). GFAP level was significantly increased in the brain beginning at 24 h after injection and continued until 14 days, whereas Iba-1 immunoreactivity was modest and not detectable until 3 days after Aβ treatment. We observed the normalisation of this protein’s level 21 days after the injection. To analyse the changes in transcription of several cytokines (IL-10, IL-6, IL-1β, and TNF-α) and inducible NO synthase (Nos2), involved in inflammatory response in murine hippocampus after Aβ icv injection, we performed a real-time PCR analysis. The levels of mRNA for all examined proteins elevated significantly 3 h after Aβ injection and then returned to the basal levels at 24 h (Fig. 3). To demonstrate that the activation of inflammatory response in the brain does not depend on introduction of foreign antigen, but it is specifically induced by Aβ1–42 oligomers, we analysed the effect of Aβ with a scrambled amino acid sequence (Aβscr) which was prepared by the same method as Aβ1–42 oligomers. We observed that in comparison with Aβ1–42, Aβscr did not exert any significant effect on gene expression of investigated proteins 3 h post-injection (Fig. 4).
Fig. 2.
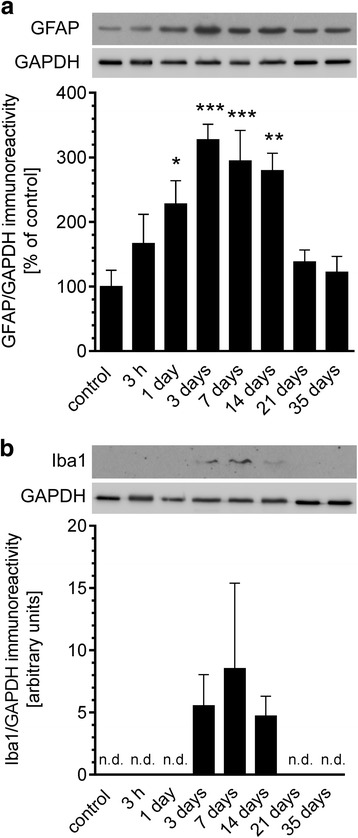
The effect of Aβ on astrocytes and glia activation in mouse hippocampus. Immunoreactivity of glial fibrillary acidic protein (GFAP) (a) and Iba1 (b) was analysed by SDS-PAGE and Western blotting in mouse hippocampus 3 h and 1, 3, 7, 14, 21 and 35 days after icv Aβ (0.5 nmol) injection. Representative pictures were shown. Results of densitometric analysis were normalised to immunoreactivity of GAPDH, as a loading control. Results are presented as the mean ± SEM from four independent experiments (n = 4) per time point. *p < 0.05; **p < 0.01; ***p < 0.001 versus control using a one-way ANOVA followed by the Bonferroni test. n.d. not detected
Fig. 3.
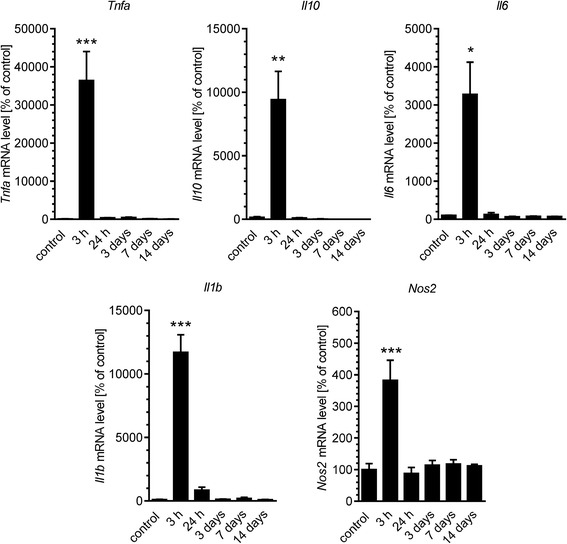
Relative changes in mRNA levels for TNF-α, IL-10, IL-6, IL-1β and iNOS in the hippocampus after Aβ treatment. Aβ (0.5 nmol) was injected intracerebroventricularly and gene expression was analysed 3 h and 1, 3, 7 and 14 days after injection by quantitative RT-PCR. Results were normalised to β-actin gene expression and are presented as the mean ± SEM from 4 to 12 independent experiments (n = 4–12). *, **, ***p < 0.05, 0.01 and 0.001 compared with the respective control using nonparametric Kruskal-Wallis followed by Dunn’s multiple comparisons test
Fig. 4.
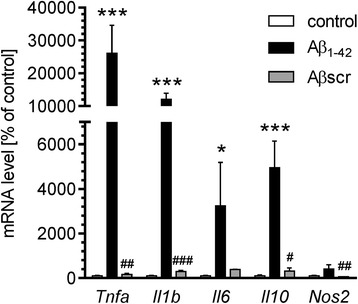
Scrambled Aβ does not change the mRNA levels for selected cytokines and iNOS in the hippocampus. Aβ (0.5 nmol) or Aβ scrambled (Aβscr, 0.5 nmol) were injected intracerebroventricularly and gene expression was analysed 3 h after injection by quantitative RT-PCR. The results were normalised to β-actin gene expression and are presented as the mean ± SEM from 6 to 12 independent experiments (n = 6–12). *, ***p < 0.05 and 0.001 compared with the respective control; #, ##, ###p < 0.05, 0.01 and 0.001 compared with the Aβ1–42-treated animals using nonparametric Kruskal-Wallis followed by Dunn’s multiple comparisons test
Subsequently, we analysed whether exogenous Aβ affects the molecular mechanisms responsible for regulation of Cdk5 activity in mice hippocampus. Our results demonstrated that icv administration of Aβ oligomers does not influence both the mRNA and protein levels of Cdk5 (Fig. 5) as well as the expression of Cdk5 regulator, p35 (Cdk5r1), at any of the investigated time points (Fig. 6a). However, the protein level of the truncated form of Cdk5 activator, p25 protein, was significantly elevated 3 h after administration of Aβ and returned to basal levels at 24 h (Fig. 6b). We also observed that truncation of p35 directly depends on the effect of Aβ oligomers, since Aβscr did not exert any significant effect on the protein level of p25 (Fig. 6c).
Fig. 5.
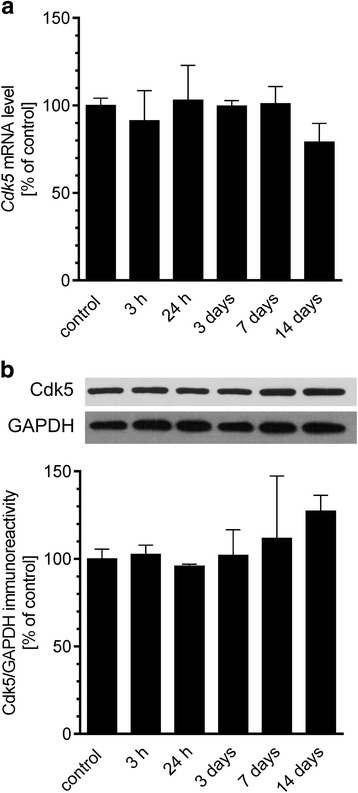
The effect of Aβ administration on Cdk5 expression and immunoreactivity in hippocampus. Aβ (0.5 nmol) was injected intracerebroventricularly. a Gene expression for Cdk5 was analysed 3 h and 1, 3, 7 and 14 days after injection by quantitative RT-PCR. b Immunoreactivity of Cdk5 was analysed 3 h and 1, 3, 7 and 14 days after injection by SDS-PAGE and Western blotting. Representative pictures were shown. Results of densitometric analysis were normalised to immunoreactivity of GAPDH, as a loading control. Results are presented as the mean ± SEM from four independent experiments (n = 4)
Fig. 6.
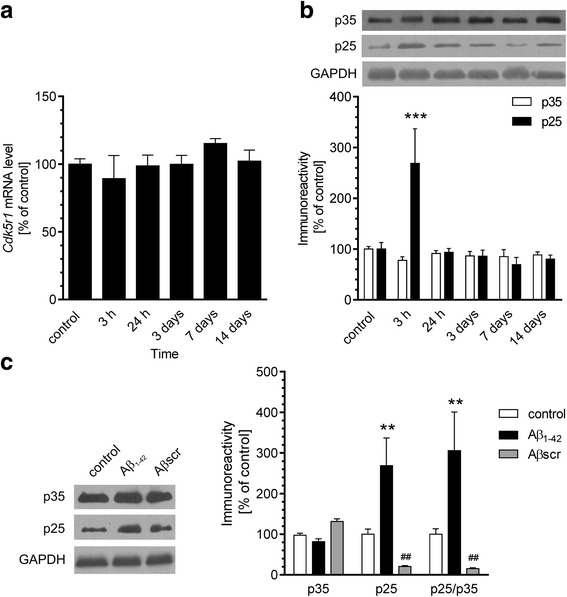
The effect of Aβ administration on p35 expression and immunoreactivity in hippocampus. Aβ (0.5 nmol) was injected intracerebroventricularly. a Gene expression for Cdk5r1 was analysed 3 h and 1, 3, 7 and 14 days after injection by quantitative RT-PCR. b Immunoreactivity of p35 and its degradation product p25 was analysed 3 h and 1, 3, 7 and 14 days after injection by SDS-PAGE and Western blotting. Representative pictures were shown. Results of densitometric analysis were normalised to immunoreactivity of GAPDH, as a loading control. The results are presented as the mean ± SEM from four independent experiments (n = 4). ***p < 0.001 compared with the respective control. c Scrambled Aβ does not change the immunoreactivity of p35 and its degradation product p25 3 h after injection. Representative pictures were shown. Results of densitometric analysis were normalised to immunoreactivity of GAPDH, as a loading control. Results are presented as the mean ± SEM from four independent experiments (n = 4). **p < 0.01 compared with the respective control (solvent injected animals), ##p < 0.01 compared with the Aβ-treated animals using a one-way ANOVA followed by the Bonferroni test
To analyse whether Cdk5 activation upon Aβ treatment may depend on deregulation of calcium homeostasis and oxidative status in neuronal or glial cells, we performed in vitro studies on SH-SY5Y and BV2 cell lines. Undifferentiated SH-SY5Y cells have been widely utilised for in vitro experiments requiring neuronal-like cells [48], whereas BV2 line has been frequently used as a substitute of primary microglia due to similar antigen pattern and phagocytic and cytotoxic activity [49, 50]. For the in vitro studies, we used the treatment paradigm, which corresponds to the applied in vivo conditions: we treated the cells with exogenous Aβ at concentration of 10 μM for 3 h. By using Fluo-4, we analysed the intracellular calcium level and found that Aβ evoked a rapid enhancement of fluorescence in both BV2 (Fig. 7a) and SH-SY5Y (Fig. 7b) cells, thus indicating an increase in the cytosolic calcium level. Interestingly, the effect of Aβ treatment on [Ca2+]i mobilisation was more pronounced in neurons than in microglia (Fig. 7c). Moreover, Aβ evoked significant enhancement of free radicals level in SH-SY5Y, but not in BV2 cells (Fig. 7d). Considering the differences in sensitivity to Aβ treatment between neuronal and glial cells, we next explored whether Aβ-evoked Cdk5 activation occurs equally within those cell types. We observed that the protein expression of Cdk5 activator p35 in BV2 cells was almost undetectable when compared to SH-SY5Y cell line (Fig. 8a), suggesting the insignificant activity of Cdk5 in glial cells. In addition, Cdk5 was immunoprecipitated to assess its activity using histone H1 as a substrate in Aβ-treated SH-SY5Y cells. As shown in Fig. 8b, Aβ treatment induced overactivation of Cdk5 kinase and the effect of Aβ was concentration dependent: while 5 and 10 μM Aβ oligomers evoked a significant increase in Cdk5 activity, Aβ at 1 μM concentration did not have any effect.
Fig. 7.
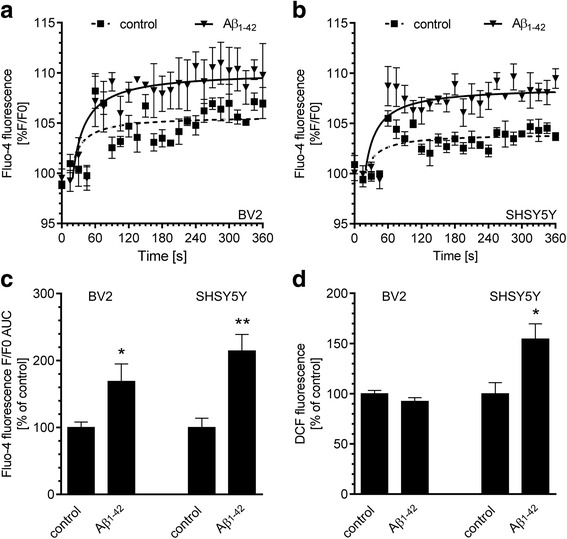
The effect of Aβ oligomers on calcium homeostasis and free radical generation in SH-SY5Y and BV2 cells. Cytoplasmic calcium level in BV2 (a) and SH-SY5Y (b) cells was measured by using Fluo-4 during 6 min after treatment with 10 μM Aβ oligomers. Data represent the mean value ± SEM for three independent experiments. c Responses of Fluo-4 were quantitated by measuring the area under the curve (AUC) value. Data represent the mean value ± SEM for three independent experiments. *, **p < 0.05 and 0.01 compared to control using Student’s t test. d Intracellular free radical level in SH-SY5Y and BV2 cells was measured by DCF fluorescence after 3 h incubation with 10 μM Aβ oligomers. Data represent the mean value ± SEM for four independent experiments. *p < 0.05 compared to control, using Student’s t test
Fig. 8.
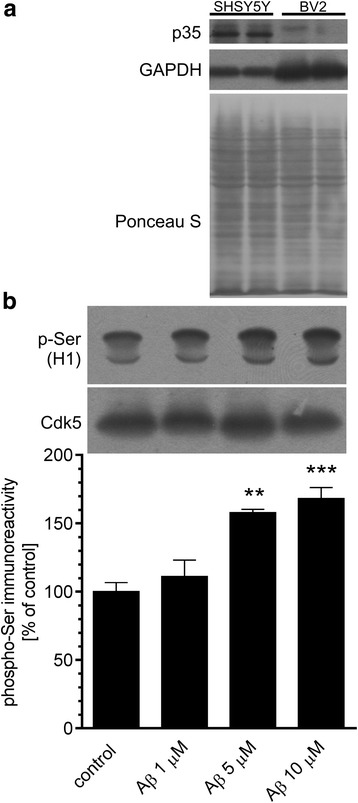
Aβ administration evokes Cdk5 activation in neuronal cells. a Immunoreactivity of p35 and GAPDH in SH-SY5Y and BV2 cells was analysed 3 h after Aβ treatment by SDS-PAGE and Western blotting. Representative pictures were shown. PonceauS staining was used as a loading control. b In SH-SY5Y cells treated with 1, 5 or 10 μM Aβ for 3 h, Cdk5 kinase activity was measured as described under “Experimental Procedures.” Results of densitometric analysis of phosphorylated histone H1 are presented as the mean ± SEM from four independent experiments (n = 4). **, ***p < 0.01 and 0.001 compared to control, using a one-way ANOVA followed by the Bonferroni test
To investigate the role of Cdk5 in Aβ-induced inflammatory signalling in the hippocampus, the effect of Cdk5 inhibition on the expression and protein level of inflammation-related proteins was studied. We used the potent Cdk5 inhibitor, roscovitine, that was previously demonstrated to be able to inhibit Cdk5 in various animal models [6, 12, 34, 51, 52]. It was previously shown that roscovitine was able to cross blood-brain barrier and to rapidly accumulate in the brain leading to transient inhibition of Cdk5 [53–56]. As shown on Fig. 9, treatment with roscovitine (50 mg/kg b.w.) significantly decreased expression of Aβ-induced genes: Tnfa, Il1b, Il10 and Nos2. We also observed the pronounced, although not significant, inhibitory effect of roscovitine on Aβ-induced elevation of Il6 mRNA level. To determine whether roscovitine treatment influences the protein level of selected cytokines, in our animal model, we used Cytometric Bead Array (CBA) analysis. The results demonstrated that the inhibition of Cdk5 significantly prevented the Aβ-dependent elevation of the brain level of TNF-α and Il-6 in mouse hippocampus (Fig. 10). Interestingly, we observed that despite of large elevation of mRNA for IL-10 after Aβ treatment, the level of this protein was not changed, and roscovitine treatment also did not affect IL-10 concentration (Fig. 10).
Fig. 9.
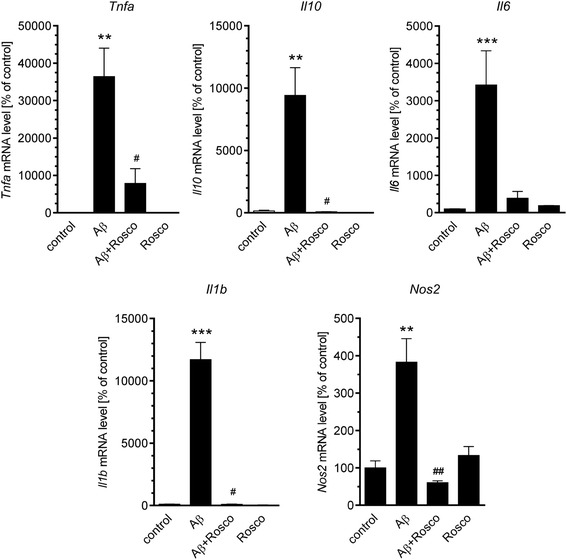
The effect of roscovitine on Aβ-induced expression of inflammation-related signalling in hippocampus. Aβ (0.5 nmol) was injected intracerebroventricularly and roscovitine (50 mg/kg b.w.) was administered intraperitoneally. Gene expression was analysed 3 h after injection by quantitative RT-PCR. Results are presented as the mean ± SEM from 3 to 12 independent experiments (n = 3–12). **, ***p < 0.01 and 0.001 compared with the respective control (solvent injected animals), #, ##p < 0.05 and 0.01 compared with the Aβ-treated animals using nonparametric Kruskal-Wallis followed by Dunn’s multiple comparisons test
Fig. 10.
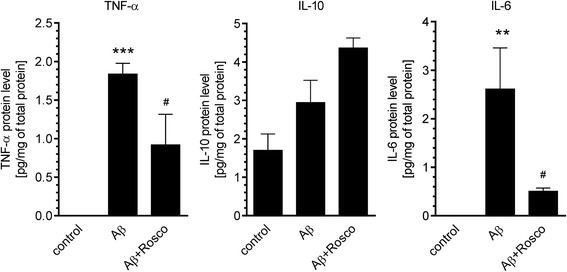
The effect of roscovitine on Aβ-induced changes in TNF-α, IL-10 or IL-6 level in hippocampus. Aβ (0.5 nmol) was injected intracerebroventricularly and roscovitine (50 mg/kg b.w.) was administered intraperitoneally. Cytokine level was analysed 3 h after injection by Cytometric Bead Array (CBA). Results are presented as the mean ± SEM from three to four independent experiments. **, ***p < 0.01 and 0.001 compared with the respective control, #p < 0.05 compared with the Aβ-treated animals using a one-way ANOVA followed by the Bonferroni test
Given the nuclear localisation of Cdk5 in neurons [57] and the various transcriptional regulators as Cdk5 substrates [58], it is possible that Cdk5-dependent deregulation of cytokines synthesis after Aβ treatment occurs on the transcriptional level. Therefore, we measured the effect of Aβ treatment on protein level and/or phosphorylation of various proteins that regulate gene expression, e.g. extracellular signal-regulated kinases (ERKs) as well as nuclear factor NFκB p65 subunit (RelA) and IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha), but we did not observe any significant changes (Additional file 1: Figure S1) what excludes the involvement of these pathways in regulation of Aβ-evoked early alterations of gene expression.
Discussion
The amyloid cascade hypothesis postulates that extracellular liberation of Aβ due to aberrant amyloid precursor protein (APP) processing plays the key role in Alzheimer’s disease (AD) pathology. Although experimental and genetic studies confirmed enhanced amyloidogenesis in AD, the amyloid theory had many critics, mainly due to the lack of a correlation between the severity of cognitive impairment and the load of senile plaques (SP) in the brain. Importantly, the attempts to develop therapeutic methods based on an anti-Aβ approach have not yielded satisfactory results [59–62]. Those observations suggest that AD pathology is more complex and involves activation of several noxious phenomena that may be connected with each other or act independently. Recent studies from preclinical and clinical studies indicate that inflammation is a powerful pathogenetic force that contributes to and drives AD pathogenesis. It was previously demonstrated that activation of microglial cells may act as a functional link between Aβ deposition and neuronal degeneration. However, the molecular mechanisms underlying Aβ induced pro-inflammatory signalling is not fully understood. In this study, we showed for the first time that activation of Cdk5 is an initiating factor of Aβ peptide-induced neuroinflammation in mouse hippocampus.
In AD brain, Aβ deposition in senile plaques is connected with prolonged and widespread activation of microglia and astrocytes [63, 64]. The majority of transgenic rodent models of AD are also characterised by extensive accumulation of activated glia and astrocytes [65, 66] long before the appearance of plaque and tangle pathology [67, 68]. Moreover, in mouse models of AD, the degree of inflammatory response correlated with development of various pathological features as well as neuronal death [67, 69]. In the present study, we used the well-characterised animal model of neurotoxicity induced by administration of Aβ1–42 into the lateral ventricle of C57BL/6 mice to determine the molecular mechanisms underlying Aβ peptide-induced neuroinflammation. Among several animal models that have been developed to investigate the course of AD pathology, the icv injection of amyloid peptides into the brain was observed to be appropriate for the analysis of potential mechanisms of inflammatory response, because it activates astrocytes and microglia [70–74] and produces profound neurodegeneration [70, 75–77]. Moreover, using this experimental model, the significant behavioural changes such as memory deficits as well as reduced locomotion and exploration were observed [42, 78]. Indeed, our data demonstrated that icv administration of Aβ1–42 resulted in the pronounced increase of the astrocytic (GFAP) and microglial (Iba-1) markers as well as induced cytokine synthesis, suggesting that this treatment significantly affects the extent of reactive gliosis. Those observations are consistent with the previous data showing that microglia and astrocytes are preferentially associated with certain amyloid plaque types [79, 80]. Amyloid peptides, their precursor protein APP and neurofibrillary tangles are potent glial activators [81, 82]. Activated microglia has a beneficial role in reducing Aβ accumulation by its phagocytosis via scavenger receptors (SRs) [83] and subsequent degradation [84]. The microglial activation is therefore a protective mechanisms promoting Aβ clearance and hinder the AD progression at the early stages of the disease. However, the persistent microglial activation stimulated by Aβ via the receptor for CD36 [85], Fc receptors, Toll-like receptors (TLRs) [86], complement and receptors for advanced glycation end-products (RAGE) [87] can increase Aβ production and decrease Aβ clearance, ultimately causing neuronal damage. Therefore, disruption of the Aβ formation delays and decreases microglial activation [88] leading to reduction of inflammatory cytokines production [89], lowering of Aβ deposition [90] and amelioration of behavioural damage [91]. Similar as microglial cells, astrocytes are known to be important for Aβ clearance and degradation, for providing trophic support to neurons and for forming a protective barrier between Aβ deposits and neurons [92]. However, astrocytes could also be a source for Aβ, because they express APP and β-secretase (BACE1), and treatment with cytokines or Aβ1–42 may activate amyloidogenic APP processing that drives feed-forward mechanism that promotes Aβ production in astrocytes [93]. It is well recognised that the activation of microglia and astrocytes is accompanied by increased production of pro- and anti-inflammatory cytokines, including interleukins (ILs), interferons (IFNs) and tumour necrosis factors (TNFs), as well as chemokines, nitric oxide (NO) and reactive oxygen species [94, 95]. In cultured astrocytes and microglia, Aβ treatment significantly increases the secretion of several inflammatory cytokines [80, 96] that can be significantly inhibited by the anti-inflammatory agents [80, 97]. Here, we show that both gene expression and protein level of selected pro-(IL-1β, TNF-α, IL-6) and anti-(IL-10) inflammatory cytokines as well as iNOS were increased following Aβ injection in murine hippocampus. This suggests that stimulation of cytokine release is a result of glia and astrocyte activation induced by Aβ. Interestingly, we observed that elevated cytokine expression significantly precedes increase in GFAP and Iba-1 immunoreactivity. These data are consistent with previous studies showing that increase in immunoreactivity of Iba-1 or GFAP as well as morphological alterations of microglia and astrocytes were delayed following the lipopolysaccharide (LPS) injection and corresponded with a resolution phase of microglia activation [98]. Also, in the study using LPS injection in a model of chronic neurodegeneration, differential cytokine induction in glia and astrocytes were detected independent of morphological differences [99].
As indicated above, chronic inflammation could be the consequence of AD pathology that further exacerbates the deleterious effects exerted by Aβ. However, there is still considerable debate over exactly what is the molecular mechanism of Aβ-induced neuroinflammation. The potential role of Cdk5 gained one of the highest scientific interests since it was suggested that Cdk5 may exist at a crossroad of inflammation and neurodegeneration [100–103]. In vitro studies showed that Cdk5 may be activated by pro-inflammatory mediators, and it plays an important role in inflammation-related signalling [31–33]. Previous reports showed that abnormal Cdk5 signalling is also an important component of the molecular mechanism of toxicity of AD-related proteins like Aβ, alpha-synuclein or Tau [45, 104–107]. Moreover, Cdk5 has a substantial role in either direct or indirect interactions of those proteins common to, and critical in, different neurodegenerative diseases [5]. It was demonstrated that Aβ treatment induces the conversion of p35 to p25 in primary cortical neurons, causing the prolonged activation of Cdk5 [15, 108]. The Cdk5-induced neurotoxicity after Aβ treatment was shown to be, at least in part, mediated by rapid nuclear dispersion and mislocalisation of Cdk5 in the nucleus, where it triggered the activation of several pro-apoptotic genes via activation of c-JUN pathway [109]. Cdk5 activation was also demonstrated to be responsible for Aβ-induced tau phosphorylation at Ser 396/404 in lipid rafts [110]. Consistently with the previous reports, our data showed that icv administration of Aβ induces the increased production of p25 in mouse hippocampus. Interestingly, the observed elevation of p25 level seems to be specific for neurons, as microglial cells display residual expression of p35 protein. This corresponds with the previous data showing that disruption of p35 gene significantly affects neuronal function, but other cell types are not affected and histologically normal [111]. Moreover, the expression of both Cdk5 activators p35 and p39 is restricted principally to the nervous system and their expression in microglia and astrocytes is very low [112]. Although some studies demonstrated the activation of Cdk5 in non-neuronal cells, this seems to be regulated by other proteins, e.g. cyclin D, cyclin E, cyclin I [113–115]. Therefore, in the mouse hippocampus, the Aβ-triggered activation of Cdk5 via p35/p25-dependent pathway occurs exclusively in neuronal cells. The accumulation of p25 in AD brain has been previously associated with inflammation and astrogliosis along with synaptic damage [12, 100, 102, 116]. Moreover, the overactivation of Cdk5 was previously demonstrated to induce the proinflammatory gene transcription and resulted in enhanced phosphorylation of tau and glycogen synthase kinase 3β during systemic inflammatory response [34]. In accordance with those data, we demonstrated that upon Aβ treatment, the activity of Cdk5 in neuronal cells is significantly increased as well as that Cdk5 inhibition resulted in significant reduction of mRNA level for TNF-α, IL-1β, IL-10 and iNOS in animals treated with Aβ. However, the most evident effect of roscovitine was observed for both gene expression and protein level of TNF-α and IL-6, which are two major cytokines involved in initiating and regulating the cytokine cascade during an inflammatory response but also have both direct and indirect neurotrophic effects as well as regulate cognitive function [117–120]. TNF-α release was previously shown to be involved in Aβ-induced learning and memory deficits in AD [121, 122]. Several anti-TNF-α treatments have prevented Aβ deposition, behavioural impairments and inflammation in AD animal models [36–39], suggesting that TNF-α is a detrimental factor in AD course and can serve as a reliable AD target. Similarly, IL-6 was reported to increase Aβ levels by stimulation of APP expression and processing in primary rat cortical neurons [123, 124]. IL-6 furthermore enhanced neuronal damage induced by Aβ [125]. On the other hand, overexpression of IL-6 in APP transgenic mice leads activation of microglia to a predominantly beneficial phenotype, which results in phagocytosis of Aβ but not its degradation [126]. In pathological conditions, both IL-6 and TNF-α may be also responsible for an increase in intraneuronal p35 level and Cdk5 activation [33, 101]. Interestingly, our data showed that although Aβ significantly elevates the expression for IL-10, the protein level for this anti-inflammatory cytokine is unchanged after Aβ treatment. Previous reports indicated that IL-10 production principally depends on mRNA stability and protein translation rather than the mRNA level [127], and rapid induction and degradation of IL-10 mRNA was shown to be mediated by the activation of pattern recognition—Toll-like receptors 2 (TLR2) [128]. Since TLR2 was demonstrated to be a primary receptor for Aβ to trigger neuroinflammatory activation in microglia [129], it is though possible that in our experimental model, the stimulation of the TLR2 resulted in inhibition of IL-10 translation. Although this hypothesis remains to be elucidated, still the lack of IL-10 response after Aβ treatment suggests the certain deficiency in microglia inhibition as well as failure of down-regulation of IL-1β, IL-6, TNF-α secretion, that is mainly mediated by this anti-inflammatory cytokine [130–132]. This seems to be consistent with the recent studies on several AD animal models, where overexpression of IL-10 weakened the phagocytosis of soluble Aβ by microglia and exacerbated Aβ deposits [133–135].
It was demonstrated that the molecular mechanism by which Aβ is able to stimulate cytokine production involves the nuclear factor-kappaB (NF-κB)-dependent activation of extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) pathways [50]. Our previous data showed that LPS-induced SIR involves Cdk5-dependent activation of NF-κB with subsequent enhancement of transcription of several proinflammatory cytokines [34]. In other studies, inhibition of Cdk5 with roscovitine decreased phosphorylation of kinase IKKβ, IκB and p65, leading to decrease of transcriptional activity of NF-κB, and, in consequence, prevented LPS-evoked expression of iNOS, COX-2, IL-1β and IL-6 [32]. For these reasons, we investigated whether Cdk5 might have a similar effect on Aβ-mediated activation of NF-κB in mouse hippocampus. Surprisingly, we did not observe any changes in NF-κB–ERK–MAPK pathway after Aβ treatment. Similarly, we did not detect any alteration in phosphorylation of glycogen synthase kinase 3β and transcription factor MEF2. Since our study demonstrated that glial cells are less sensitive to Aβ treatment and the abundance of Cdk5 co-activator, p35 protein is greatly reduced in those cells; thus, it is possible that the Aβ-induced p25 formation and Cdk5 activation in neurons may indirectly influence the glial cells to produce cytokines. While it was demonstrated that neurons can release cytokines and chemokines as well as respond to them by way of cytokine and chemokine receptors [136], the direct involvement of neurons in the inflammatory response is marginal when compared to activated glial cells that are the major source of inflammatory mediators in the brain [137]. In response to injury, neurons could produce adhesion molecules and trophic factors that recruit and activate microglial cells and astrocytes [136]. Previous reports demonstrated that p25 overexpression in neurons resulted in upregulation of cytosolic phospholipase A2 (cPLA2) and lysophosphatidylcholine (LPC) release, by which it activated the surrounding microglia and astrocytes [102]. Moreover, LPC was shown to be a potent chemoattractant for T cells [138, 139], and p25 overexpression was demonstrated to initiate the peripheral cell recruitment into the brain to exacerbate neuroinflammation [102]. Therefore, it is also possible that LPC release due to p25 formation in neurons might be also responsible for the peripheral cell recruitment. While this interesting hypothesis remains to be further elucidated in our experimental conditions, it is still most probable that Aβ-induced p25 overexpression in neurons activates the surrounding microglia, and the subsequent cytokine release is a consequence rather than primary trigger originating from p25-expressing neurons [100]. Since we have observed that cytokine expression subsided and even disappeared in time after Aβ injection, we take this as corroborating the hypothesis that Cdk5 activation in neurons leads to secretion of extracellular factors, such as LPC, that stimulate microglia, and the levels of those factors decrease in the brain as fewer neurons remain to produce them.
Conclusions
Summarising, our results clearly indicate the important role of Cdk5 in regulating early inflammation-related gene expression induced by Aβ. These might suggest Cdk5 inhibition as novel potential therapeutic targets for inhibition of brain inflammation during AD.
Acknowledgements
The authors thank Renata Wojda and Magdalena Kudlik for the technical assistance.
Funding
Financial support was provided by the National Science Centre Grant to GAC: 2011/03/B/NZ3/04549. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
Amyloid beta
- CBA
Cytometric bead array
- Cdk5
Cyclin-dependent kinase 5
- COX-2
Cyclooxygenase 2
- cPLA2
Cytosolic phospholipase A2
- EDTA
Ethylenediaminetetraacetic acid
- ERK
Extracellular signal-regulated kinase
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GFAP
Glial fibrillary acidic protein
- HBSS
Hanks’ Balanced Salt Solution
- Iba-1
Ionised calcium-binding adapter molecule 1
- IFN-γ
Interferon gamma
- IL-1
Interleukin
- iNOS
Inducible nitric oxide synthase
- LPC
Lysophosphatidylcholine
- MAP
Microtubule associated protein
- MAPK
Mitogen-activated protein kinase
- MEF2
Myocyte enhancer factor-2
- NaF
Sodium fluoride
- PE
Phycoerythrin
- PMSF
Phenylmethylsulfonyl fluoride
- RAGE
Receptors for advanced glycation end products
- RT
Room temperature
- SIR
Systemic inflammatory response
- STI
Soybean trypsin inhibitor
- TGF-β
Transforming growth factor beta
- TLR2
Toll-like receptor 2
- TNF-α
Tumour necrosis factor alpha
- TPCK
Tosyl phenylalanyl chloromethyl ketone
Additional file
The effect of Aβ administration on immunoreactivity of proteins. Aβ (0.5 nmol) was injected intracerebroventricularly. Immunoreactivity of analysed proteins was examined by SDS-PAGE and Western blotting 3 h after injection of Aβ. Results of densitometric analysis were normalised to immunoreactivity of GAPDH, as a loading control. The results are presented as the mean ± SEM from four independent experiments (n = 4). (TIFF 499 kb)
Authors’ contributions
AW and GAC conceived and designed the experiments. AW, MG, GAC, and MS performed the experiments. AW, MG, AA, GAC and MS analysed and interpreted the data. AA and GAC contributed reagents/materials/analysis tools. AW, MG, MS, AA and GAC wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study did not utilise any human participants, human data, or human tissue. The experiments on the animals were performed according to regulations following European Union directives. The experimental procedures were approved by the Local Ethics Committee for Animal Experimentation in Warsaw (permissions 62/2011 and 25/2015).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12974-017-1027-y) contains supplementary material, which is available to authorized users.
Contributor Information
Anna Wilkaniec, Email: awilkaniec@imdik.pan.pl.
Magdalena Gąssowska-Dobrowolska, Email: mgassowska@imdik.pan.pl.
Marcin Strawski, Email: marcin@chem.uw.edu.pl.
Agata Adamczyk, Email: aadamczyk@imdik.pan.pl.
Grzegorz A. Czapski, Phone: +48226086613, Email: gczapski@imdik.pan.pl
References
- 1.Masters CL, Beyreuther K. Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain. 2006;129:2823–2839. doi: 10.1093/brain/awl251. [DOI] [PubMed] [Google Scholar]
- 2.Amin J, Paquet C, Baker A, Asuni AA, Love S, Holmes C, Hugon J, Nicoll JAR, Boche D. Effect of amyloid-β (Aβ) immunization on hyperphosphorylated tau: a potential role for glycogen synthase kinase (GSK)-3β. Neuropathol Appl Neurobiol. 2015;41:445–457. doi: 10.1111/nan.12205. [DOI] [PubMed] [Google Scholar]
- 3.Dhariwala FA, Rajadhyaksha MS. An unusual member of the Cdk family: Cdk5. Cell Mol Neurobiol. 2008;28:351–369. doi: 10.1007/s10571-007-9242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133:739–749. doi: 10.1111/jnc.13037. [DOI] [PubMed] [Google Scholar]
- 5.Wilkaniec A, Czapski GA, Adamczyk A. Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: facts and hypotheses. J Neurochem. 2016;136:222–233. doi: 10.1111/jnc.13365. [DOI] [PubMed] [Google Scholar]
- 6.Crews L, Patrick C, Adame A, Rockenstein E, Masliah E. Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer's disease. Cell Death Dis. 2011;2:e120. doi: 10.1038/cddis.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lalioti V, Pulido D, Sandoval IV. Cdk5, the multifunctional surveyor. Cell Cycle. 2010;9:284–311. doi: 10.4161/cc.9.2.10466. [DOI] [PubMed] [Google Scholar]
- 8.Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22:169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Lopes JP, Agostinho P. Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol. 2011;94:49–63. doi: 10.1016/j.pneurobio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Peterson DW, Ando DM, Taketa DA, Zhou H, Dahlquist FW, Lew J. No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proc Natl Acad Sci U S A. 2010;107:2884–2889. doi: 10.1073/pnas.0912718107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez A, Toro R, Caceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/s0014-5793(99)01279-x. [DOI] [PubMed] [Google Scholar]
- 12.Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an Abeta-induced model of Alzheimer's disease: the role of Cdk5. Aging Cell. 2010;9:64–77. doi: 10.1111/j.1474-9726.2009.00536.x. [DOI] [PubMed] [Google Scholar]
- 13.Lopes JP, Oliveira CR, Agostinho P. Role of cyclin-dependent kinase 5 in the neurodegenerative process triggered by amyloid-Beta and prion peptides: implications for Alzheimer’s disease and prion-related encephalopathies. Cell Mol Neurobiol. 2007;27:943–957. doi: 10.1007/s10571-007-9224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopes JP, Oliveira CR, Agostinho P. Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle. 2009;8:97–104. doi: 10.4161/cc.8.1.7506. [DOI] [PubMed] [Google Scholar]
- 15.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 16.Tseng HC, Zhou Y, Shen Y, Tsai LH. A survey of Cdk5 activator p35 and p25 levels in Alzheimer's disease brains. FEBS Lett. 2002;523:58–62. doi: 10.1016/s0014-5793(02)02934-4. [DOI] [PubMed] [Google Scholar]
- 17.Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A. 2011;108:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu J, Nakamura T, Holland EA, McKercher SR, Lipton SA. S-nitrosylation of Cdk5: potential implications in amyloid-beta-related neurotoxicity in Alzheimer disease. Prion. 2012;6:364–370. doi: 10.4161/pri.21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura T, Ishiguro K, Hisanaga S-i. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F, Su Y, Li B, Zhou Y, Ryder J, Gonzalez-DeWhitt P, May PC, Ni B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547:193–196. doi: 10.1016/s0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- 21.Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marie I, Maurin T, Wang L, Figueroa H, Herman M, et al. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. Sci World J. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int J Neurosci. 2014;124:307–321. doi: 10.3109/00207454.2013.833510. [DOI] [PubMed] [Google Scholar]
- 24.Zheng C, Zhou X-W, Wang J-Z. The dual roles of cytokines in Alzheimer’s disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl Neurodegeneration. 2016;5:7. doi: 10.1186/s40035-016-0054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front Aging Neurosci. 2010;2:19. doi: 10.3389/fnagi.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore AH, Bigbee MJ, Boynton GE, Wakeham CM, Rosenheim HM, Staral CJ, Morrissey JL, Hund AK. Non-steroidal anti-inflammatory drugs in Alzheimer’s disease and Parkinson's disease: reconsidering the role of neuroinflammation. Pharmaceuticals. 2010;3:1812–1841. doi: 10.3390/ph3061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9:25–34. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- 28.Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012;9:151. doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51:979–984. doi: 10.3233/JAD-160152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armstrong RA. What causes Alzheimer’s disease? Folia Neuropathol. 2013;51:169–188. doi: 10.5114/fn.2013.37702. [DOI] [PubMed] [Google Scholar]
- 31.Du J, Wei N, Guan T, Xu H, An J, Pritchard KA, Shi Y. Inhibition of CDKS by roscovitine suppressed LPS-induced ·NO production through inhibiting NFκB activation and BH4 biosynthesis in macrophages. Am J Physiol Cell Physiol. 2009;297:C742–C749. doi: 10.1152/ajpcell.00138.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jhou RS, Sun KH, Sun GH, Wang HH, Chang CI, Huang HC, Lu SY, Tang SJ. Inhibition of cyclin-dependent kinases by olomoucine and roscovitine reduces lipopolysaccharide-induced inflammatory responses via down-regulation of nuclear factor κB. Cell Prolif. 2009;42:141–149. doi: 10.1111/j.1365-2184.2009.00584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Utreras E, Futatsugi A, Rudrabhatla P, Keller J, Iadarola MJ, Pant HC, Kulkarni AB. Tumor necrosis factor-α regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J Biol Chem. 2009;284:2275–2284. doi: 10.1074/jbc.M805052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Czapski GA, Gassowska M, Wilkaniec A, Chalimoniuk M, Strosznajder JB, Adamczyk A. The mechanisms regulating cyclin-dependent kinase 5 in hippocampus during systemic inflammatory response: the effect on inflammatory gene expression. Neurochem Int. 2016;93:103–112. doi: 10.1016/j.neuint.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Kitazawa M, Green KN, Caccamo A, LaFerla FM. Genetically augmenting Aβ42 levels in skeletal muscle exacerbates inclusion body myositis-like pathology and motor deficits in transgenic mice. Am J Pathol. 2006;168:1986–1997. doi: 10.2353/ajpath.2006.051232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leitch AE, Riley NA, Sheldrake TA, Festa M, Fox S, Duffin R, Haslett C, Rossi AG. The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur J Immunol. 2010;40:1127–1138. doi: 10.1002/eji.200939664. [DOI] [PubMed] [Google Scholar]
- 38.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 39.Cieslik M, Czapski GA, Strosznajder JB. The molecular mechanism of amyloid beta42 peptide toxicity: the role of sphingosine kinase-1 and mitochondrial sirtuins. PLoS One. 2015;10:e0137193. doi: 10.1371/journal.pone.0137193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ungureanu AA, Benilova I, Krylychkina O, Braeken D, De Strooper B, Van Haesendonck C, Dotti CG, Bartic C. Amyloid beta oligomers induce neuronal elasticity changes in age-dependent manner: a force spectroscopy study on living hippocampal neurons. Sci Rep. 2016;6:25841. doi: 10.1038/srep25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopes P, Xu M, Zhang M, Zhou T, Yang Y, Wang C, Ferapontova EE. Direct electrochemical and AFM detection of amyloid-beta peptide aggregation on basal plane HOPG. Nano. 2014;6:7853–7857. doi: 10.1039/c4nr02413c. [DOI] [PubMed] [Google Scholar]
- 42.Cakala M, Malik AR, Strosznajder JB. Inhibitor of cyclooxygenase-2 protects against amyloid beta peptide-evoked memory impairment in mice. Pharmacol Rep. 2007;59:164–172. [PubMed] [Google Scholar]
- 43.Wilkaniec A, Gassowska M, Czapski GA, Cieslik M, Sulkowski G, Adamczyk A. P2X7 receptor-pannexin 1 interaction mediates extracellular alpha-synuclein-induced ATP release in neuroblastoma SH-SY5Y cells. Purinergic Signal. 2017;13:347. doi: 10.1007/s11302-017-9567-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.James LR, Andrews S, Walker S, de Sousa PRS, Ray A, Russell NA, Bellamy TC. High-throughput analysis of calcium signalling kinetics in astrocytes stimulated with different neurotransmitters. PLoS One. 2011;6:e26889. doi: 10.1371/journal.pone.0026889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazmierczak A, Czapski GA, Adamczyk A, Gajkowska B, Strosznajder JB. A novel mechanism of non-Abeta component of Alzheimer’s disease amyloid (NAC) neurotoxicity. Interplay between p53 protein and cyclin-dependent kinase 5 (Cdk5) Neurochem Int. 2011;58:206–214. doi: 10.1016/j.neuint.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 46.Mitrasinovic OM, Perez GV, Zhao F, Lee YL, Poon C, Murphy GM., Jr Overexpression of macrophage colony-stimulating factor receptor on microglial cells induces an inflammatory response. J Biol Chem. 2001;276:30142–30149. doi: 10.1074/jbc.M104265200. [DOI] [PubMed] [Google Scholar]
- 47.Brooks G. Cyclin-dependent kinases and cyclin-dependent kinase inhibitors. Detection methods and activity measurements. Methods Mol Biol. 2001;124:161–170. doi: 10.1385/1-59259-059-4:161. [DOI] [PubMed] [Google Scholar]
- 48.Kovalevich J, Langford D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol Biol. 2013;1078:9–21. doi: 10.1007/978-1-62703-640-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H. An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res. 1992;31:616–621. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- 50.Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX. 2009;26:83–94. doi: 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- 51.Chagniel L, Robitaille C, Lebel M, Cyr M. Striatal inhibition of calpains prevents levodopa-induced neurochemical changes and abnormal involuntary movements in the hemiparkinsonian rat model. Neurobiol Dis. 2012;45:645–655. doi: 10.1016/j.nbd.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 52.Song H, Vita M, Sallam H, Tehranchi R, Nilsson C, Sidén Å, Hassan Z. Effect of the Cdk-inhibitor roscovitine on mouse hematopoietic progenitors in vivo and in vitro. Cancer Chemother Pharmacol. 2007;60:841–849. doi: 10.1007/s00280-007-0431-x. [DOI] [PubMed] [Google Scholar]
- 53.Hassan M, Sallam H, Hassan Z. The role of pharmacokinetics and pharmacodynamics in early drug development with reference to the cyclin-dependent kinase (Cdk) inhibitor—roscovitine. Sultan Qaboos Univ Med J. 2011;11:165–178. [PMC free article] [PubMed] [Google Scholar]
- 54.Menn B, Bach S, Blevins TL, Campbell M, Meijer L, Timsit S. Delayed treatment with systemic (S)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. PLoS One. 2010;5:e12117. doi: 10.1371/journal.pone.0012117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sallam H, Jimenez P, Song H, Vita M, Cedazo-Minguez A, Hassan M. Age-dependent pharmacokinetics and effect of roscovitine on Cdk5 and Erk1/2 in the rat brain. Pharmacol Res. 2008;58:32–37. doi: 10.1016/j.phrs.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 56.Vita M, Abdel-Rehim M, Olofsson S, Hassan Z, Meurling L, Sidén Å, Sidén M, Pettersson T, Hassan M. Tissue distribution, pharmacokinetics and identification of roscovitine metabolites in rat. Eur J Pharm Sci. 2005;25:91–103. doi: 10.1016/j.ejps.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 57.Ino H, Chiba T. Intracellular localization of cyclin-dependent kinase 5 (CDK5) in mouse neuron: CDK5 is located in both nucleus and cytoplasm. Brain Res. 1996;732:179–185. doi: 10.1016/0006-8993(96)00523-9. [DOI] [PubMed] [Google Scholar]
- 58.Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- 59.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 60.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 61.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 62.Armstrong RA. A critical analysis of the ‘amyloid cascade hypothesis’. Folia Neuropathol. 2014;52:211–225. [PubMed] [Google Scholar]
- 63.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 64.Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging. 2003;24:321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 65.Schwab C, Klegeris A, McGeer PL. Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim Biophys Acta. 1802;2010:889–902. doi: 10.1016/j.bbadis.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 66.Noble W, Hanger DP, Gallo JM. Transgenic mouse models of tauopathy in drug discovery. CNS Neurol Disord Drug Targets. 2010;9:403–428. doi: 10.2174/187152710791556131. [DOI] [PubMed] [Google Scholar]
- 67.Garwood CJ, Cooper JD, Hanger DP, Noble W. Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front Psychiatry. 2010;1:136. doi: 10.3389/fpsyt.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parachikova A, Vasilevko V, Cribbs DH, LaFerla FM, Green KN. Reductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylation. J Alzheimers Dis. 2010;21:527–542. doi: 10.3233/JAD-2010-100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Craft JM, Watterson DM, Van Eldik LJ. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia. 2006;53:484–490. doi: 10.1002/glia.20306. [DOI] [PubMed] [Google Scholar]
- 71.Scali C, Prosperi C, Giovannelli L, Bianchi L, Pepeu G, Casamenti F. Beta(1-40) amyloid peptide injection into the nucleus basalis of rats induces microglia reaction and enhances cortical gamma-aminobutyric acid release in vivo. Brain Res. 1999;831:319–321. doi: 10.1016/s0006-8993(99)01492-4. [DOI] [PubMed] [Google Scholar]
- 72.Song DK, Won MH, Jung JS, Lee JC, Kang TC, Suh HW, Huh SO, Paek SH, Kim YH, Kim SH, Suh YH. Behavioral and neuropathologic changes induced by central injection of carboxyl-terminal fragment of beta-amyloid precursor protein in mice. J Neurochem. 1998;71:875–878. doi: 10.1046/j.1471-4159.1998.71020875.x. [DOI] [PubMed] [Google Scholar]
- 73.Szczepanik AM, Ringheim GE. IL-10 and glucocorticoids inhibit Abeta(1-42)- and lipopolysaccharide-induced pro-inflammatory cytokine and chemokine induction in the central nervous system. J Alzheimers Dis. 2003;5:105–117. doi: 10.3233/jad-2003-5205. [DOI] [PubMed] [Google Scholar]
- 74.Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O'Hare E, Esler WP, Maggio JE, Mantyh PW. Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18:2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, Lynch MA. Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: a role for interleukin-1beta? J Biol Chem. 2003;278:27971–27980. doi: 10.1074/jbc.M302530200. [DOI] [PubMed] [Google Scholar]
- 76.Emre M, Geula C, Ransil BJ, Mesulam MM. The acute neurotoxicity and effects upon cholinergic axons of intracerebrally injected beta-amyloid in the rat brain. Neurobiol Aging. 1992;13:553–559. doi: 10.1016/0197-4580(92)90055-3. [DOI] [PubMed] [Google Scholar]
- 77.Kowall NW, Beal MF, Busciglio J, Duffy LK, Yankner BA. An in vivo model for the neurodegenerative effects of beta amyloid and protection by substance P. Proc Natl Acad Sci U S A. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.D'Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–683. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 80.Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of A[beta]-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167. doi: 10.1038/cddis.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, Novak M. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47. doi: 10.1186/1742-2094-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 83.Yang CN, Shiao YJ, Shie FS, Guo BS, Chen PH, Cho CY, Chen YJ, Huang FL, Tsay HJ. Mechanism mediating oligomeric Abeta clearance by naive primary microglia. Neurobiol Dis. 2011;42:221–230. doi: 10.1016/j.nbd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 84.Solito E, Sastre M. Microglia function in Alzheimer’s disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carty M, Bowie AG. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. 2011;81:825–837. doi: 10.1016/j.bcp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 87.Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.DeGiorgio LA, Shimizu Y, Chun HS, Kim YS, Sugama S, Son JH, Joh TH, Volpe BT. Amyloid precursor protein gene disruption attenuates degeneration of substantia nigra compacta neurons following axotomy. Brain Res. 2002;938:38–44. doi: 10.1016/s0006-8993(02)02483-6. [DOI] [PubMed] [Google Scholar]
- 89.Liu YY, Bian JS. Hydrogen sulfide protects amyloid-beta induced cell toxicity in microglia. J Alzheimers Dis. 2010;22:1189–1200. doi: 10.3233/JAD-2010-101002. [DOI] [PubMed] [Google Scholar]
- 90.Fleisher-Berkovich S, Filipovich-Rimon T, Ben-Shmuel S, Hulsmann C, Kummer MP, Heneka MT. Distinct modulation of microglial amyloid beta phagocytosis and migration by neuropeptides (i) J Neuroinflammation. 2010;7:61. doi: 10.1186/1742-2094-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ralay Ranaivo H, Craft JM, Hu W, Guo L, Wing LK, Van Eldik LJ, Watterson DM. Glia as a therapeutic target: selective suppression of human amyloid-beta-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci. 2006;26:662–670. doi: 10.1523/JNEUROSCI.4652-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR. Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem. 2005;92:226–234. doi: 10.1111/j.1471-4159.2004.02857.x. [DOI] [PubMed] [Google Scholar]
- 93.Zhao J, O'Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2011;8:150. doi: 10.1186/1742-2094-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 96.Tuppo EE, Arias HR. The role of inflammation in Alzheimer’s disease. Int J Biochem Cell Biol. 2005;37:289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 97.Qi Y, Zou LB, Wang LH, Jin G, Pan JJ, Chi TY, Ji XF. Xanthoceraside inhibits pro-inflammatory cytokine expression in Abeta25-35/IFN-gamma-stimulated microglia through the TLR2 receptor, MyD88, nuclear factor-kappaB, and mitogen-activated protein kinase signaling pathways. J Pharmacol Sci. 2013;122:305–317. doi: 10.1254/jphs.13031fp. [DOI] [PubMed] [Google Scholar]
- 98.Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64:300–316. doi: 10.1002/glia.22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Muyllaert D, Terwel D, Kremer A, Sennvik K, Borghgraef P, Devijver H, Dewachter I, Van Leuven F. Neurodegeneration and neuroinflammation in cdk5/p25-inducible mice : a model for hippocampal sclerosis and neocortical degeneration. Am J Pathol. 2008;172:470–485. doi: 10.2353/ajpath.2008.070693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295:245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 102.Sundaram JR, Chan ES, Poore CP, Pareek TK, Cheong WF, Shui G, Tang N, Low CM, Wenk MR, Kesavapany S. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J Neurosci. 2012;32:1020–1034. doi: 10.1523/JNEUROSCI.5177-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takahashi S, Ohshima T, Hirasawa M, Pareek TK, Bugge TH, Morozov A, Fujieda K, Brady RO, Kulkarni AB. Conditional deletion of neuronal cyclin-dependent kinase 5 in developing forebrain results in microglial activation and neurodegeneration. Am J Pathol. 2010;176:320–329. doi: 10.2353/ajpath.2010.081158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Czapski GA, Gassowska M, Wilkaniec A, Cieslik M, Adamczyk A. Extracellular alpha-synuclein induces calpain-dependent overactivation of cyclin-dependent kinase 5 in vitro. FEBS Lett. 2013;587:3135–3141. doi: 10.1016/j.febslet.2013.07.053. [DOI] [PubMed] [Google Scholar]
- 105.Czapski GA, Gassowska M, Songin M, Radecka UD, Strosznajder JB. Alterations of cyclin dependent kinase 5 expression and phosphorylation in amyloid precursor protein (APP)-transfected PC12 cells. FEBS Lett. 2011;585:1243–1248. doi: 10.1016/j.febslet.2011.03.058. [DOI] [PubMed] [Google Scholar]
- 106.Engmann O, Giese KP. Crosstalk between Cdk5 and GSK3beta: implications for Alzheimer’s disease. Front Mol Neurosci. 2009;2:2. doi: 10.3389/neuro.02.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu Y, Li T, Qureshi HY, Han D, Paudel HK. Early growth response 1 (Egr-1) regulates phosphorylation of microtubule-associated protein tau in mammalian brain. J Biol Chem. 2011;286:20569–20581. doi: 10.1074/jbc.M111.220962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alvarez A, Muñoz JP, Maccioni RB. A Cdk5–p35 stable complex is involved in the β-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res. 2001;264:266–74. [DOI] [PubMed]
- 109.Sun KH, Lee HG, Smith MA and Shah K. Direct and indirect roles of cyclin-dependent kinase 5 as an upstream regulator in the c-Jun NH2-terminal kinase cascade: relevance to neurotoxic insults in Alzheimer's disease. Mol Biol Cell. 2009;20:4611–19. [DOI] [PMC free article] [PubMed]
- 110.Hernandez P, Lee G, Sjoberg M, Maccioni RB. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): involvement of lipid rafts. J Alzheimers Dis. 2009;16:149–156. doi: 10.3233/JAD-2009-0933. [DOI] [PubMed] [Google Scholar]
- 111.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 112.Tang D, Chun AC, Zhang M, Wang JH. Cyclin-dependent kinase 5 (Cdk5) activation domain of neuronal Cdk5 activator. Evidence of the existence of cyclin fold in neuronal Cdk5a activator. J Biol Chem. 1997;272:12318–12327. doi: 10.1074/jbc.272.19.12318. [DOI] [PubMed] [Google Scholar]
- 113.Brinkkoetter PT, Olivier P, Wu JS, Henderson S, Krofft RD, Pippin JW, Hockenbery D, Roberts JM, Shankland SJ. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J Clin Invest. 2009;119:3089–3101. doi: 10.1172/JCI37978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu C, Zhai X, Zhao B, Wang Y, Xu Z. Cyclin I-like (CCNI2) is a cyclin-dependent kinase 5 (CDK5) activator and is involved in cell cycle regulation. Sci Rep. 2017;7:40979. doi: 10.1038/srep40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S, Quadros IM, Newman J, et al. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell. 2011;21:655–668. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell. 2014;13:379–390. doi: 10.1111/acel.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 118.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 119.Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8:1254–1266. doi: 10.7150/ijbs.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scheller J, Garbers C, Rose-John S. Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin Immunol. 2014;26:2–12. doi: 10.1016/j.smim.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 121.Tobinick E. Tumour necrosis factor modulation for treatment of Alzheimer’s disease: rationale and current evidence. CNS Drugs. 2009;23:713–725. doi: 10.2165/11310810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 122.Shin J-W, Cheong Y-J, Koo Y-M, Kim S, Noh C-K, Son Y-H, Kang C, Sohn N-W. α-Asarone ameliorates memory deficit in lipopolysaccharide-treated mice via suppression of pro-inflammatory cytokines and microglial activation. Biomol Ther. 2014;22:17–26. doi: 10.4062/biomolther.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ringheim GE, Szczepanik AM, Petko W, Burgher KL, Zhu SZ, Chao CC. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res Mol Brain Res. 1998;55:35–44. doi: 10.1016/s0169-328x(97)00356-2. [DOI] [PubMed] [Google Scholar]
- 124.Brugg B, Dubreuil YL, Huber G, Wollman EE, Delhaye-Bouchaud N, Mariani J. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc Natl Acad Sci U S A. 1995;92:3032–3035. doi: 10.1073/pnas.92.7.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qiu Z, Gruol DL. Interleukin-6, beta-amyloid peptide and NMDA interactions in rat cortical neurons. J Neuroimmunol. 2003;139:51–57. doi: 10.1016/s0165-5728(03)00158-9. [DOI] [PubMed] [Google Scholar]
- 126.Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Zubair AC, Dickson D, Golde TE, Das P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24:548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Post-transcriptional regulation of IL-10 gene expression allows NK cells to express immunoregulatory function. Immunity. 2008;29:295–305. doi: 10.1016/j.immuni.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Teixeira-Coelho M, Guedes J, Ferreirinha P, Howes A, Pedrosa J, Rodrigues F, Lai WS, Blackshear PJ, O'Garra A, Castro AG, Saraiva M. Differential post-transcriptional regulation of IL-10 by TLR2 and TLR4-activated macrophages. Eur J Immunol. 2014;44:856–866. doi: 10.1002/eji.201343734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, et al. TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 130.Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate a beta(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J Neuroimmunol. 2001;113:49–62. doi: 10.1016/s0165-5728(00)00404-5. [DOI] [PubMed] [Google Scholar]
- 131.Ledeboer A, Breve JJ, Wierinckx A, van der Jagt S, Bristow AF, Leysen JE, Tilders FJ, Van Dam AM. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur J Neurosci. 2002;16:1175–1185. doi: 10.1046/j.1460-9568.2002.02200.x. [DOI] [PubMed] [Google Scholar]
- 132.Pousset F, Cremona S, Dantzer R, Kelley K, Parnet P. Interleukin-4 and interleukin-10 regulate IL1-beta induced mouse primary astrocyte activation: a comparative study. Glia. 1999;26:12–21. doi: 10.1002/(sici)1098-1136(199903)26:1<12::aid-glia2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 133.Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85:519–533. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther. 2012;19:724–733. doi: 10.1038/gt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J, Jr, Leung BP, Rezai-Zadeh K, Town T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85:534–548. doi: 10.1016/j.neuron.2014.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat Inflamm. 2013;2013:20. doi: 10.1155/2013/480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vezzani A, Ravizza T, Balosso S, Aronica E. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49:24–32. doi: 10.1111/j.1528-1167.2008.01490.x. [DOI] [PubMed] [Google Scholar]
- 138.Zhang Z, Lee Y-C, Kim S-J, Choi MS, Tsai P-C, Saha A, Wei H, Xu Y, Xiao Y-J, Zhang P, et al. Production of lysophosphatidylcholine by cPLA2 in the brain of mice lacking PPT1 is a signal for phagocyte infiltration. Hum Mol Genet. 2007;16:837–847. doi: 10.1093/hmg/ddm029. [DOI] [PubMed] [Google Scholar]
- 139.Ousman SS, David S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia. 2000;30:92–104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.